
Abstract
Background
Intensive care unit (ICU) survivors often suffer from cognitive, physical and mental impairments, known as post-intensive care syndrome (PICS). ICU follow-up clinics may improve aftercare of these patients. There is a lack of evidence whether or which concept of an ICU follow-up clinic is effective. Within the PINA study, a concept for an ICU follow-up clinic was developed and will be tested in a pilot randomised controlled trial (RCT), primarily to evaluate the feasibility and additionally the potential efficacy.
Methods/design
Design: Pilot RCT with intervention and control (usual care) arms plus mixed-methods process evaluation. Participants: 100 ICU patients (50 per arm) of three ICUs in a university hospital (Regensburg, Germany), ≥ 18 years with an ICU stay of > 5 days, a sequential organ failure assessment (SOFA) score > 5 during the ICU stay and a life expectancy of more than 6 months.
Intervention: The intervention will contain three components: information, consultation and networking. Information will be available in form of an intensive care guide for patients and next of kin at the ICU and phone support during follow-up. For consultation, patients will visit the ICU follow-up clinic at least once during the first 6 months after discharge from ICU. During these visits, patients will be screened for symptoms of PICS and, if required, referred to specialists for further treatment. The networking part (e.g. special referral letter from the ICU follow-up clinic) aims to provide a network of outpatient care providers for former ICU patients. Feasibility Outcomes: Qualitative and quantitative evaluation will be used to explore reasons for non-participation and the intervention´s acceptability to patients and caregivers. Efficacy Outcomes: Health-related quality of life (HRQOL) will be assessed as primary outcome by the physical component score (PCS) of the Short-Form 12 Questionnaire (SF-12). Secondary outcomes encompass further patient-reported outcomes. All outcomes are assessed at 6 months after discharge from ICU.
Discussion
The PINA study will determine feasibility and potential efficacy of a complex intervention in a pilot RCT to enhance follow-up care of ICU survivors. The pilot study is an important step for further studies in the field of ICU aftercare and especially for the implementation of a pragmatic multi-centre RCT.
Trial registration
ClinicalTrials.gov, NCT04186468. Submitted 2 December 2019
Similar content being viewed by others

Background
Due to medical progress the number of intensive care unit (ICU) survivors increased over the past decades in industrial nations [1, 2]. As a result of the ICU stay, ICU survivors often suffer from cognitive, physical and mental impairments, also known as post-intensive care syndrome (PICS) [3,4,5]. Epidemiological data from international studies on PICS in general are very scarce. However, it is assumed that one-half or more of former ICU patients will suffer from some component of PICS after discharge from ICU [6,7,8,9,10]. Estimates of the incidence of muscle weakness in ICU survivors in the USA, for example, range from 20 to 80% [11]. 30–80% of patients have cognitive impairments and 10–50% suffer from post-traumatic stress disorders (PTSD) [12,13,14].
These impairments are associated with a higher utilization of medical services [15,16,17] and a reduced (health-related) quality of life (HRQOL) [18, 19]. In addition, former ICU patients often show up in aftercare as multi-morbid patients and it has been shown that the provision of continuity of care for multi-morbid people is a special challenge [20]. This is one of the reasons why specific follow-up services are recommended for these patients [21].
There are various aftercare models potentially addressing PICS [22, 23], including ICU follow-up-clinics [24]. Follow-up services in general vary, for example, in the way they are managed (e.g. led by nurses or intensivists) [25], the type of consultation (e.g. conducted face-to-face or by telephone) or the number of consultations (e.g. weekly or monthly). They may also differ in the criteria used to select patients (e.g. all ICU survivors or stratified risk groups). Even within the concept of ICU follow-up clinics there currently exists no uniform concept, which makes it difficult to assess effectiveness [26]. Thus, there is only some evidence of little or no positive effects on mortality, HRQOL, PTSD or depression. No negative effects of ICU follow-up clinics are reported. In the UK, the first ICU follow-up clinic was established in 1985 and by 2006 almost every third ICU was linked to an ICU follow-up clinic [27]. Also in the USA and Scandinavian countries, various models of ICU follow-up clinics or services exist [24, 26, 28], but none in Germany so far.
Following the recommendations for the development and evaluation of complex interventions [29], we formed a multi-disciplinary stakeholder group composed of researchers and health care professionals to develop a concept for a complex intervention in ICU aftercare, based on existing literature and extensive qualitative research with health care professionals, ICU survivors and their next-of-kin. The primary objective of this pilot study is to evaluate whether this concept of a pragmatic randomised controlled trial (RCT) on the effects of an ICU follow-up clinic is feasible in terms of recruitment, randomisation, intervention and follow-up. Additionally, we want to explore if the ICU follow-up clinic itself shows effects in improving physical HRQOL of ICU survivors after a prolonged ICU stay (> 5 days) and use the results for sample size planning for the future pragmatic trial.
