
Abstract
Background
ASP8232 is a potent and specific small molecule vascular adhesion protein-1 (VAP-1) inhibitor. This study evaluated the effect of ASP8232 on excess retinal thickness when given alone or in combination with ranibizumab in patients with center-involved diabetic macular edema (CI-DME).
Methods
This was a phase 2a, placebo and sham-injection controlled, double-masked, randomized, parallel-group clinical trial. Participants were patients with CI-DME and central subfield thickness (CST) ≥ 375 µm in the study eye as assessed by spectral domain optical coherence tomography. Eligible patients were randomized to (1) daily oral ASP8232 40 mg monotherapy; (2) combination therapy of daily oral ASP8232 40 mg and monthly intravitreal ranibizumab 0.3 mg; or (3) monthly intravitreal ranibizumab 0.3 mg monotherapy. The treatment period was 12 weeks. CST and best corrected visual acuity (BCVA) were assessed at baseline and at Weeks 2, 4, 8, 12, 16 and 24. The primary outcome was the mean percent change from baseline in excess CST at Week 12. Secondary outcomes were BCVA, safety and tolerability, and pharmacokinetic and pharmacodynamic characteristics of ASP8232.
Results
After 12 weeks, the mean (95% confidence interval) percent change in excess CST was 11.4% (− 15.0%, 37.8%) in the ASP8232 group, − 61.7% (− 86.1%, − 37.2%) in the ASP8232/ranibizumab group, and − 75.3% (− 94.8%, − 55.8%) in the ranibizumab group. The change from baseline in the two ranibizumab arms was statistically significant (P < 0.001) as was the difference between the ranibizumab groups and the ASP8232 group (P < 0.001). Mean (SD) increase in BCVA score from baseline was 3.1 (7.3) in the ASP8232 group, 5.2 (7.1) in the ASP8232/ranibizumab group, and 8.2 (9.5) in the ranibizumab group. The increase from baseline in BCVA score was statistically and clinically significant in the ranibizumab group compared with the ASP8232 group (P = 0.015). ASP8232 resulted in near complete inhibition of plasma VAP-1 activity whilst ranibizumab had no effect.
Conclusions
Near complete inhibition of plasma VAP-1 activity with ASP8232 had no effect on CST in patients with CI-DME. Furthermore, combination therapy did not provide additional benefit to treatment with ranibizumab alone, which significantly reduced CST and improved BCVA.
Trial registration clinicaltrials.gov; NCT02302079. Registered on November 26, 2014
Similar content being viewed by others


Background
Diabetic retinopathy (DR) is a common complication of diabetes mellitus that leads to loss of vision and blindness among working age adults [1,2,3]. During progression of DR, patients can develop diabetic macular edema (DME), which is characterized by the thickening of the macula caused by the breakdown of the blood-retinal barrier and consequent retinal vascular hyperpermeability [3]. In 2010, the global prevalence of DR among adults with diabetes mellitus aged 20–79 years was estimated to be 34.6% for any DR and 6.81% for DME [4]. DME is the leading cause of vision loss among patients with DR. It is associated with the type of diabetes, and increases with the duration and severity of disease [5, 6]. Other significant risk factors common to DR and DME include hyperglycemia and hypertension [4]. DME negatively impacts patients’ health-related quality of life and represents an economic burden due to the increased use of healthcare resources by affected patients [7, 8].
While there is no curative treatment available for DME, laser photocoagulation represents an effective treatment to preserve vision. However, this treatment modality is limited by its inability to restore vision once it has been lost [9]. The current standard of care for DME includes intravitreal anti-vascular endothelial growth factor (VEGF) antibodies and corticosteroids [9]. Clinical studies have confirmed that monthly intravitreal treatment with the anti-VEGF antibody ranibizumab can improve vision, with up to 45% of patients gaining ≥ 15 letters in best-corrected visual acuity (BCVA) after 24 months [10, 11]. Similar improvements were found after treatment with the anti-VEGF antibodies aflibercept [12,13,14] and with bevacizumab, an approved treatment for colon cancer that is commonly used off label for DME [9, 15]. Despite the proven efficacy of VEGF inhibitors, the requirement of frequent injections causes a high rate of treatment discontinuation among patients with DME and represents a major limitation [16]. Moreover, the presence of potential side effects and the significant proportion of patients who do not respond to treatment [10, 12] suggest that there remains a need for the development of improved therapies for DME.
Vascular adhesion protein-1 (VAP-1) belongs to the family of copper-containing amine oxidase/semicarbazide-sensitive amine oxidases that catalyze the oxidative deamination of primary amines with subsequent production of aldehyde, ammonium, and hydrogen peroxide, which are involved in oxidative stress and are cytotoxic. VAP-1 is expressed in vascular endothelium (including renal and retinal capillaries), smooth muscle cells, hepatic stromal cells and adipocytes [17, 18]. The enzymatic action stimulates leukocyte trafficking to the interstitium and therefore has a pro-inflammatory action [17, 18]. After inflammatory stimulus, a soluble form of VAP-1, which retains its enzymatic activity, is released into circulation from the endothelial cells. Elevated levels of soluble VAP-1 have been found in the serum of diabetic patients [19] and in the vitreous fluid of patients with proliferative DR (PDR) compared with non-PDR patients [20, 21]. Recently, it was shown that VEGF induces soluble VAP-1 release in retinal capillaries and thereby induces increased production of reactive oxygen species [22]. In experimental models of uveitis and DR in rats, VAP-1 was suggested to play a significant role in leucocyte recruitment into retinal endothelium [23,24,25]. Such evidence suggests that inhibition of VAP-1 may represent a novel therapeutic strategy for DR and DME.
