
Abstract
Background
Pikes, members of genus Esox, are widespread freshwater predators of the northern hemisphere, and important sport fish also. From the Carpathian basin only one species, the northern pike (E. lucius) is noted. At the same time the pike stocks living in this area show high level of phenotypic variance (e.g. various body pattern) and its growth varies highly both among and within populations. These features usually explained by the environmental diversity of the area. Whereas we think that genetic reasons -e.g. the appearance of other/new pike species in the area- may also be responsible for these observed features. Since as no detailed information have been published from the pike populations of this area, so far; we conducted phylogenetic and morphological assay on 88 pike specimens, collected from 49 Middle Danubian sampling sites.
Results
Our phylogenetic surveys showed that the northern pike appear in the study area solely, but all the three of its major lineages (Northern, Circumpolar, Southern) were indicated. Only six specimens represent the Northern lineage, collected from the western part of the study area. The Circumpolar and Southern lineages were common in the Carpathian basin, but the Southern lineage showed higher levels of haplotype diversity than the Circumpolar clade. Which indicates that only the Southern lineage is native in the area, while the other two groups could have appeared in the Middle Danubian system either spontaneously or by human introduction. Moreover, the different clades appeared in the same populations, suggesting the opportunity of inter-lineage hybridisation. From the studied morphologicalal features, the number of scales on the lateral line and the head length showed significant differences among the lineages. At the same time the body pattern of the studied individuals seems to be rather influenced by the ontogenic changes than phylogeny.
Conclusions
The high phenotypic variability of Middle Danubian northern pike populations may be due that all of its three major clades appeared and came in secondary contact in the area. In the within watershed spread of the non-native lineages the human stocking/transfer may play a considerable role.
Similar content being viewed by others

Background
Pikes (Actinopterygii: Esocidae) belong to the most characteristic piscivorous fish of the northern hemisphere [1]. This ancient teleost family consists of only seven species, from which three can be found only in North America and three others appear in Eurasia [2]. The eponymous species of the Esociformes genus, the northern pike (Esox lucius Linnaeus, 1758) has a circumpolar distribution and wide ecological tolerance [3,4,5]. Northern pike is one of the most widespread predators of the European freshwaters; moreover, it is one of the most important game and commercial fish species in the area [6,7,8]. Hence, this species has both great ecological and economical importance. Additionally, in the recent years, pike has become a preferred model organism of ecological and evolutionary biological studies, therefore its scientific importance has been continuously increasing [9,10,11].
The northern pike was known for a long time as a widely distributed species in the northern hemisphere, and the only Esox species in Europe [12]. However, it is characterised by high phenotypic variability. For example, the number of scales on the lateral line and the number of vertebrae vary in a wide range, 105–148 and 56–65 respectively [3]. Studies using both molecular and morphological methods published in recent years have modified all the genetic, taxonomic and distribution features of this taxon greatly. The phylogenetic investigations of European Esox populations showed that pikes are among those taxa whose distribution and phylogeny had been considerably affected by paleoclimatic changes [13]. For example, the Atlantic areas of Europe, which were glaciated in the ice ages, host genetically much more uniform stocks than the South European drainages [14]. In the last comprehensive study, Skog et al. analysed samples from the whole distribution area of E. lucius [15]. Their results showed that this species separated into three major lineages (Circumpolar, Northern, and Southern) only in the geological near past, 0.18–0.26 Myr ago. The existence of the latter two lineages suggests that the postglacial colonization of Europe set off from two independent refugia, situated in Western Europe and in the Danube region. Moreover, it is proved that the circumpolar is the only northern pike lineage of Eastern Asia [16].
In the last decade, two new European pike species have been described based on the results of detailed phylogenetic works [17,18,19]. The E. cisalpinus Bianco & Delmastro, 2011 [17], and E. aquitanicus [19] supposedly survived the last glaciations in refugia situated in the Appennine peninsula and Southwestern France respectively. These two recently described species are characterised by specific body colour and pattern and lower scale number on the lateral line. Moreover the E. aqutanicus, has shorter preorbital region, and it has less vertebrae than the E. lucius [18, 20, 21].
Our study area, the Carpathian basin, is located in the middle Danubian drainage. This basin was never glaciated during the ice ages [22], therefore it might have served as an extra-Mediterranean refugium and source of recolonisation to northern European areas for many plant and animal taxa [23, 24]. As many suitable lowland aquatic habitats can be found in the Carpathian basin, the pike is one of the most frequent piscivorous fish species in this area, and additionally, it is one of the most popular game fish in Hungary [25, 26]. At the same time, Hungarian pike populations express a high degree of phenotypic plasticity [27]. Moreover, the growth and length–weight relationships of Hungarian pike populations vary greatly among and within sites [28, 29], which is generally explained by the highly variable environmental conditions of the area [30]. Furthermore, their colour and body patterns are highly diverse, and the number of scales on the lateral line shows high variability but often less than 120 [31], which makes these individuals similar to the newly described European Esox species. Parallelly, scarce genetic information is available regarding the pikes from the Carpathian basin. Only a few specimens were analysed in the work of Nicod et al. [14], and these samples showed high levels of phylogenetic variability. Moreover, some of the indicated haplotypes were highly different from the ones that originated from Western Europe.
