
Abstract
Clinical practice has shown that Parkin is the major causative gene found in an autosomal recessive juvenile parkinsonism (AR-JP) via Parkin mutations and that the Parkin protein is the core expression product of the Parkin gene, which itself belongs to an E3 ubiquitin ligase. Since the discovery of the Parkin gene in the late 1990s, researchers in many countries have begun extensive research on this gene and found that in addition to AR-JP, the Parkin gene is associated with many diseases, including type 2 diabetes, leprosy, Alzheimer’s, autism, and cancer. Recent studies have found that the loss or dysfunction of Parkin has a certain relationship with tumorigenesis. In general, the Parkin gene, a well-established tumor suppressor, is deficient and mutated in a variety of malignancies. Parkin overexpression inhibits tumor cell growth and promotes apoptosis. However, the functions of Parkin in tumorigenesis and its regulatory mechanisms are still not fully understood. This article describes the structure, functions, and post-translational modifications of Parkin, and summarizes the recent advances in the tumor suppressive function of Parkin and its underlying mechanisms.
Similar content being viewed by others

Background
Parkin gene, also called PARK2, is located on chromosome 6q25.2-q27, contains 12 exons, and has a length of about 1.5 Mb [1]. It is widely expressed in various tissues and is mainly found in the brain and muscles [2]. Since 1998, Kitada et al. [3] were the first to discover the Parkin gene mutation in a Japanese family diagnosed with Parkinson. To date, many studies have confirmed that Parkin has very broad roles, in addition to Parkinson’s disease, and is also associated with many diseases, such as type 2 diabetes, Alzheimer’s disease, multiple sclerosis [3,4,5,6]. There is a certain correlation between Parkin and the occurrence and development of tumors according to genetic studies of many cancer patients. Many studies have shown that the in vivo loss of chromosomal region fragments is associated with malignant tumors, such as p53, Rb fragments and fragile sites [2]. The Parkin gene is located near the fragile site FRA6E [3]. FRA6E is located in an unstable region on chromosome 6q26, which is susceptible to mutate under external stimuli and then promotes tumor formation in normal cells [3]. Parkin is also a class of molecules that exhibits high variability under different signal induction. Various stimuli can modulate Parkin’s activities through different post-translational modifications [7] which play a very important role in life activities. Through the post-translational modification, the structure of the protein becomes more complicated, the function is enhanced, the regulation is more refined, and the effect is more distinctive [8]. Recent studies have demonstrated that the expression level of Parkin is low in cancers and its dysfunctions or loss has certain relationships with many cancers [4]. Therefore, an in-depth study of Parkin to clarify its connection with cancers will help provide new drug targets and strategies for cancer treatment.
Overview: structure, regulation, and functions of Parkin
Structural domains of Parkin
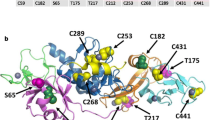
The Parkin gene encodes 465 amino acids to form a protein with a molecular weight of about 52 kDa, namely the Parkin protein [3]. Parkin is a multi-domain protein, and its C-terminus consists of the ring structure (RING1 and RING2) on both sides and the in-between RING (IBR) in the middle to form the RING1-IBR-RING2 structure [3, 9, 10]. In the N-terminal ubiquitin-like domain (UBL), there are 76 amino acids homologous to ubiquitin, which is a ubiquitin-like structural region with a typical ubiquitin folding [3], so Parkin protein is considered to be involved in the activities of ubiquitin–proteasome system (UPS) as E3 ubiquitin ligase [11] (Fig. 1a, b).

The two-dimensional structure and three-dimensional structure of human Parkin. a The two-dimensional structure of the Parkin protein, the letters in the column indicate the domain. b Three-dimensional structure of Parkin protein, based on the datasets in cBioPortal (http://www.cbioportal.org). UBL ubiquitin-like domain, RING loop finger domain, IBR in-between RING, cysteine-rich domain, REP repressor element of RING
Functions of Parkin
Parkin has E3 ubiquitin ligase activity
Ubiquitination refers to the process in which ubiquitin molecules classify proteins in cells under the action of E1, E2 and E3 enzymes, select target protein molecules, and specifically modify target proteins [3, 8]. In addition to degradation by the proteasome, ubiquitination can also act as a signal for autophagy degradation by lysosomes and alter the activity or location of the substrate protein [12]. As an E3 ligase, Parkin can ubiquitinate the substrate delivered by E2 binding enzyme and further deliver the ubiquitinated substrate to the proteasome, which is degraded into small molecules by proteasome action for recycling of intracellular substances [2], including synphilin-1, cyclin E, P38 tRNA synthetase, SP22 (22-kDa glycosylated form of α-synuclein), and more [8] (Fig. 2a).

Function of Parkin. a Proteasome degradation pathway. b Pathway of PINK1 activation of Parkin leading to autophagy of depolarized mitochondria. c Degradation pathway of unfolded or misfolded proteins. ub ubiquitin, OMM outer mitochondrial membrane, P phosphorylation, CCCP carbonyl cyanide 3-chlorophenylhydrazone
The role and mechanism of Parkin in mitochondrial autophagy
Mitochondrial autophagy is a physiological process that removes damaged or excessive mitochondria through a degradation pathway in autophagosomes [13]. The Parkin/PTEN-induced kinase 1 (PINK1) pathway is the most typical mitochondrial autophagy pathway [14]. In damaged mitochondria, depolarization of the mitochondrial membrane results in the immobilization of PINK1 on the outer membrane of the mitochondria and activation by autophosphorylation [15]. Activated PINK1 phosphorylates many substrates, including Parkin and ubiquitin, and experiments have shown that the combination between phospho-ubiquitination (p-Ub) and phosphorylated Parkin has a high affinity that causes Parkin to produce a conformational change. As a result, the recruitment of E2 is promoted, thus activating Parkin [16]. Parkin rapidly catalyzes the ubiquitination of large amounts of mitochondrial proteins, followed by ubiquitinated mitochondrial proteomes linked to autophagic machinery and initiation of selective autophagy [17] (Fig. 2b).