ASP8232 is a potent and specific small molecule VAP-1 inhibitor. In a streptozocin-induced rat model of DME, ASP8232 inhibited plasma VAP-1 activity and improved retinal hyperpermeability and plasma total antioxidant status. In combination with intravitreal anti-rat VEGF antibody, ASP8232 was more effective in reducing ocular hyperpermeability compared with either the anti-ratVEGF antibody or ASP8232 alone (data on file).
In multiple pharmacology and safety/toxicity experimental studies, ASP8232 had low acute toxicity and showed no effect on central nervous system, cardiovascular, and respiratory functions or fertility and early embryonic development, and had no genotoxic or teratogenic potential. ASP8232 was tested in two phase 1 studies in healthy subjects, patients with renal impairment, and patients with type 2 diabetes mellitus. ASP8232 was administered in doses up to 6000 mg as single dose and up to 800 mg as multiple doses in healthy subjects. In subjects with renal impairment and diabetes, ASP8232 was administered in a dose of 150 mg for 4 weeks (unpublished data from first-in-man study, NCT02218099). ASP8232 was safe and well-tolerated, and pharmacokinetic and pharmacodynamic modelling and simulations suggested that a daily dose of 40 mg would be safe and would deliver complete inhibition of VAP-1.
The VIDI study (VAP-1 Inhibition in DME) was a phase 2 study designed to evaluate ASP8232 safety and effect on excess retinal thickness when given alone or in combination with the anti-VEGF agent, ranibizumab, in patients with CI-DME.
Methods
Study design
The VIDI study was a proof of concept, phase 2a, randomized, placebo capsule and sham-intravitreal injection controlled, double-masked clinical trial (www.clinicaltrials.gov; NCT02302079) conducted at 21 centers in the US from 12 Jan 2015 to 12 Aug 2016.
Institutional Review Board (IRB)/Ethics Committee approval was obtained and the study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and the Health Insurance Portability and Accountability Act; all patients provided informed consent. An independent academic reading center, the Ocular Imaging Research and Reading Center (OIRRC, Sunnyvale, California), served as the Reading Center for the VIDI study; the OIRRC received, stored, processed, and graded all images from the study.
The study was divided into 3 periods: a screening period of 1–4 weeks; a treatment period of 12 weeks, and a follow-up period of 12 weeks. Patients were randomized (1:1:1) to one of three treatment groups: (1) ASP8232 monotherapy: oral ASP8232 40 mg once daily + sham intravitreal injections once per month; (2) combination therapy of ASP8232 and ranibizumab: oral ASP8232 40 mg once daily + ranibizumab (0.3 mg) intravitreal injections once per month; and (3) ranibizumab monotherapy: oral ASP8232-matched placebo once daily + ranibizumab (0.3 mg) intravitreal injections once per month.
During the treatment period, study participants received ASP8232 (40 mg) or placebo once daily from Day 1–84 and an intravitreal injection of ranibizumab or sham on Days 1, 29, and 57. The dose of 40 mg ASP8232 was selected based on previous studies that showed that this dose was safe and was predicted to achieve maximal VAP-1 inhibition over a 24-h period with once daily dosing. All study participants and site staff were masked to all treatments, except for the injecting ophthalmologist, who was not involved in any other study assessments at the study site.
Participants
Patients were eligible if they were aged 18–85 years with type 1 or type 2 diabetes mellitus with the following specific characteristics: glycated hemoglobin (HbA1c) ≤ 12%; central subfield thickness (CST) ≥ 375 µm in the study eye; early treatment diabetic retinopathy study (ETDRS) BCVA letter score ≤ 73 (Snellen 20/40) and ≥ 24 (Snellen 20/320) in the study eye. Patients with any of the following characteristics were excluded from study participation: macular edema or decrease in BCVA due to a cause other than DME; presence of any other ocular disease in the study eye that may have caused substantial reduction in BCVA; significant macular ischemia or other retinal inflammatory or active periocular or ocular infection; history of noninfectious uveitis, high myopia (− 8 diopter or more correction), pars plana vitrectomy, any ocular surgery, YAG (yttrium–aluminum-garnet) capsulotomy, panretinal scatter photocoagulation, or focal laser within 3 months of study enrollment; history of intravitreal, subtenon, or periocular, non-sustained release, steroid therapy within 3 months before the study; history of intravitreal sustained release dexamethasone therapy within 6 months of study enrollment; history of intravitreal sustained release fluocinolone within 3 years of study enrollment or history of prior treatment with intravitreal VEGF treatment within 8 weeks of study enrollment.