Additionally to the confusing phylogenetic background, we experienced high phenotypic variability during regular sampling campaigns. Therefore, the presence of at least two or more phylogenetically and morphologicalally distinct pike clades were hypothesized in the study area. However, it is still not clarified if the assumed clades are identical to the lineages noted by Skog et al. [15] or if an unknown (relict?) pike taxon that could be found in the study area. Moreover, as the E. cisalpinus has already been noted from the River Danube [32], it cannot be ruled out that any of the newly described pike species may be present in our study area. Therefore, the present study aims to clarify the phylogenetic features of pike populations of the Carpathian basin and to reveal if the observed genetic differences may also manifest in phenotypic traits.
Results
Phylogenetic features
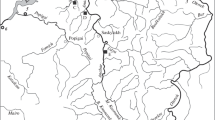
In the case of D-loop and CytB loci, 436 bp and 1088 bp long sequences were analysed respectively. In these mtDNA sequences altogether 5 and 14 polymorphic (segregating) sites, with no gaps or missing data were indicated. The 88 analysed samples were classified into four (D-loop) and eight (CytB) haplotypes (Table 1, Supplementary table 1). Nucleotide diversity for the entire assemblage ranged from 0.00381 (D-loop) to 0.00418 (CytB). Pairwise genetic differences (uncorrected p-distances) among the indicated haplotypes ranged between 0.0023 and 0.0092, and between 0.0009 and 0.0083 for the D-loop, and CytB loci respectively concerning substitutions (Table 2). Analyses using the MegaBlastN online software [33] showed that all the indicated D-loops, and five out of the eight CytB haplotypes, show a complete match with the ones that can be found in the GenBank database (Table 1). The three newly revealed CytB sequences have been uploaded into GenBank under the accession numbers MZ822418-19–20. The ML trees showed the monophyletic origin of the revealed haplotypes in both cases (Fig. 1), with only slight differences between the D-loop haplotypes. At the same time, the CytB haplotypes were separated into three distinct groups, and findings are reinforced by the results of network analyses (Fig. 2). The individuals showed uneven distribution among the three detected groups (lineages). Only six individuals (6.8%) showed the haplotypes of the Northern lineage while individuals belonging to the Circumpolar and Southern lineage dominated the samples. These two latter lineages are generally distributed in the area. And their abundances differ considerably in the Tisza (Tisa) and Száva (Sava) subdrainages only (Fig. 3). In the former 69% of the individuals, while in the latter 64% of the stock showed Southern and Circumpolar haplotypes respectively. But even in these subdrainages the different lineages may also appear on closely situated sites. Moreover, on nine sample sites stocks with mixed lineage origin were detected (Fig. 3, Supplementary table 1). The Circumpolar and Southern lineages did not show any differences by their geographic distribution patterns and altitude. While the individuals belonging to the Northern lineage appeared from sites situated to more western longitudes (Kruskal–Wallis test, H = 8.752, p < 0.01) and from significantly higher altitudes (Kruskal–Wallis test, H = 10.17, p < 0.01).

Evolutionary analysis of the indicated CytB (A) and D-loop (B) haplotypes by using the Maximum Likelihood method and Tamura-Nei model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The haplotypes appeared during our survey are highlighted by bold letter type. The lineage membership are shown by different colors (blue: Northern, red: Circumpolar, green: Southern). ML trees were built using E. reichertii and E. cisalpinus haplotypes as outgroup sequences, their Genbank accession numbers are indivcated in brackets. Colour figure online

Median-Joining network of CytB (A) and D-loop (B) sequence data of the investigated 88 northern pike individuals. Line length refers to the genetic distances of haplotypes. Each vertical line is one mutation step. Small black circles represent missing or theoretical haplotypes. Haplotypes appeared from the study area marked by bold letter type. Haplotypes which were not indicated from the area are marked by white circles. The different lineages are emphasized with different coloured frames (blue: Northern, red: Circumpolar, green: Southern). The size and coloration of pie charts refer to the number of individuals carriing the certain haplotype and to their hydrographic origin (C). For more information see Table 1 and 2. Colour figure online

Distribution of the 49 sample sites in the inner area of the Carpathian Basin. (A) and the location of the studied area in Europe (B). The circles’ area corresponds with the number of the sampled individuals. The inner area and the frame of the circles show the D-loop and CytB haplotypes, respectively. The colors and shades of circles indicate their lineage membership (blue shades: Northern, red: Circumpolar, green: Southern). Country borders are shown by grey dotted lines. Base map was generated by QGIS software using layers freely available from European Environment Agency (EEA). Color online only
Phenotypic features
All the investigated phenotypic and morphometric data are available in the Supplementary table 1. The standard body length of the analysed individuals ranged between 103 and 630 mm (mean ± SD: 289.9 ± 110.6 mm). The number of scales on the lateral line ranged between 105 and 130 (mean ± SD: 117.9 ± 5.8). The measured and counted phenotypic features of the three lineages are presented in Fig. 4. In most cases the recorded variables showed high level of within group variations. However, the largest individuals belonged to the circumpolar clade the overlapping SL data showed that no significant differences can be found in bodysize of the three lineages. From the compared phenotypic features only two showed significant differences. The standardized head length (HL/SL) of the Southern lineage and the scale number on the lateral line of the Northern lineage (Kruskal–Wallis test, H = 5.048, p < 0.05) showed significantly higher values than that of the other two lineages (Fig. 4.). The body pattern of the sampled individuals showed high level of variation (Fig. 5) but they could be classified into four major groups. The body pattern of 38 individuals showed diagonal bars. In case of 23 pike specimens, the body was dotted but these dots were arranged into stripes. More 26 individuals had dotted body pattern, while only one specimen showed longitudinal bars on its body. The body pattern proportion of the investigated specimens classified into different size groups and clades are shown in Fig. 6.