In addition to PINK1/Parkin-mediated mitochondrial autophagy, autophagy-mediated by the B-cell lymphoma-2 (Bcl-2) and adenovirus E1B 19 kDa-interacting protein 3 (BNIP3) and NIP3-like protein X (NIX) signaling pathways also plays a key role in autophagy [18, 19]. Studies have found that BNIP3 and NIX can directly link to the microtubule-associated protein light chain 3 (LC3) protein and recruit autophagosomes to degrade targeted proteins and NIX can also directly modulate ubiquitination of Parkin substrates to mediate mitochondrial autophagy [19]. However, whether PINK1/Parkin-mediated mitochondrial autophagy pathway is associated with this pathway will be a new field for future studies of mitochondrial autophagy.
Parkin as an important monitoring system in the cell
When intracellular proteins are misfolded, the ubiquitin–proteasome system can remove or degrade these proteins in time, thereby effectively reducing the cytotoxic load caused by excessive accumulation of misfolded proteins [3, 7] (Fig. 2c). This mechanism has important protective effects on cells. When the endoplasmic reticulum undergoes a stress response, the protein is unfolded or misfolded, and Parkin’s E3 ubiquitin ligase activity is lost, resulting in the accumulation of a large number of mitochondrial proteins and many other substrates, and ultimately induces endoplasmic reticulum stress-mediated cell death [3, 4]. For instance, regarding the Peal receptor protein, studies have confirmed that it has dopamine neurotoxicity, which can cause stress in endoplasmic reticulum and cytoplasm of the cell, thereby inducing dopaminergic neurons death in the substantia nigra of the brain [20]. The inactivation of Parkin protein magnifies these damaging effects. Parkin proteins are dephosphorylated and more active when cells are exposed to stress caused by Parkinson’s disease-associated folding proteins [21]. Phosphorylation and dephosphorylation of Parkin can rapidly and efficiently regulate its functions and activities when proteins are misfolded or threatened by cell survival.
Post-translational modification of Parkin
Post-translational modification (PTM) is a fundamental process to regulate protein functions [7]. Different types of modifications affect the conformation and stability of the protein and ultimately its function [22, 23]. Phosphorylation, methylation, ubiquitination, acetylation, sumoylation, neddylation, glycosylation and sulfation are all common post-translational modifications of proteins (Table 1). Parkin’s activity can be regulated by various types of PTM, for example, phosphorylation, ubiquitination, sumoylation and neddylation [24, 25]. These reversible PTMs cause Parkin to translocate, affecting its DNA binding affinity, and altering the transcriptional activity pattern of a particular target gene locus [26]. When cells are subjected to environmental stress or signal stimulation, certain functions can be obtained or lost through specific post-translational modification, thereby producing specific effects [27].
Parkin’s post-translational modifications do not exist in isolation, but rather have intricate connections with each other, forming a complex post-translational modification control network. The phosphorylation of Parkin not only inhibits ubiquitination but also acetylation [7, 28]. Therefore, Parkin may have some post-transcriptional modifications, and no interactions between various modifications have been detected. The importance of these modifications in specific tumorigenesis remains to be elucidated.
Mechanism of Parkin activation by phosphorylation
Different protein kinases can recognize and modify different sites of different proteins, which expands the complexity of phosphorylated protein research [29]. The molecular mechanisms of protein phosphorylation have considerable guiding significance for the study of major diseases such as cancer and have become one of the hotspots in the field of biology. The primary mechanism for modulating Parkin activity and its target genes is to control Parkin’s translocation between the nucleus and cytoplasm by phosphorylation of a series of kinases [30]. There are a variety of proteins involved in Parkin phosphorylation, of which PINK1 is the most studied protein [31, 32]. Kim et al. reported that Parkin’s activity and mitochondrial localization depended on PINK1 kinase-activity [32]. Two research reports [33, 34] also indicated that Parkin translocation and stress-induced mitochondrial autophagy requires the PINK1-dependent phosphorylation of Ser65 in the UbL domain [35]. The initiation of phosphor-ubiquitin makes Parkin easier to PINK1-mediated Ser65 phosphorylation [36]. So, in a nutshell, the phosphorylation of PINK1 is necessary for Parkin activation and target recognition [14, 36, 37].
Parkin’s ubiquitination and deubiquitination
Protein ubiquitination is a fundamental post-translational modification that controls cell fate and function [7]. It has been reported that Parkin mediates its own ubiquitination through K48 protein-dependent ubiquitin chain formation, thereby affecting the stability of its own proteins [7, 38]. Durcan and colleagues identified the deubiquitinating enzyme (DUB) Ataxin-3 as a ligand for Parkin, which interacts with Parkin’s UbL and IBR-RING2 domains and promotes Parkin’s β-dimerization [39]. Mutant Ataxin-3, a polyglutamine dilatation associated with the onset of Machado-Joseph neurodegenerative disease, promotes Parkin degradation by autophagy and leads to a decrease in Parkin levels in in vivo [40]. In a subsequent study, it was shown that Ataxin-3 binds to the E2 ubiquitin ligase Ubc7 instead of Parkin and promotes Parkin de-ubiquitination only when Parkin itself is ubiquitinated [41]. Collectively, these highlight the complex regulation of Parkin ubiquitination, involving the coordinated activities of Parkin, DUB and E2 ubiquitin ligase [42, 43]. It is known that when Parkin is ubiquitinated in cells, it degrades in a proteasome-dependent manner, effectively inactivating Parkin [44]. In conclusion, the ubiquitination process involved in Parkin protein plays a key role in protein localization and degradation [45].