Study drug
ASP8232 was provided as 40 mg capsules and ASP8232-matched placebo was provided as matching capsules of microcrystalline cellulose. The capsules were taken in the morning with or without food. Ranibizumab was administered as intravitreal injections of 0.05 mL of a 6 mg/mL ranibizumab aqueous solution containing 10 mM histidine HCl, 10% α,α-trehalose dihydrate, 0.01% polysorbate 20 with pH 5.5. Sham intravitreal for ranibizumab was supplied as an empty vial. An anesthetic was administered in the study eye before each intravitreal injection (including the sham intravitreal injection). The sham injection consisted of the (unmasked) injecting ophthalmologist pressing an empty syringe against the surface of the eye to mimic an intravitreal injection. Ranibizumab was injected intravitreally according to the instructions of the manufacturer. Rescue therapy could be considered, under the discretion of the evaluating investigator, if a patient experienced a decrease in ETDRS-BCVA of ≥ 15 letters from baseline or a decrease in ETDRS-BCVA of ≥ 10 letters and an increase in CST of ≥ 75 µm. Rescue therapy in the study eye could only be considered after completion of the assessments at Week 4 and was managed by the injecting ophthalmologist. Patients who received rescue therapy with laser photocoagulation were permitted to continue the study; patients who required any other rescue therapy were discontinued from the study treatment. The randomization list and study medication mask were generated and maintained by an interactive website response system.
Assessments
Both spectral domain-optical coherence tomography (SD-OCT) and ETDRS-BCVA were evaluated at screening, at all visits (Days 1, 15, 29, 57, 85) during the treatment period, during follow-up (Day 113), and at the end of study visit (Day 169). CST was assessed by the Heidelberg Spectralis (Heidelberg Engineering, Heidelberg, Germany) SD-OCT machine. BCVA was assessed according to the ETDRS protocol and all assessments were performed by trained assessors. Fluorescein angiography (FA) and fundus photography were conducted at screening, and on Days 85 and 169. All FA, fundus photography, and SD-OCT images were sent to an independent academic centralized reading center, the OIRRC, for grading and analyses conducted by masked graders. Samples for the assessment of ASP8232 concentrations in plasma were collected during visits on Days 15, 29, 57 prior to oral dosing (approximately 24 h after the previous day dose), and on Days 85, 113, and 169 (approximately 24 h, 4 weeks, and 12 weeks post-dose). Samples for the assessment of ASP8232 concentrations in aqueous humor from the anterior chamber of the study eye were collected on Days 1, 28, and 85. Concentrations of ASP8232 were measured using a validated liquid chromatography-tandem mass spectrometry method. Samples for VAP-1 concentrations and VAP-1 activity were collected during each visit.
Outcome parameters
The primary efficacy endpoint was the percent change from baseline (%CFB) to Week 12 [end of treatment (EoT)] in excess CST in the study eye, as assessed by SD-OCT. Excess CST was defined as the difference between measured CST and the predefined “normal” value of 320 μm [26, 27]. Secondary endpoints included the absolute change from baseline in CST in the study eye at each visit, and the change from baseline in ETDRS-BCVA score in the study eye at each visit. Pharmacokinetic and pharmacodynamic end points included ASP8232 concentrations in plasma and anterior chamber aqueous humor and VAP-1 concentration and activity in plasma and in aqueous humor. Safety and tolerability of ASP8232 was assessed by monitoring the nature, frequency, and severity of systemic and ocular treatment-emergent adverse events (TEAEs), as well as vital signs, clinical laboratory assessments, and electrocardiograms (ECGs).
Statistical methods
The VIDI study was designed to reject that the %CFB in excess CST is ≤ 10%, assuming a true %CFB in excess CST of 40% with a standard deviation of 65%. Assuming a drop-out rate of 15%, the total number of patients to be randomized to have an 80% power was planned to be 84 (28 per group). The efficacy analyses were conducted using the full analysis set (FAS), which included all randomised participants who received at least one dose of the study drug and had an efficacy measurement at baseline and at least one at post baseline. The safety analysis set included all participants who received at least one dose of study drug and was used for the demographics and baseline characteristics and also the safety analyses. The pharmacokinetic analysis set included participants from the safety analysis set population for whom sufficient plasma concentration data were collected (i.e. at least one plasma concentration with a recorded dosing time prior to the sample collection). All data analyses were conducted using Statistical Analysis Software® version 9.3 on UNIX.
Efficacy analysis was performed on %CFB in excess CST and change from baseline in ETDRS-BCVA score at 12 weeks. The hypothesis test on the %CFB in excess CST was based on the upper bound of a 2-sided 80% confidence interval (CI) constructed using the t-distribution. If this upper bound was lower than − 10%, then the null hypothesis of no significant effect was rejected, and the correspondent alternative hypothesis was accepted. In addition, a secondary analysis was performed using a mixed model for repeated measures including treatment group, visit and treatment group by visit interaction, as fixed class factors, and baseline excess CST as continuous covariate. Observations after treatment discontinuation were excluded from the primary analysis but reported separately as part of the post-treatment follow-up period.