Phenotypic features of the three pike lineages (blue: Northern, red: Circumpolar, green: Southern). SL—Standard lengths (A), HL/SL—Head length in the proportion of Standard Lengths (B), PA / SL—Preanal distances in the proportion of the Standard Length values (C), PPL / SL—Prepelvic distances in the proportion of the Standard Length valuess (D), PPEC / SL—Prepectoral distances in the proportion of Standard Lengths (E), PRE / HL—Preorbital distances in the proportion of the Head Length values (F), ED / HL—Eye diameter in the proportion of the Head Length values (F) and the number of scales on the lateral line (H). Each box represents the 25% and 75% quartiles of the dataset, the band in the box is the median. The whiskers are drawn from the top of the box up to the largest data point less than 1.5 times the box height from the box (the"upper inner fence"), and similarly below the box. Values outside the inner fences are shown as circles values further than three times the box height from the box (the "outer fences") are shown as black stars. Datasets marked by red asterisk differ significantly from the others by the results of non-parametric Kruskal–Wallis pairwise comparisons (p˂0.05). Colour figure online

Coloration and body patterns of some representatives of the Southern (A-B), the Northern (C-D), and the Circumpolar (E–F) lineages. Codes beside the presented individuals showing their D-loop and CytB haplotypes, while the No refer to their collection sites, for more details see Table 1. Color figure online. All photos were taken by the authors

The body pattern features in different phylogenetic clades and lenght group categories. Distribution of individuals characterised by the same body pattern in different phylogenetic clades (A) and in certain size categories (B). Body pattern categories: Light grey: diagonal bars on the body; dark grey: dots arranged to stripes; black dotted: dotted body pattern; black: longitudinal bars on the body
Discussion
Notwithstanding that the Carpathian area served as an extra-Mediterranean refugium for numerous species and many endemic species live in this area [34,35,36], we did not find any unknown pike lineages. All sampled individuals belonged to E. lucius, and no other European Esox species were identified. At the same time, our results show that all the three known E. lucius lineages mentioned by Skog et al. [15] occur in the natural waters of the Carpathian basin (Figs. 1 and 2).
The genetic diversity of the two mitochondrial loci shows considerable differences. For D-loop, only four (two Northern, one Circumpolar, and one Southern) haplotypes were revealed from the study area. Parallelly, eight CytB haplotypes were detected from our samples. This finding might be unusual since the D-loop—although barely half as long as the other sequence analysed—is known to be more variable because it is a noncoding locus [37]. The haplotype diversity of the CytB locus showed variation among the three lineages. The Northern and the Southern lineage had two and five haplotypes, while the Circumpolar clade, in spite of its wide range, had only one haplotype. The haplotype diversity and their specific distribution feature may refer to the origin of the detected lineages. Due to the high genetic variability and the distribution data of the haplotypes of the Southern lineage, it might be the only native lineage in this area.
The other two lineages presumably appeared in the area by natural migration and/or human introduction. The Northern lineage is present mostly in the westernmost area of the Carpathian basin which assumes the recent spread of this lineage eastwards from the Alpin area. At the same time, the fact that the few individuals carrying these haplotypes appeared from distant and isolated places (e.g. Lake Velencei, or Koppány stream) presuppose that anthropogenic effects (introductions) may also play an important role in the river basin spread of this lineage. The fact that this lineage appeared from higher altitudes can be explained by the geographic characteristics of the area (Fig. 3), namely that the westernmost part of the basin is mostly highland.
The Circumpolar lineage is likely to be an introduced one in the Carpathian basin also. This assumption is reinforced by the genetic uniformity of this clade. Moreover, its wide range distribution suggest that this clade might have appeared in the area earlier than the Northern lineage. Considering the international relations of the socialist countries in the field of aquaculture in the mid twentieth century [38, 39], it can be rightly assumed that this lineage introduced from the Soviet union into one or more countries can be found in this area. And these pike stocks exploited the characteristics of the Carpathian water system [40] which allowed the natural spread of this clade in the whole basin. At the same time, the importance of pike in aquaculture and angling, might have facilitated the human-assisted spread of this nonnative lineage also in the study area. Moreover, the stockings are still ongoing in these years also. There are several natural water bodies (e.g. Lake Tisza, Lake Balaton, etc.) in Hungary, where hundreds of thousands of pike juveniles are stocked in every year for angling purposes [41, 42]. Recently we have assumptions only why these, nonnative lineages are stocked in the study area. One possible reason for the human-facilitated spread of the Circumpolar and Northern lineages can be, that they grow larger and/or faster than Southern clade. And these characteristics provide an advantage in the selection of aquaculture breeding lineages [43]. But to test this hypothesis, further studies are required.