Sumoylation modification
In recent years, many proteins similar to ubiquitin sequences have been discovered, one of which is the ubiquitin-related analogue SUMO (small ubiquitin-related modification) [46]. SUMO is a highly conserved family of proteins widely found in eukaryotes. There are three SUMO genes in vertebrates called SUMO-1, -2, -3, which are very similar to ubiquitin in secondary structure and catalytically modified [46, 47]. This modification plays an important role in stabilizing protein conformation and regulating protein subcellular localization [7, 48]. Studies have shown that non-covalent binding of Parkin protein to SUMO-1 enhances Parkin’s nuclear translocation and increases its own ubiquitination, but no significant Parkin protein level difference was detected after the overexpression of SUMO-1, indicating that an increase in autoubiquitination activity does not necessarily result in the protease-dependent degradation of Parkin [48]. Therefore, a positive regulator of Parkin, such as SUMO-1, may simply disintegrate the self-inhibiting conformation of Parkin protein or enhance the binding of E2 to the substrate without causing degradation of the Parkin protein [49], thereby causing apoptosis of cancer cells. Recent studies have found that sumoylation is also involved in the repair of DNA damage and the regulation of mitochondrial division, genomic stability, ion channels, and biological rhythms. In addition, disorders of the SUMO-modifying function can cause certain diseases to occur [49]. The function of many oncogenes and tumor suppressor genes is regulated by SUMO modification, such as P53, IRF-1 (interferon regulatory factor 1) [46]. Studies have shown that SUMO1 modification can inhibit the activity of the P53 gene and promote the occurrence, development, and metastasis of cancer [50, 51]. IRF-1 is a tumor suppressor and inhibits phenotypic changes. The SUMO modification level of IRF-1 was significantly increased in tumor cells by screening for SUMO protein. SUMO modification of IRF-1 increases the stability of this protein in tumors [52].
Neddylation modification
Neural precursor cell-expressed developmentally downregulated 8 (NEDD8) is a class of molecules with similar structure to ubiquitin proteins, called neddylation, which can be involved in the post-translational modification of proteins. Like ubiquitin, NEDD8 is also expressed in most tissue types [53, 54]. Recent studies have shown that protein neddylation modification abnormalities are closely related to the occurrence and development of a variety of tumors [55]. Enzymes involved in neddylation modification are higher in tumors than normal adjacent tissues. Neddylation modification has become a new anti-tumor therapeutic target that can exert its anti-tumor effect by ubiquitin ligase regulating the neddylation modification process [55, 56]. Studies have shown that Parkin binds to the ubiquitin-like protein NEDD8 [57], indicating that NEDD8 is linked to Parkin to increase E3 ligase activity by increasing the affinity to E2 ubiquitin ligase Ubiquitin-conjugating Enzyme H8 (UbcH8) and putative substrate aminoacyltransferase p38 subunit, thereby inhibiting the development of the tumor. Walden et al. reported that neddylation enhanced the interaction of Parkin with UbcH8 and its putative substrate, the p38 subunit of the amino acyltransferase, which enhances the activity of ubiquitin ligase [11]. Nedd8 is capable of bidirectional regulation of ubiquitination. When Nedd8 modifies the Cullin E3 enzyme by neddylation, it changes its enzyme configuration, making E3 easier to bind to the E2 binding enzyme, and the ubiquitination-modifying enzyme activity of E3 is promoted [53]. However, when neddylation competes with the ubiquitination modification for the same modification site, it can also inhibit the ubiquitination of the substrate. RING box proteins (RBXs), a component of the ubiquitin ligase Cullin-RING complex, is the most studied neddylation modified ligase, and further studies have found that ubiquitin ligases MDM2 (murine double minute 2), Smurf1 (Smad ubiquitin regulatory factor 1) and NEDL2 (NEDD4-like ubiquitin ligase 2) can also act as neddylation modified ligases [7].
Parkin protein S-nitrosylation and cancers
S-nitrosylation is a reversible post-translational modification involving the covalent attachment of a NO (nitric oxide) group to a cysteine residue to form an S-nitrosothiol species that stabilizes the structure of the protein [58, 59]. Numerous studies have shown that abnormal S-nitrosylation is associated with the development and progression of cancer and the response to certain therapies [58]. S-nitrosylation abnormalities are key events in cancer episodes and may significantly increase cancer risk [60]. S-nitrosylation regulates the biological activities of a variety of proteins in the body and is involved in key processes in the cell life cycle, including transcriptional regulation, DNA repair and apoptosis [58, 60]. Parkin is rich in cysteine and coordinates 8 zinc atoms to ensure proper folding of Parkin. Therefore, the S-nitrosylation of any zinc-coordinated cysteine affects Parkin’s function [61]. However, it is controversial that S-nitrosylation regulating Parkin’s function. On one hand, the effect of S-nitrosylation on the mitochondrial degradation of Parkin function in human neuroblastoma cells (SH-SY5Y) by Ozawa group [62] found that S-nitrosylation of Parkin protein increases E3 ligase activity after mitochondrial depolarization to induce mitochondrial aggregation and degradation, in addition, Cys323 in Parkin is S-nitrosylated key site. On the other hand, Ted Dawson’s team [58] found that the degree of S-nitrosylation of parkin protein was increased in the human brain and the S-nitrosylation of Parkin protein attenuated E3 ligase activity after mitochondrial depolarization. In addition, studies have demonstrated that the functional regulation of Parkin protein S-nitrosylation is bidirectional and undergo self-ubiquitination. S-nitrosylation will increase first and then decrease [58]. Therefore, the specific mechanism by which S-nitrosylation regulates Parkin’s function requires further study.