Results
Patient disposition and baseline characteristics
All study patients were consecutively screened and randomized across the participating sites. Of 240 patients who signed informed consent, 96 were randomized to receive ASP8232 monotherapy (N = 32), ASP8232/ranibizumab combination therapy (N = 33), or ranibizumab monotherapy (N = 31). A total of 29 patients (90.6%) in the ASP8232 group, 31 patients (93.9%) in the ASP8232/ranibizumab group and 27 patients (87.1%) in the ranibizumab group completed the study. All 96 randomized patients were included in the safety analysis set; 95 patients were included in the FAS (ASP8232 group, n = 32; ASP8232/ranibizumab group, n = 32; ranibizumab group, n = 31). Table 1 and Additional file 1: Table S1 summarize the patient demographics and baseline characteristics.
Overall, 15.6% and 18.8% of patients in the safety analysis set received previous and concomitant eye medications to the study eye, respectively. Treatment groups were well-balanced with respect to clinically important medical history, and the median treatment duration was similar across treatment groups and ranged between 84.0–84.5 days. A minority of patients also had a fellow eye enrolled (ASP8232 group, n = 7; ASP8232/ranibizumab group, n = 3; ranibizumab group, n = 4). The mean absolute CST values at baseline in the FAS were 535.8 µm, 511.8 µm, and 501.6 µm in the ASP8232, ASP8232/ranibizumab and ranibizumab groups, respectively. The mean ETDRS-BCVA scores at baseline were 59.0, 59.8 and 57.7, respectively.
Primary endpoint: percent change from baseline in excess CST
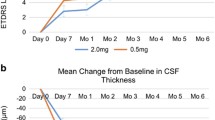
In the ASP8232 group, the mean %CFB in excess CST to Week 12/EoT in the study eye was 11.4% (95% CI, − 15.0%, 37.8%; P = 0.108), and therefore the study did not meet the primary endpoint. The %CFB in the ranibizumab group was − 75.3% (95% CI, − 94.8%, − 55.8%; P < 0.001) after 12 weeks. The mean %CFB in excess CST in the study eye in the ASP8232/ranibizumab group showed a somewhat lesser treatment response compared with the ranibizumab group [− 61.7% (95% CI, − 86.1%, − 37.2%); P < 0.001] (Fig. 1). ANCOVA analysis of the %CFB in excess CST at Week 12/EoT showed no significant difference between the ASP8232/ranibizumab and the ranibizumab group; however, both groups showed a significant difference compared with the ASP8232 group (P < 0.001). In the fellow eye, the %CFB was not statistically significant in any of the 3 treatment groups (data not shown).

Percent change (95% CI) from baseline in excess CST at EoT visit (FAS). CI, confidence interval; CST, central subfield thickness; EoT, end of treatment; FAS, full analysis set
Change in absolute CST
The mean (SD) absolute CST change from baseline was − 123.1(112.3) μm in the ranibizumab group after 12 weeks of treatment, whereas no change was observed in the ASP8232 group. The change from baseline to Week 12 in the ASP8232/ranibizumab group was comparable with the ranibizumab group (Table 2).
CST decreased quickly after the first injection of ranibizumab in the study eye in both ranibizumab and ASP8232/ranibizumab groups, whereas no changes were observed in the ASP8232 treated eyes at any visit. CST values remained stable for up to 12 weeks after the last injection in the ranibizumab group, whereas values tended to return to baseline levels in the ASP8232/ranibizumab group (Fig. 2, Table 3).

Mean (95% CI) change from baseline in absolute CST values in the study eye (FAS). CI, confidence interval; CST, central subfield thickness; FAS, full analysis set; LDD, last dose date; LOCF, last observation carried forward
The mean change from baseline in absolute CST values in the qualified fellow eye at Week 12/EoT was − 82 μm, − 15.3 μm and − 55 μm in the ASP8232, ASP8232/ranibizumab, and ranibizumab groups, respectively (Fig. 3).

Mean (95% CI) change from baseline in absolute CST values in the fellow eye (FAS). CI, confidence interval; CST, central subfield thickness; FAS, full analysis set; LDD, last dose date; LOCF, last observation carried forward
Effect on ETDRS-BCVA
The mean (SD) change from baseline in ETDRS-BCVA score to Week 12/EoT was 3.1 (7.3) in the ASP8232 group, 5.2 (7.1) in the ASP8232/ranibizumab group, and 8.2 (9.5) in the ranibizumab group. The change in ETDRS-BCVA score was significantly higher in the ranibizumab group than in the ASP8232 group (P = 0.015) (Fig. 4). In the ranibizumab treated eyes, ETDRS-BCVA showed an increase by Week 2 and then continued to improve until week 12/EoT. In the ASP8232 group a small increase in ETDRS-BCVA was observed at week 2, and then ETDRS-BCVA remained at the same level in the ASP8232 group. During the follow-up period, ETDRS-BCVA remained stable until 12 weeks after the last injection in the ranibizumab group and returned to baseline levels in the two remaining treatment groups. At week 2, the number of patients with 5-, 10-, and 15-letter gain in the ETDRS-BCVA was highest in the ranibizumab groups and at week 12, the percentage of patients with ≥ 5-letter gain in ETDRS-BCVA was 48.1%, 64.5%, 69.0% in the ASP8232, ASP8232/ranibizumab, and ranibizumab group, respectively.