The body pattern of the studied individuals showed high variation, but it did not related to clade membership. At the same time it seems to be in correspondence with body size. Namely the Carpathian pike juveniles, similarly to other Northern pike stocks, are mainly striped, and during the ontogeny these transversal stirpes are broken to separate dots, and most of the adults show dotted body pattern [1]. Besides, only two of the studied morphometric traits showed considerable differences among the three lineages. The standardized head length (HL/SL) of the Southern, and the number of scales on the lateral line of the European lineage, showed significantly higher values than the others. While the preorbital distance (PRE/HL), an important discriminating feature of the newly described European pike species [19], and the other studied morphometric variables did not show significant differences among lineages (Fig. 4.). These findings show the indicated three northern pike lineages may be distinguished using certain phenotypic traits, but here we have to emphasize that due to uneven group sizes (6–41-41) and the considerable overlap of these values, these features may only be used if sufficient numbers of individuals are available. Additionally, here we have to note that only mitochondrial markers were used in this study. These are widely used in phylogenetic studies [37, 44,45,46,47], although they do not provide any information if hybridisation has taken place between certain lineages. Whereas hybridisation is highly plausible in the study area, since several sampled populations show mixed-lineage origin (see Fig. 3 and Supplementary table 1). The highly variable growth and length–weight relationships of Hungarian pike populations [28, 29] were generally argued to be the consequence of the highly variable environmental conditions of the area. However, knowing these results we can reasonably assume that the detected growth differences may be at least partially due to genetic effects (e.g. the patchy distribution of certain lineages, possible hybridisation). Thus, our results suggest that allopatric pike lineages separated only in the geological near past [15] similarly to other Middle Danubian fish taxa [48, 49], came to secondary contact. This feature provides a potential for rapid adaptation to new environmental conditions [50], at the same time it hinders or reverses the long-term speciation process in its initial steps. In many cases, the introductions through the appearance of new forms and gene variants may even increase the local genetic variability [51]. However, this change may lead to a decrease in infraspecific genetic diversity, so it can be considered as a form of biotic homogenization [52, 53]. Therefore, the human-facilitated introductions of non-native lineages may also contribute to biodiversity loss.
Conclusions
The geographic distribution and the haplotype diversity data are in correspondence with the suggestion that the Danube drainage could have served as a glacial refugium for the Southern lineage of the northern pike [15]. Although the isolation of the lineages took place in the geological near past only, it has already manifested in some slight phenotypic differences. Additionally, our results suggest that not just the newly described pike species [20, 54] but the slightly isolated northern pike lineages are also threatened by human-facilitated stockings. These human-induced secondary contacts interfere with the million-year-scale speciation processes of evolution. Moreover, results also draw attention to the fact that in the case of economically exploited species the frequency and the spatial resolution of sampling can greatly influence the results of investigations.
Materials and methods
Sampling
We collected samples from 49 sites in the Carpathian basin (from Hungary, the Republic of Serbia, and Romania) by electrofishing between 2017 and 2019 (Supplementary table 1, Fig. 1). During our field collections, we paid special attention to the smaller-bodied individuals with unusual body color and pattern. Altogether 88 northern pike individuals’ fin clips were sampled for genetic investigations and stored in 96% ethanol at -20 °C until DNA extraction. After tissue sampling, all specimens were placed flat, and their left sides photographed from a perpendicular angle using a digital camera with a fixed zoom range. All individuals were returned to their original habitats after the sampling procedure.
Mitochondrial, phylogenetic investigations
DNA was isolated from 10–20 mg of fin tissue per individual using DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s instructions. The quality and quantity of the extracted DNA were checked by using a NanoDrop 2000c Spectrophotometer (Thermo Scientific, USA). The mitochondrial D-loop and Cytochrome B (CytB) loci were sequenced for taxonomic and phylogenetic analysis. Amplification of both sequences was performed using Phusion Hot Start II High Fidelity DNA Polymerase (1U) in Phusion Green HF buffer (ThermoFisher Scientific) containing 500–500 nM primers. The primers for D-loop PCR were M13CDL-D (5’ GTAAAACGACGGCCAGTAGCTCCCAAAGCTAAGATTC 3’) and M13PIDL (5’ CAGGAAACAGCTATGACGCTTGGTGGGTAACGAGTC 3’). The PCR protocol was the following: 98 °C for 1 min, then 35 cycles of 98 °C for 10 s, 60 °C for 30 s, 72 °C for 20 s, followed by a final extension at 72 °C for 5 min. Partial CytB region of mitochondrial DNA was amplified using primers LCbEluc14263 (5’ GTCATAATTCTACTCGGACTCTAACC 3’) and RCbEluc15503 (5’ CCTCCAACTTCCGGATTACAAAACCGGCGCTC 3’) [16]. The PCR protocol was 98 °C for 1 min, followed by 35 cycles of 98 °C for 10 s, 63 °C for 30 s and 72 °C for 39 s, and a final extension at 72 °C for 5 min. Following electrophoresis, the PCR product was purified from agarose gel using High Pure PCR Product Purification Kit (Roche) according to the manufacturer’s instruction. Bi-directional sequencing of the amplicons was performed by capillary electrophoresis (ABI 3130 Genetic Analyzer Device, AppliedBiosystems) using the BigDye Terminator v3.1 Cycle Sequencing Kit. The primers used for sequencing were M13F (5’ GTAAAACGACGGCCAG) and M13R (5’ CAGGAAACAGCTATGAC 3’) for D-loop amplicon, while the cytochrome B amplicon was sequenced with primers used for amplification.