Methods/design
Reporting of this study is based on the SPIRIT Checklist, a guidance for content of clinical trial protocols [30, 31].
Scientific hypothesis
A pilot study of a pragmatic RCT on the effects of an ICU follow-up clinic shows to be feasible in terms of recruitment, randomisation, intervention and follow-up. The pilot study indicates improved physical HRQOL of ICU survivors being treated at the ICU follow-up clinic.
Trial design
This study is a pragmatic, single-centre, superiority, two-armed pilot RCT to determine feasibility of a future trial comparing usual care with an ICU follow-up clinic. To explore acceptability and feasibility, we will also conduct a mixed-methods process evaluation as part of the pilot RCT. Additionally, as basis for sample size planning for the future trial, we want to assess patient reported outcomes to investigate potential efficacy. Figure 1 shows an overview of the study design and data collection procedures. Figure 2 reports the schedule for enrolment, interventions and assessment according to the SPIRIT template. The pilot trial started in December 2019 and is scheduled to last until October 2020. Patients will be followed-up for 6 months after ICU discharge.

Overview of the study design, participant flow and a coarse description of the intervention components

Schedule of enrolment, interventions, and assessments based on the SPIRIT recommendation
Participants
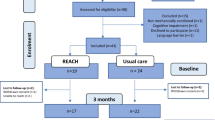
We will recruit participants meeting the inclusion criteria from three intensive care units (two medical and one surgical ICU) at the university hospital Regensburg, Germany. There will be 50 participants each in the intervention and control group. Eligible patients are screened on a daily basis by the ICU follow-up clinic team (study physician and nurse). Patients will be approached at the end of their ICU stay and will receive no incentive. The physician will explain the pilot trial, hand out the study information flyer and will be available to answer questions. Written informed consent will be obtained at the end of the ICU stay or shortly afterwards, when patients are transferred to a normal ward. Figure 3 illustrates the participant flow in the presented study. If the selected participant refuses to participate in the study, we will record reasons for non-participation.

Planned participant flow in the PINA study
Inclusion criteria
Patients will be eligible to participate if they meet the following criteria: 18 years or older, duration of ICU stay more than 5 days, SOFA (sequential organ failure assessment) score greater than five at any time of the ICU stay and expected survival time greater than 6 months estimated by intensivists.
Exclusion criteria
Patients will be excluded if they are less than 18 years old, gave no written informed consent (unable or unwilling), are not expected to survive 6 months after hospital discharge, are unable to complete questionnaires or have insufficient German language skills.
Sample size
We chose a total of 100 participants for the pilot study, i.e. 50 per arm. This sample size is expected in literature to allow for a detailed analysis of feasibility and to be sufficient for an estimation of the difference in HRQOL (continuous variable) between intervention and control group which will be used for sample size estimation for the future pragmatic trial [32, 33].
Randomisation
We will use computer-generated permuted block randomisation with blocks of size 10 to ensure balance between groups over short time spans, such as shifts and days of the week, as well as over the entire course of the study [34]. Treatment assignments (ICU follow-up clinic, usual care) will be placed in opaque, sequentially numbered envelopes prepared by the study team who has no contact with participants. The responsible study physician will only know the assignment after informed consent of the participant. The study physician will inform the participant about the result of the randomisation. Blinding of the study physician or participants after randomisation will not be possible. However, the research team performing data analysis will be blinded with regard to the participant’s allocation to intervention or control group.
Intervention
The intervention was developed in a participatory process. The participatory development process of the intervention included one-on-one interviews with former intensive care patients (n = 26) and with next of kin of former intensive care patients (n = 23, not necessarily belonging to the interviewed patients). Six focus group discussions (n = 41, average duration of 97 min) and six expert interviews were conducted to capture also the perspective of the health care professionals. In total, persons from nine different professions were interviewed, with one third of the participants being physicians, followed by nurses (23%) and physiotherapists (17%). In addition, a short online survey with 15 items was sent to the health care professionals (n=46).
This information was evaluated, summarised and integrated with evidence from the literature and evidence from claims data analysis to create a first draft of the intervention [35]. The further development of the intervention was then carried out in 2 workshops with 23 and 21 participants each, composed of different health care professionals (physicians, nurses, therapists) and scientists (involved authors of the paper: MR, SB, CB, CF, VB, MM, CA).