Mean (95% CI) change from baseline in ETDRS calculated BCVA score (FAS). FAS, full analysis set; LDD, last dose date; LOCF, last observation carried forward
Pharmacokinetic and pharmacodynamic end points
Median trough plasma levels of ASP8232 ranged from 24.1 to 43.10 ng/mL during the treatment period. ASP8232 was still measurable in plasma 4 and 12 weeks after the last dose (Fig. 5). The median ASP8232 levels in the aqueous humor at 12 weeks was 2.98 ng/ml ranging from 0 to 31.1 ng/ml. The median plasma VAP-1 protein levels did not change throughout the study across treatment groups and ranged from 443 to 569 ng/ml. The median VAP-1 protein levels in aqueous humor did not change significantly from baseline to week 12 in any of the 3 treatment groups and ranged from 0.72 to 1.21 ng/ml across treatment groups and time points. In the ASP8232 groups, plasma VAP-1 activity was nearly completely inhibited throughout the treatment period, with median inhibition from baseline to week 12 ranging from 96.8% in the ASP8232/ranibizumab group to 97.3% in the ASP8232 group, indicating that ASP8232 fully inhibits VAP-1 with or without ranibizumab. VAP-1 activity was ~ 50% of baseline within 4 weeks after the last dose of ASP8232 and remained between 70% and 85% of the baseline value within 12 weeks after the last dose. Plasma VAP-1 activity did not change in the ranibizumab group (Fig. 6). In the aqueous humor, the median inhibition of VAP-1 activity was 53.2% in the ASP8232 group compared with 35.4% and 27.9% in the ASP8232/ranibizumab and ranibizumab groups, respectively.

Mean plasma concentration of ASP8232 (pharmacokinetic analysis set). LDD, last dose date

Mean (SD) VAP-1 activity in plasma (pharmacodynamic analysis set). LDD, last dose date
Safety results
The incidence of overall and ocular TEAE was similar across treatment groups, and most were mild-to-moderate in severity (Table 4; Additional file 2: Table S2). The most common TEAEs were conjunctival hemorrhage [ASP8232/ranibizumab, n = 3 (9.1%)], worsening of type 2 diabetes mellitus [ASP8232, n = 2 (6.3%); ASP8232/ranibizumab, n = 3 (9.1%)] and diabetic retinal edema [ASP8232, n = 3 (9.4%); ASP8232/ranibizumab, n = 1 (3.0%); ranibizumab, n = 2 (6.5%)] (Table 4). A small number of patients reported drug-related TEAE [ASP8232, n = 2 (6.3%); ASP8232/ranibizumab, n = 3 (9.1%); and ranibizumab, n = 3 (9.7%)]. Drug-related ocular TEAEs included retinal disorder [ASP8232, n = 1 (3.1%); ASP8232/ranibizumab, n = 1 (3.0)], metamorphopsia [ranibizumab, n = 1 (3.2%)] and photophobia [ranibizumab, n = 1 (3.2%)]. The number of patients reporting serious TEAEs was generally low [ASP8232 n = 3 (9.4%); ASP8232/ranibizumab, n = 1 (3.0%); and ranibizumab, n = 3 (9.7%)] and none of these events were considered to be related to the study drug.
Three patients experienced TEAEs leading to discontinuation of the study drug. In the ASP8232 group, one patient experienced progression of DME that required rescue treatment and was considered possibly drug-related, and another discontinued due to a diagnosis of prostate cancer diagnosed 1 month after randomization, which was considered not related to the study drug. In the ASP8232/ranibizumab group, one patient discontinued due to progression of metastatic malignant melanoma that had been diagnosed 3 years prior to the start of the trial. The patient had been in remission at enrollment and the condition progressed during the study; this TEAE was not considered to be related to the study drug. No clinically significant differences were observed in the change from baseline for clinical laboratory assessments, vital signs, and ECGs between treatment groups, and no deaths occurred during the study. Intraocular pressure was assessed as a safety measure across the treatment arms. Median levels were comparable across treatment groups and time points and ranged from 14 to 17 mmHg with no significant changes (> 10 mmHg after baseline) reported.
Discussion
VAP-1 is an endothelial adhesion molecule with semicarbazide-sensitive amine oxidase activity that is expressed in the endothelial cells of the retinal vessels [19]. During inflammation, VAP-1 acts with other leukocyte adhesion molecules to recruit inflammatory cells [19]. Since the VAP-1 enzymatic activity results in the production of toxic metabolites including hydrogen peroxide and aldehydes, which are involved in cellular oxidative stress, it has been postulated that increased levels of VAP-1 may contribute to the development of DR.