Sequences were trimmed manually using FinchTV 1.4.0 (Geospiza) and aligned using MUSCLE [55] as implemented in MEGA X software [56]. Calculation of sequence polymorphism and haplotype detachment was performed using FaBox online software [57]. The obtained sequences were compared with the ones uploaded to GenBank using Blast online software [33]. Gene and nucleotide diversity were computed by DnaSP 6 [58] software. The phylogenetic analyses were made separately for the two loci. To reveal the phylogenetic relationships of our sequences, we analysed them together with already known haplotypes [14, 15, 59, 60]. Therefore, these analyses involved 21 and 13 sequence variants for the CytB and D-loop analyses respectively (Table 2). Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach based on the Tamura-Nei model [61], and then selecting the topology with superior log likelihood value. The trees are drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated (complete deletion option). At both loci, ML trees were built using the close relative pike species, E. reichertii and E. cisalpinus haplotypes (GenBank acc. numbers: KM28145, NC02326, AJ609613, and KR053430) as outgroup sequences. Phylogenetic detachments were tested by 100 bootstrap replications. Sequence divergence was calculated with the maximum composite likelihood method also in MEGA X [56]. Besides median-joining networks were constructed—using 11 (D-loop) and 19 (CytB) haplotypes—in Network 10.2.0.0. software [62]. The possible geographic and altitudinal separation of the detected phylogenetic groups were tested by non-parametric Kruskal–Wallis tests.
Phenotypic investigations
On the digital images taken from the sampled individuals the standard length (SL), head length (HL), preanal (PA), prepelvic (PPL), prepectoral (PPC) and preorbital distances (PRE) and the eye diameter (ED) were measured, and the number of scales of the lateral line was counted. We chose these variables because these are the most reliable ones in the separation of the European Esox species [17,18,19]. The measurements and counting were carried out in imageJ software [63] for each individual. The individuals were classified into groups by the results of phylogenetic investigations. The measured values were presented in the proportion of SL (HL, PA, PPL, and PPEC) or HL (ED and PRE). The among group differences of the recorded phenotypic variables were tested by non-parametric Kruskal–Wallis tests. The body pattern of the studied individuals was charecterised according to Lucientini et al. [18] and sorted into groups. We used these groups to show the relationships among body pattern, clade memberships, and body size categories.
Availability of data and material
All meristic and phenotype data used or analysed during the current study are available in a supplementary file attached to the publication. The newly indicated haplotypes’ sequence data are available in the Genbank on the following accession numbers: MZ822418-19–20. All other analysed sequence data (see: Table 1) are available in the Genbank also.
References
Craig JF. Pike: Biology and exploitation. Fish and Fisheries series Vol. 19. London: Chapman and Hall; 2013. https://books.google.hu/books?hl=hu&lr=&id=XbPyCAAAQBAJ&oi=fnd&pg=PP13&dq=Craig,+J.+(Ed.).+(2013).+Pike:+biology+and+exploitation+(Vol.+19).+Springer+Science+%26+Business+Media.&ots=QDeR1SpWqX&sig=L35CU6AxEnqTjgR8DgGBoPxN17s&redir_esc=y#v=onepage&q&f=fals. Accessed 23 Sept 2021.
López J, Chen W, Copeia GO-, 2004 U. Esociform phylogeny. Copeia. 2004;3:449–64. https://meridian.allenpress.com/copeia/article-abstract/2004/3/449/125383. Accessed 23 Sept 2021.
Kottelat M, Freyhof J. Handbook of European freshwater fishes. 2007. http://aflimno.free.fr/fichiers/AIDEGESTION/Kottelat.pdf.
Jørgensen AT, Hansen BW, Vismann B, Jacobsen L, Skov C, Berg S, et al. High salinity tolerance in eggs and fry of a brackish Esox lucius population. Fish Manag Ecol. 2010;17:554–60.
Réalis-Doyelle E, Pasquet A, Fontaine P, Teletchea F. Effects of temperature on the survival and development of the early life stages of northern pike ( Esox lucius ). Knowl Manag Aquat Ecosyst. 2022;423:10.
Crane D, Miller L, Diana J, Casselman J, Farrell J, Kapuscinski K, et al. Muskellunge and Northern Pike ecology and management: important issues and research needs. Fisheries. 2015;40:258–67. https://doi.org/10.1080/03632415.2015.1038382.
Steffens W, Winkel M. Current status and socio-economic aspects of recreational fisheries in Germany. Fish Cent Res. 1999;7:130–3. http://fisheries.sites.olt.ubc.ca/files/2016/09/7-2.pdf#page=133. Accessed 23 Sept 2021.
Skov C, Lucas MC, Jacobsen L. Pike population size and structure: Influence of density-dependent and density-independent factors. 2018.
Forsman A, Tibblin P, Berggren H, Nordahl O, Koch-Schmidt P, Larsson P. Pike Esox lucius as an emerging model organism for studies in ecology and evolutionary biology: a review. J Fish Biol. 2015;87:472–9. https://doi.org/10.1111/jfb.12712.
Pan Q, Feron R, Yano A, Guyomard R, Jouanno E, Vigouroux E, et al. Identification of the master sex determining gene in Northern pike (Esox lucius) reveals restricted sex chromosome differentiation. PLoS Genet. 2019;15(8):e1008013.
Laskowski KL, Monk CT, Polverino G, Alós J, Nakayama S, Staaks G, et al. Behaviour in a standardized assay, but not metabolic or growth rate, predicts behavioural variation in an adult aquatic top predator Esox lucius in the wild. J Fish Biol. 2016;88:1544–63.