Parkin’s relationship with cancer and its regulatory mechanism
Tumor suppressor gene—Parkin
There is increasing evidence that Parkin is a tumor suppressor, mutations in the Parkin gene have been reported in many cancers, although the frequency of these mutations is relatively low [4]. Analyses of the database from cBioportal (http://www.cbioportal.org) indicate that Parkin gene mutation rate is ~ 5% in cervical cancer, ~ 5% in lung squamous cell carcinoma, and 2%–6% in colorectal cancer [63]. Studies have confirmed that Parkin’s deletion of the long arm of chromosome 6 is associated with several solid tumors, including ovarian cancer, breast cancer, kidney cancer, lung cancer, melanoma, and hematological cancer [4]. A number of missing regions were identified by analysis of 6q21-q23, 6q25.1-q25.2, and 6q25-q27. In addition, a loss of 6q27 was found in benign ovarian tumors. Later studies identified a homozygous deletion of exon 2 in lung adenocarcinoma [4, 64]. Parkin’s loss of heterozygosity and loss of copy number were observed in breast cancer [65]. With in-depth studies on Parkin, it was found that its overexpression inhibits the proliferation of cancer cells, while Parkin’s inactivation promotes the proliferation of cancer cells, demonstrating that Parkin acts as a tumor suppressor [63, 66]. Parkin gene deletions and mutations often occur in lung cancer, and the inactivation of the Parkin gene increases the incidence of lung cancer. By analyzing the cancer genome map, it was found that about one-quarter of the glioblastoma samples had heterozygous or homozygous loss of the Parkin gene and point mutation [67]. Experiments have shown that mice lacking the Parkin gene are more prone to pancreatic cancer [68]. The reduction of the Parkin gene enhances the proliferation and migration of pancreatic cancer cells. When the Parkin gene is overexpressed, the migration and invasion ability of cancer cells is weakened, indicating that Parkin has the potential to inhibit pancreatic cancer, and its expression level is positively correlated.
Parkin-mediated tumor suppression and underlying mechanisms
Anti-apoptosis
Apoptosis is the balance between multicellular organisms to maintain cell stability. The active physically death process of cells controlled by genes is a natural obstacle to the development of cancer [3, 17]. Recent studies have found that Parkin seems to promote cancer cell apoptosis. Parkin has been reported to induce apoptosis by promoting mitochondrial depolarization [69]. Parkin promotes the ubiquitination and degradation of myeloid cell leukemia-1 (Mcl-1), a member of the B-cell leukemia/lymphoma 2 (Bcl-2) family, and open the Bax/Bak channel, making cells sensitive to apoptosis [69]. In the Michigan Cancer Foundation 7 (MCF7) human breast cancer cells, Parkin binds to the outer surface of microtubules to increase the interaction of paclitaxel with microtubules, increasing cell sensitivity to apoptosis [70]. Parkin also promotes histone deacetylases (HDAC) inhibitors to induce apoptosis in hepatocellular carcinoma through a mechanism that is poorly understood. In conclusion, Parkin can promote cancer cell apoptosis through different pathways.
Anti-cell proliferation
The ability to maintain chronic proliferative signals is the most important feature of cancer cell survival. Previous studies have shown that Parkin plays an important role in inhibiting cell cycle progression. Parkin regulates the stability of G1/S cyclins and maintains the coordination of different cyclins, thus acts as a major regulator of the cell cycle. Interestingly, Parkin’s loss is mutually exclusive with the amplification of cyclin D1, cyclin E1 [60, 71] and cyclin-dependent kinase 4 (CDK4) genes, suggesting that Parkin and these cell cycle components interact in a common pathway [72]. In MCF7 breast cancer cells, Parkin is reported to regulate the mRNA levels of CDK6 (cyclin-dependent kinase 6) [11], which leads to cell cycle arrest and growth inhibition [73]. Thus, Parkin mutation abolishes its ligase activity and impairs its ability to ubiquitinate cyclins, which in turn leads to amplification of G1/S [72] phase cyclin turnover, hyperproliferative signaling and ultimately cancer [74].
Anti-cell metastasis
Tumor invasion and metastasis are the most critical steps in defining the aggressive phenotype of human cancer [75]. As a potential tumor suppressor protein, the increased expression of Parkin may be related to the viability of invasion and metastasis. Parkin helps microtubule polymerization through its three separate microtubule/tubulin-binding domains and cooperates with paclitaxel treatment to increase their stability [63]. In breast cancer, a decrease in Parkin’s cytoplasmic expression may be helpful in predicting paclitaxel treatment outcomes [73]. In addition, it was found that when parkin is overexpressed, the migration and invasion ability of various cancer cells is weakened [73]. Since microtubule dynamics are closely related to cell migration and metastasis, Parkin has some negative regulation on cancer cell metastasis through its microtubule-stabilizing activity [76]. Collectively, these studies demonstrated the potential role of Parkin in the tumor microenvironment [63].
Anti-angiogenesis
Cancer cells require adequate nutrition and oxygen to maintain and assess the ability to metabolize waste. To achieve this, as early as 1971, American scholar Folkman put forward the theory that “tumor growth depends on angiogenesis” [77]. Vascular endothelial growth factor (VEGF) is one of the most potent stimulating factors found in angiogenesis, affecting endothelial cell proliferation, motor and vascular permeability [78, 79]. Interestingly, it was observed that Parkin significantly affected the expression of vascular endothelial growth factor receptor-2 (VEGFR-2). In U87-Parkin cells (Glioma cells stably expressing Parkin), the expression of VEGFR-2 was found to be nearly 4-fold lower than the control group [11]. In most invasive tumors, the production and secretion of VEGF are usually observed, a phenomenon that seriously affects the prognosis of cancer patients [80]. In a study of glioma cells, the negative relation between Parkin function and VEGFR-2 has been shown to be a key factor in promoting angiogenesis. Thus, Parkin-mediated inhibition of glioma cell proliferation involves the regulation of the VEGFR-2 pathway [11].