The VIDI study assessed the clinical efficacy of the specific VAP-1 inhibitor ASP8232 on excess retinal thickness when administered alone or in combination with the anti-VEGF drug ranibizumab in patients with CI-DME. After 12 weeks of treatment, a statistically significant decrease from baseline in mean excess CST of 75.3% was observed in the ranibizumab group whilst no change was observed in the ASP8232 group. The effect in the combination treatment group was numerically less (but not statistically significant) compared with ranibizumab alone and ASP8232 did not provide any additional benefits on CI-DME. Similarly, changes from baseline in absolute CST of 123.1 μm and 137.8 μm were observed in the ranibizumab and ASP8232/ranibizumab group, respectively, whereas no change was observed in the ASP8232 group.
After 12 weeks of treatment, the mean ETDRS-BCVA score in the study eye increased from baseline by 3.1, 5.2, and 8.2 letters in the ASP8232, ASP8232/ranibizumab, and ranibizumab groups, respectively. These data confirm results from previous studies of ranibizumab and demonstrated that treatment with ranibizumab is effective in improving ETDRS-BCVA in DME. ASP8232 alone provided limited improvement in ETDRS-BCVA of only three letters, which is not considered to be of clinical relevance. Interestingly, the outcomes in the combination group were less favorable compared with the ranibizumab group, which confirms that ASP8232 does not provide relevant improvement in ETDRS-BCVA. The minor effects in the qualified fellow eye suggest some effect on CST but because of the very low number of eyes, these data should be interpreted with caution.
In the ASP8232 groups, VAP-1 activity in plasma was greatly inhibited throughout the treatment period and remained below the baseline value within 12 weeks after the last dose, whereas ranibizumab had no effect on VAP-1 activity, as expected. This confirms that the dose of 40 mg ASP8232 once daily was sufficient to completely inhibit VAP-1 in plasma. VAP-1 protein levels and activity in the aqueous humor were much lower than plasma VAP-1 protein levels and activity; the median concentration of ASP8232 in the aqueous humor was only about 11.5% of plasma trough levels.
It is not known whether VAP-1 plays a role in DR at the level of the retinal vasculature or at the vitreo-retinal interface. We measured VAP-1 levels and activity in the anterior chamber as a possible representation of the activity in the vitreous. However, in this study, VAP-1 activity in the aqueous humor was below the detection limits at all points. Therefore, we cannot draw any conclusions regarding its activity.
Although the VIDI Study did not meet its primary endpoint, we have demonstrated that VAP-1 activity can be inhibited with ASP8232. Recently, VAP-1 inhibition demonstrated a significant benefit in diabetic nephropathy, an end-organ diabetic complication of diabetes (manuscript accepted). Therefore, it would not be appropriate to disregard the VAP-1 pathway in diabetic macular edema and/or diabetic retinopathy based solely on the VIDI study results. Further studies evaluating different modes of delivery and/or concentrations of VAP-1 inhibition for eye diseases such as DME and DR may be warranted to further elucidate this potentially important target of pathophysiology.
Other studies evaluating combination treatment have yielded similar results, suggesting that VEGF is the major driver in the pathophysiology of DME [28]. Another VAP-1 inhibitor is currently being investigated in patients with DR in the absence of center-involved macular edema (www.clinicaltrials.gov; NCT03238963). It is conceivable that the presence of significant CI-DME requires inhibition of VEGF, whilst DR could be reduced by VAP-1 inhibition alone.
Our study had several strengths. Patients were randomly assigned to treatments, and all staff at the investigative sites and the sponsor were masked to treatment allocation. A trained and experienced injecting ophthalmologist performed all intravitreal and intravitreal sham injections and had no other involvement in the trial. Standardized and state-of-the-art measures of efficacy and safety were assessed, and study staff were trained before activation of the site. All images were reviewed and assessed by an independent and experienced reading center and formalized reading protocols were utilized. One potential limitation of our study was that the study duration may have been too short to show additional benefit of the combination treatment. Moreover, VAP-1 protein levels and activity were only measured in the aqueous humor and not in the vitreous.
Conclusions
ASP8232 was not efficacious in reducing CI-DME in patients with DME. ASP8232 was able to inhibit VAP-1 activity; however, addition of ASP8232 to VEGF inhibition did not provide any added benefits in eyes with DME. ASP8232 was well tolerated and no serious unexpected adverse events were reported. VAP-1 inhibition may not impact DR efficacy; future studies investigating this will provide more insight. The proper route of delivery of ASP8232 for ocular diseases should also be reassessed, as local delivery may be more appropriate than systemic administration.