The first workshop was used for a rough conceptualisation based on intervention mapping, while the second workshop was used for further specification of the concept according to the design-thinking approach. The results of both workshops were distributed to all participants and interested parties for correction. The concept was then finalised and written down by the interdisciplinary project team consisting of physicians, scientists and nurses. This extensive development process was assumed to possibly increase the effectiveness of the intervention.
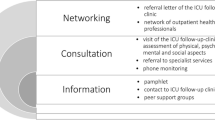
The intervention will contain three main components: information, consultation and networking (see Fig. 4). The design and timing of the appointment(s) has been determined by focus group interviews and an online survey with health care professionals. Information to participants will be provided by a pamphlet (developed by ICU steps [36] and translated by Deutsche Sepsishilfe and German Society of Skilled Nursing and Functional Services e.V. (DGF) [37]). Further, patients will have the possibility to contact the ICU follow-up clinic team by telephone at any time during the intervention. Study participants and their next of kin will be encouraged to join two self-help group meetings. A self-help group meeting will include a short information talk on a topic that has previously been shown in the consultations in the ICU follow-up clinic to be important to most participants. The lecture will be given by a member of the ICU follow-up clinic or by an outpatient health care professional, depending on the selected topic. Afterwards, both the participants and their next of kin will have the opportunity to get to know each other and to share their experiences. Patients and their next of kin will meet in separate rooms. This is assumed to facilitate disclosure of possibly difficult situations patients or next of kin are experiencing during the caregiving process.

Main components of the ICU follow-up clinic
Consultation will be provided in appointments at the ICU follow-up clinic at least once at about 2 months post-ICU discharge: initially, a checklist will be completed to assess physical, mental, cognitive and social functioning. The checklist will be based on standardised questionnaires (Mini-Cog [38], PHQ-8 [39], GAD-7 [40], PTSS-10 [59]), physical examination (chair rise test [41] and hand grip strength [42]), medical check-up (including blood pressure and body weight) and structured questions asked by the study physician (including symptoms of dysphagia and neuropathy). Results from this screening checklist will be used by the study physician of the ICU follow-up clinic to guide further treatment and thus to select the appropriate medical specialties, for example. Later, participants and next of kin will have the opportunity to discuss their ICU experience and to visit the ICU. This procedure has the potential to prevent posttraumatic stress by reintegrating traumatic memories. Where indicated, participants will be referred to outpatient medical specialists and/or outpatient therapists (ergotherapists, psychotherapist, logopedics, physiotherapists). After each clinic visit, a referral letter of the ICU follow-up clinic will be written primarily to the treating general practitioner and discussed with the participant. All intervention treatments will use the standard health care system processes. If the participant is not mobile, the ICU follow-up clinic team will visit him or her at home. All parts of the assessment can also be conducted at the patients’ home. Only the study physician’s letter and any referrals are then sent to the patient by mail in case of a home visit. Following the appointment, participants will be monitored by phone (one or more times depending on their needs) by the study physician. The physician will first explore the patient’s general state of health and then will ask to what extent the recommended interventions have been accepted and implemented. Any physical, mental or cognitive impairment will be inquired according to several items addressed during the visit in the ICU follow-up clinic. Based on monitoring results, participants may be encouraged to visit the ICU follow-up clinic once more or to contact their general practitioner. The third component concerns the establishment of a network of health care professionals who agree to care for participants of the ICU follow-up clinic or to provide information to other health care professionals. Since PICS can affect all health-related domains of former intensive care patients, the members of the network should represent different professions, disciplines and health care sectors. The referral letter of the ICU follow-up clinic is also intended to promote cooperation with the physicians and therapists providing further treatment.
ICU follow-up clinic personnel
The ICU follow-up clinic will be led by a physician (ICU specialist) with support from an intensive care nurse. The nurse will coordinate the appointments, e.g. via telephone calls, and will take care of collecting the questionnaires. The physician will provide medical information during the ICU stay in present and via telephone during the follow-up period. They will focus on consultation, which will include interpreting the questionnaires, assessing physical, mental and social functioning and the referral to specialists. To improve adherence to the intervention, both a study manual and an intervention manual will be provided to all who are involved. The manuals contain the following information (Table 1):
Control treatment
Participants in the control group will receive the usual care without any additional information or consultation (no intensive care follow-up after hospital discharge), since there are not any ICU follow-up clinics in Germany. In addition, there is insufficient evidence on the effectiveness of ICU follow-up clinics in other countries; Schofield-Robinson et al. therefore propose to consider only the ICU follow-up clinic compared to standard care [24].