Anti-inflammation
How inflammation induces tumors is an important scientific issue in the international frontier. Previous studies have demonstrated that many tumors are induced by inflammation [81]. Inflammatory mediators cause genetic and epigenetic changes such as DNA methylation, tumor suppressor gene point mutations, and post-translational modifications, which cause changes in intracellular homeostasis and occurrence of tumors [81, 82]. With in-depth research, inflammatory mediators involved in the occurrence and development of tumors have been identified [83]. The expression of inflammatory markers interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) was abnormally increased in Parkin-deficient cells [84, 85], while IL-6 level was significantly higher in Parkin knocked-out mice than in wild-type [67]. A recent study has suggested that inflammation and genomic instability caused by Parkin deficiency may be a trigger in lung cancer [86]. In the absence of any stimulation, a decrease in Parkin expression leads to an increase in nuclear factor kappa B (NF-κB) localization [67]. NF-κB is a widely expressed transcription factor that induces cytokine and immunoglobulin gene expression in chronic obstructive pulmonary disease-associated inflammation [86]. These results proved that Parkin has anti-inflammatory properties, while Parkin deficiency may aggravate inflammation.
Conclusions and perspectives
In recent years, evidence from cultured cells and Parkin knockout mice experiments, as well as clinical studies have shown that Parkin is an important tumor suppressor that is abnormally expressed in many malignancies, including colorectal cancer, lung cancer, and endometrial cancer [7]. As a tumor suppressor gene, little is known about the way that parkin inhibits tumor growth, as well as the mechanisms of the parkin promoter region methylation and parkin mutation leading to tumorigenesis.
The role of Parkin in Parkinson’s disease has been established, and the association between Parkinson’s disease and cancer risk seems complicated, and many epidemiological studies have shown a connection between Parkinson’s disease and the risk of developing gastric cancer, uterine cancer, lung cancer, and breast cancer [87]. Previous studies have shown that the incidence of most cancer in Parkinson’s patients is lower than in patients without Parkinson’s disease [55, 58]. In patients with Parkinson’s disease, the risk of smoking-related cancer is reduced, such as lung cancer, bladder cancer, and laryngeal cancer [5]. However, the risk of malignant melanoma and breast cancer in patients with Parkinson’s disease has increased significantly [55]. Therefore, future research should consider whether the risk of cancer in patients with Parkinson’s disease is higher than in patients with non-Parkinson’s disease, and the potential roles of Parkin mutations in regulating the relationship between Parkinson’s disease and cancer risk.
Post-translational modifications can control the activity, conformation, solubility, and cofactor interactions required for Parkin activation, substrate affinity and specificity. When cells are subjected to environmental stress or changes in the internal environment, post-translational modifications can occur rapidly to regulate various activities of the cell. In recent years, researchers in many countries have been focusing on the role of Parkin as a tumor suppressor [65]. However, little is known about post-translational modifications of Parkin participates in the development of tumors. Future research should explore the effects of post-translational modification on tumors and whether it can be used as a new approach to prevent tumorigenesis by regulating post-translational modification of Parkin.
Availability of data and materials
Not applicable.
Abbreviations
- AR-JP:
-
autosomal recessive juvenile parkinsonism
- RBR:
-
RING1-IBR-RING2
- UBL:
-
ubiquitin-like domain
- UPS:
-
ubiquitin–proteasome system
- SP22:
-
22-kilodalton glycosylated form of α-synuclein
- PINK1:
-
PTEN-induced kinase 1
- p-Ub:
-
phospho-ubiquitination
- BNIP3:
-
Bcl-2 and adenovirus E1B19 kDa-interacting protein 3
- NIX:
-
NIP3-like protein X
- LC3:
-
microtubule-associated protein light chain 3
- PTM:
-
post-translational modification
- DUB:
-
deubiquitinating enzyme
- SUMO:
-
small ubiquitin-related modifier
- NEDD8:
-
neural precursor cell-expressed developmentally downregulated 8
- RBXs:
-
RING box proteins
- MDM2:
-
murine double minute 2
- Smurf1:
-
mad ubiquitin regulatory factor 1
- NEDL2:
-
NEDD4-like ubiquitin ligase 2
- NO:
-
nitric oxide
- Mcl-1:
-
myeloid cell leukemia-1
- SH-SY5Y:
-
human neuroblastoma cells
- MCF7:
-
Michigan Cancer Foundation 7
- Bcl-2:
-
B-cell leukemia/lymphoma 2
- HDAC:
-
histone deacetylases
- CDK4:
-
cyclin-dependent kinase 4
- CDK6:
-
cyclin-dependent kinase 6
- VEGF:
-
vascular endothelial growth factor
- VEGFR-2:
-
vascular endothelial growth factor receptor-2
- IL-1β:
-
interleukin-1β
- TNF-α:
-
tumor necrosis factor-α
- NF-κB:
-
nuclear factor kappa B
- Ig:
-
immunoglobulin
- COPD:
-
chronic obstructive pulmonary disease
References
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605. https://doi.org/10.1038/33416.
Stichel CC, Augustin M, Kühn K, Zhu XR, Engels P, Ullmer C, et al. Parkin expression in the adult mouse brain. Eur J Neurosci. 2000;12(12):4181. https://doi.org/10.1046/j.1460-9568.2000.01314.x.