Abbreviations
- %CFB:
-
percent change from baseline
- BCVA:
-
best corrected visual acuity
- BMI:
-
body mass index
- CI:
-
confidence interval
- CI-DME:
-
center-involved diabetic macular edema
- CST:
-
central subfield thickness
- DME:
-
diabetic macular edema
- DPP-4:
-
dipeptidyl peptidase 4 inhibitors
- DR:
-
diabetic retinopathy
- ECG:
-
electrocardiogram
- ETDRS:
-
early treatment diabetic retinopathy study
- EOT:
-
end of treatment
- FA:
-
fluorescein angiography
- FAS:
-
full analysis set
- OIRRC:
-
Ocular Imaging Research and Reading Center
- PDR:
-
proliferative diabetic retinopathy
- SD-OCT:
-
spectral domain-optical coherence tomography
- TEAE:
-
treatment-emergent adverse event
- VAP-1:
-
vascular adhesion protein-1
- VEGF:
-
vascular endothelial growth factor
- VIDI:
-
VAP-1 Inhibition in DME
- YAG:
-
yttrium–aluminum-garnet
References
Ciulla TA, Amador AG, Zinman B. Diabetic retinopathy and diabetic macular edema: pathophysiology, screening, and novel therapies. Diabetes Care. 2003;26(9):2653–64.
Ding J, Wong TY. Current epidemiology of diabetic retinopathy and diabetic macular edema. Curr Diabetes Rep. 2012;12(4):346–54.
Wang W, Lo ACY. Diabetic retinopathy: pathophysiology and treatments. Int J Mol Sci. 2018;19(6):1816–30.
Yau JW, Rogers SL, Kawasaki R, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35(3):556–64.
Boyer DS, Hopkins JJ, Sorof J, Ehrlich JS. Anti-vascular endothelial growth factor therapy for diabetic macular edema. Ther Adv Endocrinol Metab. 2013;4(6):151–69.
Klein R, Klein BE, Moss SE, et al. The Wisconsin epidemiologic study of diabetic retinopathy. IV. Diabetic macular edema. Ophthalmology. 1984;91(12):1464–74.
Chen E, Looman M, Laouri M, et al. Burden of illness of diabetic macular edema: literature review. Curr Med Res Opin. 2010;26(7):1587–97.
Shea AM, Curtis LH, Hammill BG, et al. Resource use and costs associated with diabetic macular edema in elderly persons. Arch Ophthalmol. 2008;126(12):1748–54.
Ford JA, Lois N, Royle P, et al. Current treatments in diabetic macular oedema: systematic review and meta-analysis. BMJ Open. 2013;3(3):e002269.
Nguyen QD, Brown DM, Marcus DM, et al. Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology. 2012;119(4):789–801.
Brown DM, Nguyen QD, Marcus DM, et al. Long-term outcomes of ranibizumab therapy for diabetic macular edema: the 36-month results from two phase III trials: RISE and RIDE. Ophthalmology. 2013;120(10):2013–22.
Brown DM, Schmidt-Erfurth U, Do DV, et al. Intravitreal aflibercept for diabetic macular edema: 100-week results from the VISTA and VIVID studies. Ophthalmology. 2015;122(10):2044–52.
Wykoff CC, Le RT, Khurana RN, et al. Outcomes with as-needed aflibercept and macular laser following the phase III VISTA DME trial: ENDURANCE 12-month extension study. Am J Ophthalmol. 2017;173:56–63.
Midena E, Gillies M, Katz TA, et al. Impact of baseline central retinal thickness on outcomes in the VIVID-DME and VISTA-DME studies. J Ophthalmol. 2018;2018:3640135.
Haritoglou C, Kook D, Neubauer A, et al. Intravitreal bevacizumab (Avastin) therapy for persistent diffuse diabetic macular edema. Retina. 2006;26(9):999–1005.
VanderBeek BL, Shah N, Parikh PC, Ma L. Trends in the care of diabetic macular edema: analysis of a national cohort. PLoS ONE. 2016;11(2):e0149450.
Trivedi PJ, Tickle J, Vesterhus MN, et al. Vascular adhesion protein-1 is elevated in primary sclerosing cholangitis, is predictive of clinical outcome and facilitates recruitment of gut-tropic lymphocytes to liver in a substrate-dependent manner. Gut. 2018;67(6):1135–45.
Weston CJ, Shepherd EL, Claridge LC, et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest. 2015;125(2):501–20.
Luo W, Xie F, Zhang Z, Sun D. Vascular adhesion protein 1 in the eye. J Ophthalmol. 2013;2013:925267.
Murata M, Noda K, Fukuhara J, et al. Soluble vascular adhesion protein-1 accumulates in proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 2012;53(7):4055–62.
Abu El-Asrar AM, Alam K, Garcia-Ramirez M, et al. Association of HMGB1 with oxidative stress markers and regulators in PDR. Mol Vis. 2017;23:853–71.
Yoshida S, Murata M, Noda K, et al. Proteolytic cleavage of vascular adhesion protein-1 induced by vascular endothelial growth factor in retinal capillary endothelial cells. Jpn J Ophthalmol. 2018;62(2):256–64.
Noda K, Miyahara S, Nakazawa T, et al. Inhibition of vascular adhesion protein-1 suppresses endotoxin-induced uveitis. FASEB J. 2008;22(4):1094–103.