Nelson JS, Grande TC, Wilson MV. Fishes of the World. New Jersey: John Wiley & Sons Inc.; 2016. https://books.google.com/books?hl=hu&lr=&id=E-MLDAAAQBAJ&oi=fnd&pg=PT43&dq=Nelson,+J.+S.,+Grande,+T.+C.,+%26+Wilson,+M.+V.+(2016).+Fishes+of+the+World.+John+Wiley+%26+Sons.&ots=uVi3GMq0nN&sig=yQhprgxBjIqHId29rZErp6MUbPY. Accessed 23 Sept 2021.
Bernatchez L, Wilson C. Comparative phylogeography of Nearctic and Palearctic fishes. Mol Ecol. 1998;7:431–52. https://doi.org/10.1046/j.1365-294x.1998.00319.x. Accessed 23 Sept 2021.
Nicod JC, Wang YZ, Excoffier L, Largiadèr CR. Low levels of mitochondrial DNA variation among central and southern European Esox lucius populations. J Fish Biol. 2004;64:1442–9.
Skog A, Vøllestad LA, Stenseth NC, Kasumyan A, Jakobsen K. Circumpolar phylogeography of the northern pike (Esox lucius) and its relationship to the Amur pike (E. reichertii). Front Zool. 2014;11:1–8.
Bachevskaja LT, Pereverzeva VV, Agapova GA, Grunin SI. Genetic Diversity of the Population of Northern Pike Esox lucius L. from the Rivers of the Northeastern Part of Russia. Biol Bull. 2019;46:154–60.
Bianco PG, Delmastro GB. Recenti Novità Tassonomiche Riguardanti i Pesci D’acqua Doice Autoctoni in Italia e Descrizione di Una Muova Specie di Luccio. In: Filippo G. (Ed.) Researches on Wildlife Conservation 2 (Suppl.). Italia: IGF Publishing; 2011. https://scholar.google.com/scholar?hl=hu&as_sdt=0%2C5&q=Recenti+Novità+Tassonomiche+Riguardanti+iPesci+D’acqua+Doice+Autoctoni+in+Italia+e+Descrizione+di+Una+Muova+Specie+di+Luccio.+In+Researches+on+Wildlife+Conservation+2&btnG. Accessed 23 Sept 2021.
Lucentini L, Puletti ME, Ricciolini C, Gigliarelli L, Fontaneto D, Lanfaloni L, et al. Molecular and phenotypic evidence of a new species of genus Esox (Esocidae, Esociformes, Actinopterygii): The southern pike. Esox flaviae PLoS One. 2011;6:e25218.
Denys GPJ, Dettai A, Persat H, Hautecoeur M, Keith P. Morphological and molecular evidence of three species of pikes Esox spp. (Actinopterygii, Esocidae) in France, including the description of a new species. Comptes Rendus - Biol. 2014;337:521–34.
Bertoli M, Manfrin C, Bonzi L, Pizzul E, Pallavicini A. FIRST TAXONOMICAL ANALYSES OF PIKE POPULATIONS (ESOCIDAE, ESOX) IN FRIULI VENEZIA GIULIA (NORTHEAST ITALY)/PRIME INDAGINI TASSONOMICHE A CARICO DELLE POPOLAZIONI DI LUCCIO (ESOCIDAE, ESOX) IN FRIULI VENEZIA GIULIA (NORDEST ITALIA). Ann Ser Hist Nat. 2016;26:41. https://search.proquest.com/openview/63a619191cd6711b324f6339c9088c09/1?pq-origsite=gscholar&cbl=75964. Accessed 23 Sept 2021.
Jeanroy C, Denys G. Morphological traits allow distinguishing their hybrids from the Northern pike, Esox lucius, and the Aquitanian pike, Esox aquitanicus (Actinopterygii, Esociformes). Cybium. 2019;43:227–32. https://doi.org/10.26028/cybium/2019-433-003.
Hewitt G. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc. 1996;58:247–76. https://academic.oup.com/biolinnean/article-abstract/58/3/247/2662810. Accessed 23 Sept 2021.
Varga Z. Extra-Mediterranean refugia, post-glacial vegetation history and area dynamics in Eastern Central Europe. In: Relict species. Berlin: Springer; 2009. https://link.springer.com/chapter/10.1007/978-3-540-92160-8_3.
Schmitt T, Varga Z. Extra-Mediterranean refugia: The rule and not the exception? Front Zool. 2012;9:1–12.
Demeter E. Lehalászás jelentés, 2020. év XXVI(1):1–30. Agrárközgazdasági Intézet/online at: https://www.aki.gov.hu/product/lehalaszas-jelentes-2006-2020/. 2020:1–59.
Harka Á, Sallai Z. Magyarország halfaunája. Szarvas: Nimfea Természetvédelmi Egyesület; 2004. https://scholar.google.com/scholar?hl=hu&as_sdt=0%2C5&q=Harka+Sallai+2004&btnG. Accessed 23 Sept 2021.
Berinkey L. Halak-Pisces. Budapest: Akadémiai Kiadó; 1966. https://scholar.google.com/scholar?hl=hu&as_sdt=0%2C5&q=Berinkey+L.%2C+%281966%29+Halak-Pisces%2C+Fauna+Hungarie%2C+Akadémiai+Kiadó+%2C+Budapest%2C+132+p.&btnG. Accessed 23 Sept 2021.