Seirafi M, Kozlov G, Gehring K. Parkin structure and function. FEBS J. 2015;282(11):2076–88. https://doi.org/10.1111/febs.13249.
Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, Mcadams H, et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci USA. 2003;100(10):5956. https://doi.org/10.1073/pnas.0931262100.
Liu J, Zhang C, Hu W, Feng Z. Parkinson’s disease-associated protein Parkin: an unusual player in cancer. Cancer Commun. 2018;38(1):40. https://doi.org/10.1186/s40880-018-0314-z.
Xiang RL, Huang Y, Zhang Y, Cong X, Zhang ZJ, Wu LL, et al. Type 2 diabetes-induced hyposalivation of the submandibular gland through PINK1/Parkin-mediated mitophagy. J Cell Physiol. 2020;235(1):232–44. https://doi.org/10.1002/jcp.28962.
Chakraborty J, Basso V, Ziviani E. Post translational modification of Parkin. Biol Direct. 2017;12(1):6. https://doi.org/10.1186/s13062-017-0176-3.
Zhang Y, Gao J, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA. 2000;97(24):13354–9. https://doi.org/10.1073/pnas.240347797.
Zarate-Lagunes M, Gu WJ, Blanchard V, Francois C, Muriel MP, Mouatt-Prigent A, et al. Parkin immunoreactivity in the brain of human and non-human primates: an immunohistochemical analysis in normal conditions and in Parkinsonian syndromes. J Comp Neurol. 2001;432(2):184–96. https://doi.org/10.1002/cne.1096.
Shridhar V, Staub J, Huntley B, Cliby W, Jenkins R, Pass HI, et al. A novel region of deletion on chromosome 6q23.3 spanning less than 500 Kb in high grade invasive epithelial ovarian cancer. Oncogene. 1999;18(26):3913. https://doi.org/10.1038/sj.onc.1202756.
Yeo CW, Ng FS, Chai C, Tan JM, Koh GR, Chong YK, et al. Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Can Res. 2012;72(10):2543. https://doi.org/10.1158/0008-5472.CAN-11-3060.
Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. 2014;15(4):403–11. https://doi.org/10.1016/j.chom.2014.03.012.
Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;2(12 Suppl):1542–52. https://doi.org/10.1038/sj.cdd.4401765.
Vivesbauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107(2):378–83. https://doi.org/10.1073/pnas.0911187107.
Seok Min J, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125(4):795–9. https://doi.org/10.1242/jcs.093849.
Kahori SF, Tsuyoshi I, Nobutaka H, Yuzuru I. PINK1-mediated phosphorylation of Parkin boosts Parkin activity in Drosophila. PLoS Genet. 2014;10(6):e1004391. https://doi.org/10.1371/journal.pgen.1004391.
Hang L, Thundyil J, Lim KL. Mitochondrial dysfunction and Parkinson disease: a Parkin-AMPK alliance in neuroprotection. Ann N Y Acad Sci. 2015;1350(1):37–47. https://doi.org/10.1111/nyas.12820.
Fatima Zahra C, Stéphanie SP, Jaclyn Nicole LG, Annick F, Régis DM, Gilles D, et al. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy. 2010;6(4):495–505. https://doi.org/10.4161/auto.6.4.11819.
Youngil L, Hwa-Youn L, Hanna RA, Gustafsson ÅB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;301(5):1924–31. https://doi.org/10.1152/ajpheart.00368.2011.
Yasuko K, Yuzuru I, Kentaro O, Ayane K, Toshio I, Mariko S, et al. Pael receptor induces death of dopaminergic neurons in the substantia nigra via endoplasmic reticulum stress and dopamine toxicity, which is enhanced under condition of parkin inactivation. Hum Mol Genet. 2007. https://doi.org/10.1093/hmg/ddl439.
Ayako Y, Arno F, Yuzuru I, Ryosuke T, Kahle PJ, Christian H. Parkin phosphorylation and modulation of its E3 ubiquitin ligase activity. J Biol Chem. 2005;280(5):3390. https://doi.org/10.1074/jbc.M407724200.
Berndsen C, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21(4):301–7. https://doi.org/10.1038/nsmb.2780.
Liu Y, Ao X, Ding W, Ponnusamy M, Wu W, Hao X, et al. Critical role of FOXO3a in carcinogenesis. Mol Cancer. 2018. https://doi.org/10.1186/s12943-018-0856-3.
Wang X, Wang X, Hu S, Hu S, Liu L, Liu L. Phosphorylation and acetylation modifications of FOXO3a: independently or synergistically? Oncol Lett. 2017;13(5):2867–72. https://doi.org/10.3892/ol.2017.5851.
Sanphui P, Biswas SC. FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis. 2013;4(5):e625. https://doi.org/10.1038/cddis.2013.148.
Daitoku H, Sakamaki JI, Fukamizu A. Regulation of FoxO transcription factors by acetylation and protein–protein interactions. Biochim Biophys Acta. 2011;1813(11):1954–60. https://doi.org/10.1016/j.bbamcr.2011.03.001.
Yang XJ. Multisite protein modification and intramolecular signaling. Oncogene. 2005;24(10):1653–62. https://doi.org/10.1038/sj.onc.1208173.
Gupta A, Anjomani-Virmouni S, Koundouros N, Dimitriadi M, Choo-Wing R, Valle A, et al. PARK2 depletion connects energy and oxidative stress to PI3K/Akt activation via PTEN S-nitrosylation. Mol Cell. 2017;65(6):999. https://doi.org/10.1016/j.molcel.2017.02.019.
Avraham E, Rott R, Liani E, Szargel R, Engelender S. Phosphorylation of Parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. J Biol Chem. 2007;282(17):12842. https://doi.org/10.1074/jbc.M608243200.