Noda K, Nakao S, Zandi S, et al. Vascular adhesion protein-1 regulates leukocyte transmigration rate in the retina during diabetes. Exp Eye Res. 2009;89(5):774–81.
Matsuda T, Noda K, Murata M, et al. Vascular adhesion protein-1 blockade suppresses ocular inflammation after retinal laser photocoagulation in mice. Invest Ophthalmol Vis Sci. 2017;58(7):3254–61.
Grover S, Murthy RK, Brar VS, Chalam KV. Normative data for macular thickness by high-definition spectral-domain optical coherence tomography (spectralis). Am J Ophthalmol. 2009;148(2):266–71.
Ibrahim MA, Sepah YJ, Symons RC, et al. Spectral- and time-domain optical coherence tomography measurements of macular thickness in normal eyes and in eyes with diabetic macular edema. Eye (Lond). 2012;26(3):454–62.
Campochiaro PA, Khanani A, Singer M, et al. Enhanced benefit in diabetic macular edema from AKB-9778 Tie2 activation combined with vascular endothelial growth factor suppression. Ophthalmology. 2016;123(8):1722–30.
Acknowledgements
Editorial and logistical support was provided by OPEN Health Medical Communications (Chicago, IL).
Authors’ contributions
Concept and design: QDN, YJS, BB, DB, YS, RWR. Data acquisition: QDN, YJS, BB, DB, DVD, AGH, SP, FMR, YS, RWR. Data analysis/interpretation: QDN, YJS, BB, DB, AGH, YS, RWR. Drafting of the work or revising for important intellectual content: QDN, YJS, BB, DB, DVD, AGH, SP, FMR, YS, RWR. Final approval of the work to be published: QDN, YJS, BB, DB, DVD, AGH, SP, FMR, YS, RWR. Agreement to be accountable for all aspects of the work: QDN, YJS, BB, DB, DVD, AGH, SP, FMR, YS, RWR. All authors read and approved the final manuscript.
Funding
This study was funded by Astellas Pharma, Inc., Northbrook, IL. The funder of the study had a role in the study design, and was responsible for the collection, analysis, and interpretation of data, writing the report, and the decision to submit the paper for publication. All authors had full access to the study data in preparing the manuscript and the corresponding author had final responsibility for the decision to submit for publication.
Availability of data and materials
Access to anonymized individual participant level data collected during the trial, in addition to supporting clinical documentation, is planned for trials conducted with approved product indications and formulations, as well as compounds terminated during development. Conditions and exceptions are described under the Sponsor Specific Details for Astellas on www.clinicalstudydatarequest.com. Study-related supporting documentation is redacted and provided if available, such as the protocol and amendments, statistical analysis plan and clinical study report. Access to participant level data is offered to researchers after publication of the primary manuscript (if applicable) and is available as long as Astellas has legal authority to provide the data. Researchers must submit a proposal to conduct a scientifically relevant analysis of the study data. The research proposal is reviewed by an Independent Research Panel. If the proposal is approved, access to the study data is provided in a secure data sharing environment after receipt of a signed Data Sharing Agreement.
Ethics approval and consent to participate
This study was approved by the Ethics Committee and all patients provided informed consent.
Consent for publication
Not applicable.
Competing interests
QDN: chaired the Steering Committee for the RISE and RIDE studies of ranibizumab for diabetic macular edema; Grants from Astellas, Genentech, and Regeneron; scientific advisory boards for Astellas, Genentech, and Regeneron. YJS: Research Support from Astellas, Genentech, Optovue; Personal Fees from Genentech/Roche and Optos. BB: Research Support from Genentech, Regeneron, Allegro. DB: Grants from Astellas and Genentech/Roche, Regeneron, Bayer, Adverum, Allergan, Regenxbio, Clearside Biomedical, OHR, Heidelberg, Novartis, Thrombogenics, Ophthotech, NEI, Kanghong Biotech, Samsung, Ophthea, Apellis; Personal Fees from Genentech/Roche, Adverum, Allergan, Regenxbio, Clearside Biomedical, OHR, Bayer, Heidelberg, Novartis, Optos, Zeiss, Thrombogenics, Santen, Senju, Kanghong Biotech, Samsung, Apellis. DVD: Grants from Genentech, Regeneron, Santen. AGH: Personal Fees from Astellas; Employee of Astellas. SP: Grants from Genentech, Novartis, Ophthotech, Allergan, Opthea, Clearside, Samsung, Aerpio, Regeneron, Graybug and Taiwan Liposome Company. FMR: Nothing to disclose. YS: advisory board and research support from Castle Biosciences. RWR: Employee of Astellas.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table
S1. Baseline characteristics (safety analysis set).
Additional file 2: Table
S2. Ocular TEAEs (safety analysis set).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nguyen, Q.D., Sepah, Y.J., Berger, B. et al. Primary outcomes of the VIDI study: phase 2, double-masked, randomized, active-controlled study of ASP8232 for diabetic macular edema. Int J Retin Vitr 5, 28 (2019). https://doi.org/10.1186/s40942-019-0178-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40942-019-0178-7