Harka Á. Growth of pike (Esox lucius L.) in the section of the Tisza river at Tiszafüred. Tiscia. 1983;18:105–14. http://digit.bibl.u-szeged.hu/00000/00066/00018/tiscia_018.pdf#page=110. Accessed 23 Sept 2021.
Takács P, Kovács B, Farkas A, Dévai G. A csuka (Esox lucius Linnaeus, 1758) populációinak növekedésvizsgálata különböző környezeti adottságú halastavakban és természetes vízterekben. Halászatfejlesztés. 2003;28:33–40. http://real.mtak.hu/74862/. Accessed 23 Sept 2021.
Haraszthy L. Natura 2000 fajok és élőhelyek Magyarországon. Csákvár: Pro Vértes Közalapítvány; 2014. https://scholar.google.com/scholar?hl=hu&as_sdt=0%2C5&q=Haraszthy%2C+L.+%282014%29.+Natura+2000+fajok+és+élőhelyek+Magyarországon.+Pro+Vértes+Közalapítvány%2C+Csákvár%2C+954.&btnG. Accessed 23 Sept 2021.
Takács P, Bánó B, Czeglédi I, Ferincz Á, Kern B, Preiszner B, et al. Hány csukafaj él a Kárpát-medencében?= How many Pike (Esox) species live in the Carpathian Basin? Pisces Hungarici. 2018;12:67–70. http://real.mtak.hu/90303/1/Takacs_et.al_Pisces.Hungarici_2018_csuka.pdf. Accessed 23 Sept 2021.
Szabó T. A csuka biológiája és tenyésztése. Gödöllő: Szent István Egyetemi Kiadó; 2016. http://real.mtak.hu/118810/1/AWETH202002146155_doi.pdf. Accessed 23 Sept 2021.
Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schäffer AA. Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24:1757–64.
Antal L, László B, Kotlík P, Mozsár A, Czeglédi I, Oldal M, et al. Phylogenetic evidence for a new species of Barbus in the Danube River basin. Mol Phylogenet Evol. 2016;96:187–94. https://www.sciencedirect.com/science/article/pii/S1055790315003796. Accessed 23 Sept 2021.
Hurdu B, Puşcaş M, Turtureanu P, Ninetić M, Coldea G, Zimmermann N. Patterns of plant endemism in the Romanian carpathians (South-Eastern Carpathians). Contrib Bot. 2012;47:25–38. https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.707.2306&rep=rep1&type=pdf. Accessed 23 Sept 2021.
Marić S, Stanković D, Wanzenböck J, Šanda R, Erős T, Takács P, et al. Phylogeography and population genetics of the European mudminnow (Umbra krameri) with a time-calibrated phylogeny for the family Umbridae. Hydrobiologia. 2017;792:151–68.
DeSalle R, Schierwater B, Hadrys H. MtDNA: The small workhorse of evolutionary studies. Front Biosci. 2017;22:873–87. https://doi.org/10.2741/4522.
Ribiánszky M. Magyar-szovjet halászati együttműködés. Halászat. 1969;15(2):33. https://scholar.google.com/scholar?hl=hu&as_sdt=0%2C5&q=Ribiánszky+M+%281969%29+Magyar-szovjet+halászati+együttműködés+Halászat+15%282%29%3A33.+%28in+Hungarian%29&btnG. Accessed 23 Sept 2021.
Tahy B. A magyar halászat kapcsolatai. Csehszlovákia. Halászat. 1975;68(2):37. https://doi.org/10.1007/s10750-017-3147-x.
Dövényi Z. A Kárpát-medence földrajza. Budapest: Akadémiai Kiadó; 2012.
URL1. http://www.hirbalaton.hu/250-ezer-csukat-telepitettek-a-balatonba-tvkeszthely-hu/. Accessed 14 Apr 2022.
URL2. http://sporthorgasz.eu/2019/05/08/lezajlott-a-csuka-ivadekok-telepitese/. Accessed 14 Apr 2022.
Chavanne H, Janssen K, Hofherr J, Contini F, Haffray P, Aquatrace Consortium, et al. A comprehensive survey on selective breeding programs and seed market in the European aquaculture fish industry. Aquac Int. 2016;24:1287–307.
Kitamura T, Takemura A, Watabe S, Taniuchi T, Shimizu M. Mitochondrial DNA analysis for the cytochrome b gene and D-loop region from the bull shark Carcharhinus leucas. Fish Sci. 1996;62:21–7. https://www.jstage.jst.go.jp/article/fishsci1994/62/1/62_1_21/_article/-char/ja/. Accessed 23 Sept 2021.
Aboim MA, Menezes GM, Schlitt T, Rogers AD. Genetic structure and history of populations of the deep-sea fish Helicolenus dactylopterus (Delaroche, 1809) inferred from mtDNA sequence analysis. Mol Ecol. 2005;14:1343–54. https://doi.org/10.1111/j.1365-294X.2005.02518.x.
Lalitha R, Chandavar VR. Intraspecific variations in Cyt b and D-loop sequences of Testudine species, Lissemys punctata from south Karnataka. J Adv Res. 2018;9:87–95. https://www.sciencedirect.com/science/article/pii/S209012321730111X. Accessed 23 Sept 2021.
Sun C, Xuan Z, Liu H, Jiang T, Science JY-RS in M, 2019 U. Cyt b gene and D-loop sequence analyses of Coilia nasus from the Rokkaku River of Japan. Reg Stud Mar Sci. 2019;32:100840. https://www.sciencedirect.com/science/article/pii/S235248551930180X. Accessed 12 Sept 2021.