Finnberg N, El-Deiry WS. Activating FOXO3a, NF-kappaB and p53 by targeting IKKs: an effective multi-faceted targeting of the tumor-cell phenotype? Cancer Biol Ther. 2004;3(7):614–6. https://doi.org/10.4161/cbt.3.7.1057.
Sha D, Chin LS, Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-κB signaling. Hum Mol Genet. 2010;19(2):352–63. https://doi.org/10.1093/hmg/ddp501.
Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377(3):975–80. https://doi.org/10.1016/j.bbrc.2008.10.104.
Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2(5):120080. https://doi.org/10.1098/rsob.120080.
Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2(12):1002. https://doi.org/10.1038/srep01002.
Poole AC, Thomas RE, Andrews LA, Mcbride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA. 2008;105(5):1638–43. https://doi.org/10.1073/pnas.0709336105.
Kazlauskaite A, Martínez-Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep. 2015;16(8):939–54. https://doi.org/10.15252/embr.201540352.
Chen Y, Ii GWD. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340(6131):471–5. https://doi.org/10.1126/science.1231031.
Pinto MJ, Pedro JR, Costa RO, Almeida RD. Visualizing K48 ubiquitination during presynaptic formation by ubiquitination-induced fluorescence complementation (UiFC). Front Mol Neurosci. 2016;9:43. https://doi.org/10.3389/fnmol.2016.00043.
Durcan TM, Fon EA. Mutant ataxin-3 promotes the autophagic degradation of parkin. Autophagy. 2011;7(2):233–4. https://doi.org/10.4161/auto.7.2.14224.
Durcan TM, Kontogiannea M, Thorarinsdottir T, Fallon L, Williams AJ, Djarmati A, et al. The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum Mol Genet. 2011;20(1):141. https://doi.org/10.1093/hmg/ddq452.
Durcan TM, Kontogiannea M, Bedard N, Wing SS, Fon EA. Ataxin-3 deubiquitination is coupled to Parkin ubiquitination via E2 ubiquitin-conjugating enzyme. J Biol Chem. 2012;287(1):531–41. https://doi.org/10.1074/jbc.M111.288449.
Varshavsky A. The ubiquitin system. Nat Med. 1998;67(67):1–17. https://doi.org/10.1146/annurev.biochem.67.1.425.
Hegde AN. The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem. 2010;17(7):314. https://doi.org/10.1101/lm.1504010.
Federica M, Mark VDM, Suzan VDL, Domenico G. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol Neurodegener. 2011;6(1):34.
Nagy V, Dikic I. Ubiquitin ligase complexes: from substrate selectivity to conjugational specificity. Biol Chem. 2010;391(2/3):163–9. https://doi.org/10.1515/bc.2010.021.
Müller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol. 2001;2(3):202–10. https://doi.org/10.1038/35056591.
Hay RT. Protein modification by SUMO. Trends Biochem Sci. 2001;26(5):332–3. https://doi.org/10.1016/S0968-0004(01)01849-7.
Won UJ, Kwang Chul C. Functional modulation of parkin through physical interaction with SUMO-1. J Neurosci Res. 2010;84(7):1543–54. https://doi.org/10.1002/jnr.21041.
Guerra de Souza AC, Prediger RD, Cimarosti H. SUMO-regulated mitochondrial function in Parkinson’s disease. J Neurochem. 2016;137(5):673–86. https://doi.org/10.1111/jnc.13599.
Watson IR, Irwin MS. Ubiquitin and ubiquitin-like modifications of the p53 family. Neoplasia. 2006;8(8):655–66. https://doi.org/10.1593/neo.06439.
Bartosz W, Alicja Z, Maura W, Ted H, Maciej Z. MDM2 chaperones the p53 tumor suppressor. J Biol Chem. 2007;282(45):32603–12. https://doi.org/10.1074/jbc.M702767200.
Junsoo P, Kwangsoo K, Eun-Ju L, Yun-Jee S, Si-Nae L, Kyoungsook P, et al. Elevated level of SUMOylated IRF-1 in tumor cells interferes with IRF-1-mediated apoptosis. Proc Natl Acad Sci USA. 2007;104(43):17028–33. https://doi.org/10.1073/pnas.0609852104.
Dil KA, Kito K, Abe Y, Shin RW, Kamitani T, Ueda N. NEDD8 protein is involved in ubiquitinated inclusion bodies. J Pathol. 2003;199(2):259–66. https://doi.org/10.1002/path.1283.
Ji WU, Han KA, Im E, Oh Y, Lee K, Chung KC. Neddylation positively regulates the ubiquitin E3 ligase activity of parkin. J Neurosci Res. 2012;90(5):1030–42. https://doi.org/10.1002/jnr.22828.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Sharmila A, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–6. https://doi.org/10.1038/nature07884.
Yongchao Z, Morgan MA, Yi S. Targeting neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid Redox Signal. 2014;21(17):2383–400. https://doi.org/10.1089/ars.2013.5795.
Choo YS, Vogler G, Wang D, Kalvakuri S, Iliuk A, Tao WA, et al. Regulation of parkin and PINK1 by neddylation. Hum Mol Genet. 2012;21(11):2514–23. https://doi.org/10.1093/hmg/dds070.
Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, et al. S-nitrosylation of Parkin regulates ubiquitination and compromises Parkin’s protective function. Science. 2004;304(5675):1328–31. https://doi.org/10.1126/science.1093891.
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, et al. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324(5923):102–5. https://doi.org/10.1126/science.1171091.
Selvaraju V, Taylor BS, Shasha M, Fang F, Emrullah Y, Igor V, et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet. 2010;42(1):77–82. https://doi.org/10.1038/ng.491.