Measures/outcomes
Participant characteristics
At baseline, sociodemographic data (e.g. age, gender, socioeconomic and marital status) as well as disease- and therapy-related characteristics will be extracted from the patient data management system. Disease-related characteristics include reason for admission to the ICU, as well as main and secondary diagnoses. Furthermore, the severity of disease will be captured by the Sequential Organ Failure Assessment (SOFA) score and the Simplified Acute Physiology Score (SAPS). Therapy-related characteristics will include characteristics of the ICU stay (e.g. duration, invasive ventilation and use of extracorporeal life support). HRQOL at baseline will only be assessed in form of the EQ-5D Visual Analogue Scale (VAS) [43] in order to minimise the burden on the participants during the acute phase of disease.
Feasibility outcomes
We will include process evaluation to explore aspects of feasibility and acceptability of both the trial procedures and the intervention. We will explore aspects of implementation, namely how intervention delivery is achieved and what is delivered (including dose, fidelity, reach and adaptions) [29]. Furthermore, we will investigate mechanisms of impact, as well as unanticipated pathways and consequences. We will use a logic model (Fig. 5) to frame our evaluation questions [44].

Overview of the components of the process evaluation in the PINA study according to the logic model
The process evaluation will include qualitative and quantitative components [45]. Figure 5 shows an overview of the process evaluation components and contains exemplary questions. The implementation of the trial and the intervention are influenced by context factors, which will also be documented and considered in the evaluation. The quantitative data will be collected during the intervention period, focusing on intervention fidelity and dose, and will be evaluated by frequencies. This will include, for example, the calculation of recruitment and dropout rate. Furthermore, after each visit, the study physician or nurse will take notes on characteristics of the visit in a standardised observation sheet. Acceptability of the study as well as of the intervention is in part also quantitatively recorded (e.g. via response rates). The qualitative data will be collected at the end of the intervention. Semi-structured one-to-one interviews will be conducted with health care professionals who delivered the intervention, participants (control and intervention group), next of kin and outpatient health care professionals, who will be involved in the treatment of the participants. In some circumstances, paired interviews (participants and next of kin) may be appropriate. We aim to conduct qualitative interviews with 10 participants in the control and the intervention group, respectively. In order to achieve heterogeneity among the participants in the interviews, we will purposively contact participants with different sociodemographic and disease related characteristics (e.g. age, sex, severity of disease). A researcher with qualitative research experience will conduct the interviews. The researchers are part of the PINA study team. We will design a topic guide to explore the following aspects: general opinions towards the study including acceptability and the willingness to undergo randomisation (all participants), general opinions towards the intervention, views on the intervention components, facilitators and barriers for attendance and adherence, the perceived impact of the intervention and possible improvements (intervention group). All interviews will be audio recorded, transcribed verbatim and de-identified with regard to the identity of the interviewee and mentioned clinicians. Content analysis will be performed using a computer-assisted qualitative data analysis software package (Atlas.ti).
In order to evaluate feasibility, we will consider, e.g. consent rate, attrition rate, adherence rate to the intervention, percentage of missing values in connection with the results of the qualitative interviews on feasibility and acceptability of the study and its implementation. We have deliberately decided against certain thresholds because there will not be a single threshold above which the RCT is not feasible anymore. Rather, our intention is to adapt individual parts of the pilot study to make a later large-scale study possible.
Potential efficacy outcomes
Primary outcome
Primary efficacy outcome will be the physical health-related quality of life at 6 months after informed consent/ICU discharge. We chose this outcome as primary outcome because physical impairments are very common at 6 months. For example, a study from the UK reported mobility problems 6 months after the ICU stay in 64% of ICU survivors [46]. A prospective multi-centre study in Germany also showed that the limitation of the physical component is more pronounced at 6 months compared to the mental component [47]. Physical health-related quality of life will be assessed by the physical component scale (PCS) of the Short Form-12 self-report questionnaire (SF-12) [48]. This comprehensive, generic questionnaire comprises overall twelve items resulting in a physical (PCS) and mental component scale (MCS, see secondary outcomes), which will be scored according to published algorithms (German norm values; resulting in a standard score with mean = 50 and standard deviation = 10) [49]. Scores can range between 0 and 100, with higher values indicating higher HRQOL. The questionnaire has been used in critically ill patients before [23, 50], takes only minutes to complete and can be self-completed or interviewer assessed. Psychometric properties have not been tested in former critically ill patients so far, but studies on validity and reliability of the SF-12 in populations of older patients, which also applies to most former intensive care patients, show acceptable measurement characteristics [51, 52]. In our study we will use the German translation with a 1-week recall period [53].