Takács P, Ferincz Á, Imecs I, Kovács B, Nagy AA, Ihász K, et al. Increased spatial resolution of sampling in the Carpathian basin helps to understand the phylogeny of central European stream-dwelling gudgeons. BMC Zool. 2021;6:1–11.
Zangl L, Daill D, Gessl W, Friedrich T, Koblmüller S. Austrian gudgeons of the genus Gobio (Teleostei: Gobionidae): A mixture of divergent lineages. J Zool Syst Evol Res. 2020;58:327–40.
Stemshorn KC, Reed FA, Nolte AW, Tautz D. Rapid formation of distinct hybrid lineages after secondary contact of two fish species (Cottus sp.). Mol Ecol. 2011;20:1475–91.
Clarke GM, Young AG. Genetics, demography and viability of fragmented populations. Cambridge: Cambridge University Press; 2000. https://ci.nii.ac.jp/naid/10016734293/. Accessed 23 Sept 2021.
Rahel FJ. Homogenization of freshwater faunas. Annu Rev Ecol Syst. 2002;33:291–315.
Olden J. Biotic homogenization: a new research agenda for conservation biogeography. J Biogeogr. 2006;33:2027–39. https://doi.org/10.1111/j.1365-2699.2006.01572.x.
Denys GP, Lauga T, Delamstro G, Dettaï A. S7 characterization of Western European pikes Esox spp (Actinopterygii, Esociformes). Cybium Rev Int d’Ichtyologie. 2018;42:221–8. https://doi.org/10.26028/cybium/2018-423.
Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Villesen P. FaBox: An online toolbox for FASTA sequences. Mol Ecol Notes. 2007;7:965–8.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–302. https://academic.oup.com/mbe/article-abstract/34/12/3299/4161815. Accessed 23 Sept 2021.
Grande T, Laten H, López JA. Phylogenetic relationships of extant esocid species (Teleostei: Salmoniformes) based on morphological and molecular characters. Copeia. 2004;2004:743–57. https://meridian.allenpress.com/copeia/article-abstract/2004/4/743/114919. Accessed 23 Sept 2021.
Javadian O, Hazaie K, Yousefian M, Lalolie F. Genetic diversity of pike (Esox lucius) in the south Caspian Sea using mtDNA sequences. Breeding and Aquaculture Sciences Quarterly. 2013;1:35–40. https://www.sid.ir/en/Journal/ViewPaper.aspx?ID=347337. Accessed 23 Sept 2021.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–26.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Rasband WS. ImageJ. Bethesda: National Institutes of Health; 2012.
Acknowledgements
We would like to thank editor Lisa Heinke and the three anonymous reviewers for their helpful comments and discussion.
Funding
Open access funding provided by Balaton Limnological Research Institute. Péter Takács was supported by the OTKA FK140902 grant and the Bolyai Fellowship of the Hungarian Academy of Sciences. Balázs Kovács has received funding from the EFOP-3.6.3-VEKOP-16–2017-00008 grant, co-financed by the European Union and the European Social Fund, and the Higher Education Institutional Excellence Program FEKUT2019: TUDFO/ 47138/2019-ITM awarded by the Ministry of Human Capacities (Hungary). Attila András Nagy was supported by the Collegium Talentum Programme of Hungary. Eszter Csoma was supported by the Bolyai Fellowship of the Hungarian Academy of Sciences. Árpád Ferincz was supported by the Ministry of Innovation and Technology within the framework of the Thematic Excellence Programme 2020, National Challenges Subprogramme (TKP2020- NKA-16). Ádám Staszny was supported by the OTKA PD138612 grant. Krisztián Nyeste was supported by the New National Excellence Program of the Ministry for Innovation and Technology in Hungary (ÚNKP-21–4) and by the project no. TKP2020-IKA-04 which has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the 2020–4.1.1-TKP2020 funding scheme.
The research presented in this article was carried out with the support of the NKFIH–872–3/2020 project. The funders provided only the monetary and material background of our study; they had no role in study design, data collection, analysis, publication or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
The study was designed by PT, PT, BB, IC, TE, ÁF, BG, BBK, AAN, KNy, VL, BP, SS, ÁS, ZV, and AW collected the samples. BK, and ECs conducted the laboratory works. PT and BK made the statistical analyses. PT wrote the article with critical feedback from all co-authors The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Our research complies with institutional, national, and international guidelines, and approved by the Fishery Department of the Ministry of Agriculture (Hungary) (reg. numb.: HHgF/230–4/2016, OKTF-KP/517–2/2016, HaGF/64/2020).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary table 1. General, geographic, genetic, phenotype and morphometric data of the analysed 88 pike individuals. Name of waterbody and sample site, their coordinates, subdrainage, country of origin, altitude above seal level (Alt. asl). Codes and colors of haplotypes (blue: Northern, red: Circumpolar, green: Southern) correspond with Fig 1-2-3, and Table 1. The collectors’ inicials are identical with the authors’ initials of the ms. Sites which Esox population is charecterised by mixed phylogenetic origin are emphasized with bold letter type.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Takács, P., Bánó, B., Czeglédi, I. et al. The mixed phylogenetic origin of northern pike (Esox lucius Linnaeus 1758) populations in the Middle Danubian drainage. BMC Zool 7, 28 (2022). https://doi.org/10.1186/s40850-022-00129-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40850-022-00129-6