Hristova VA, Beasley SA, Rylett RJ, Shaw GS. Identification of a novel Zn2+-binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J Biol Chem. 2009;284(22):14978. https://doi.org/10.1074/jbc.m808700200.
Ozawa K, Komatsubara AT, Nishimura Y, Sawada T, Kawafune H, Tsumoto H, et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci Rep. 2013;3(29):2202. https://doi.org/10.1038/srep02202.
Wahabi K, Perwez A, Rizvi MA. Parkin in Parkinson’s disease and cancer: a double-edged sword. Mol Neurobiol. 2018;55(8):6788–800. https://doi.org/10.1007/s12035-018-0879-1.
Denison SR, Fang W, Becker NA, Birgitt S, Norman K, Phillips LA, et al. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene. 2003;22(51):8370. https://doi.org/10.1038/sj.onc.1207072.
Liu J, Zhang C, Zhao Y, Yue X, Wu H, Huang S, et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat Commun. 2017;8(1):1823. https://doi.org/10.1038/s41467-017-01947-w.
Stichel CC, Augustin M, Kühn K, Zhu XR, Engels P, Ullmer C, et al. Parkin expression in the adult mouse brain. Eur J Neurosci. 2010;12(12):4181–94.
Lee SB, She J, Bo D, Kim JJ, Andrade MD, Jie N, et al. Multiple-level validation identifies PARK2 in the development of lung cancer and chronic obstructive pulmonary disease. Oncotarget. 2016;7(28):44211–23. https://doi.org/10.18632/oncotarget.9954.
Xiaodong S, Min L, Jihui H, Dengwen L, Youguang L, Xiuchao W, et al. Parkin deficiency contributes to pancreatic tumorigenesis by inducing spindle multipolarity and misorientation. Cell Cycle. 2013;12(7):1133–41. https://doi.org/10.4161/cc.24215.
Carroll R, Hollville E, Martin S. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 2014;9(4):1538–53. https://doi.org/10.1016/j.celrep.2014.10.046.
Wang H, Liu B, Zhang C, Peng G, Liu M, Li D, Gu F, et al. Parkin regulates paclitaxel sensitivity in breast cancer via a microtubule-dependent mechanism. J Pathol. 2010;218(1):76–85. https://doi.org/10.1002/path.2512.
Ikeuchi K, Hfujiwara M. Attenuation of proteolysis-mediated cyclin E regulation by alternatively spliced Parkin in human colorectal cancers. Int J Cancer. 2010;125(9):2029–35. https://doi.org/10.1002/ijc.24565.
He S, Yang S, Niu M, Zhong Y, Dan G, Zhang Y, et al. HMG-box transcription factor 1: a positive regulator of the G1/S transition through the Cyclin-CDK-CDKI molecular network in nasopharyngeal carcinoma. Cell Death Dis. 2018;9(2):100. https://doi.org/10.1038/s41419-017-0175-4.
Tay SP, Yeo CC, Chua PJ, Tan HM, Ang AX, Yip DL, et al. Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J Biol Chem. 2010;285(38):29231. https://doi.org/10.1074/jbc.M110.108241.
Lee SB, Kim JJ, et al. Parkin regulates mitosis and genomic stability through Cdc20/Cdh1. Mol Cell. 2015;60(1):21–34. https://doi.org/10.1016/j.molcel.2015.08.011.
Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3(1):55. https://doi.org/10.1038/nrc967.
Kaverina I, Straube A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 2011;22(9):968–74. https://doi.org/10.1016/j.semcdb.2011.09.017.
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010. https://doi.org/10.1002/ijc.25516.
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182. https://doi.org/10.1056/NEJM197111182852108.
Rs K, Ba K. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4(6):423. https://doi.org/10.1038/nrc1369.
Alitalo K, Carmeliet P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell. 2002;1(3):219–27. https://doi.org/10.1016/s1535-6108(02)00051-x.
Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004;14(6):433–9. https://doi.org/10.1016/j.semcancer.2004.06.006.
Mitsutoshi K, Hiroki Y, Hideki K, Takashi W, Wade PA, Eling TE. DNA methylation-mediated silencing of nonsteroidal anti-inflammatory drug-activated gene (NAG-1/GDF15) in glioma cell lines. Int J Cancer. 2011;130(2):267–77. https://doi.org/10.1002/ijc.26082.
Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2010;121(11):2373–80. https://doi.org/10.1002/ijc.23173.
Danesh J, Kaptoge S, Mann AG, Sarwar N, Wood A, Angleman SB, et al. Long-term interleukin-6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS Med. 2008;5(4):e78. https://doi.org/10.1371/journal.pmed.0050078.
Popa C, Netea MG, van Riel PL, Jw VDM, Stalenhoef AF. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res. 2007;48(4):751–62. https://doi.org/10.1194/jlr.R600021-JLR200.
Rom O, Avezov K, Aizenbud D, Reznick AZ. Cigarette smoking and inflammation revisited. Respir Physiol Neurobiol. 2013;187(1):5–10. https://doi.org/10.1016/j.resp.2013.01.013.
Freedman DM, Pfeiffer RM. Factors in association between parkinson disease and risk of cancer in Taiwan. JAMA Oncol. 2016;2(1):144. https://doi.org/10.1001/jamaoncol.2015.4151.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (81622005), and Beijing Natural Science Foundation (7172213).
Author information
Authors and Affiliations
Contributions
DD and JW conceived the theme and structure of the article and wrote the manuscript. DD, XA, and YW edited figures, and DD, JW, XA, YL, YW, HF and MW edited and revised the article, JW, YH provided materials and funding. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ding, D., Ao, X., Liu, Y. et al. Post-translational modification of Parkin and its research progress in cancer. Cancer Commun 39, 77 (2019). https://doi.org/10.1186/s40880-019-0421-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40880-019-0421-5