Secondary outcomes
Secondary efficacy outcomes focus on the most relevant sequelae of ICU patients and encompass physical, mental and social impairments. The mental component summary score (MCS) of the SF-12 questionnaire will be the first of our measured secondary outcomes. Activities of Daily Living (ADL) will be assessed by the Barthel-Index [54, 55], which evaluates ten everyday functions. The degree of independence or care dependence can be assessed on a score ranging between 0 (complete need for care) and 100 points (independence). The Chair Rise Test [41, 56] and measurement of the hand grip strength will be used to assess participants‘ physical functioning and muscular strengths. In the Chair Rise Test, anyone who cannot get up from a chair at normal height five times in 11 s or less without supporting himself with his arms is considered to be at a higher risk of falling. The hand grip strength as an indicator of overall muscle strength will be assessed using a digital dynamometer (Jamar Plus+ Digital Hand Dynamometer) [42, 57]. We will measure the grip strength of both hands and use the maximum of these values for comparison with the standard values of Dodds et al. [58] stratified by age and sex expressed in percentiles, mean and standard deviation. The assessment of psychopathological symptoms comprises anxiety and panic disorder, depression and post-traumatic stress disorder (PTSD). PTSD is measured by the German translation: (Maercker, A. (1998) Posttraumatische Stress Skala-10 (PTSS-10) – deutsche Version modifiziert nach Schüffel u. Schade, unpublished manuscript, Universität Zürich, Klinische Psychologie II) of the Post-Traumatic Stress Syndrome 10-Questions Inventory (PTSS-10) [59], which consists of two parts. The first part assesses memories of traumatic experiences during the ICU stay (e.g. nightmares) and the second part measures the intensity of ten PTSD symptoms (e.g. emotional numbing) experienced presently by the participants. Each symptom is rated from one (never) to seven (always). A total score of more than 35 predicts a likely diagnosis of PTSD [60]. The other psychopathological symptoms will be measured by using modules from the German version of the Short Form of the Patient Health Questionnaire (PHQ-D) [61]. This brief screening instrument with 15 items is designed to establish DSM-IV (Diagnostic and Statistical Manual of Mental Disorders) [62] criteria-based psychiatric diagnoses. We will use the questions on depression and anxiety and panic disorders. The extent of ambulatory and stationary health care use among the former ICU patients will be assessed by self-reported contacts with health services using a questionnaire. In addition, HRQOL of next of kin will be assessed using the SF-12 questionnaire (MCS and PCS).
Outcome assessment
Outcomes will be assessed at 6 months after discharge from ICU. Study participants will be asked to visit the study centre for outcome assessment. Trained study personnel will hand out self-report questionnaires, provide standardised instructions and perform physical measurements. Missing data will be minimised by having study personnel available at all times to check the completeness of the questionnaires or to answer participants´ questions. If participants cannot visit the study centre, home visits will be scheduled. If a participant discontinues the trial before outcome assessment, only the baseline data and the reasons for discontinuation are included in the analysis.
Data analysis
A Data use and Access Committee, consisting of the three consortium partners, is part of our study, which will monitor the entire planned use of the data and releases it for analysis (see Fig. 6). We will write a statistical analysis plan before doing any data analysis. Group allocation will be masked by our trust centre during analysis of the primary outcome. The treatment effect (ICU follow-up clinic) on HRQOL as primary outcome and the secondary outcomes will be assessed using analysis of covariance according to the intention-to-treat-principle. Thus, all randomly assigned participants will be included in a complete case analysis. Multiple imputation will be considered for missing follow-up data as part of sensitivity analysis [63]. Descriptive statistics will be used to determine participant characteristics and to check their distribution at baseline in the intervention and control group. For sensitivity analysis, a per-protocol analysis will be performed additionally. The primary outcome physical HRQOL will be compared 6 months post randomisation. We will calculate point and interval estimates with the respective confidence intervals for the difference in medians. Secondary analysis will be performed depending on scale level and for descriptive purposes only. No formal hypothesis testing will be performed.

Flow of study data in the PINA project
Ethical principles and description of risks
The institutional Ethics Committee of the University of Regensburg (19-1522-101) approved the study protocol. The study is planned and conducted in accordance with the declaration of Helsinki, the medical professional codex, the European General Data Protection Regulation (DS-GVO) and the Federal Data Protection Act (BDSG). Participants (and next of kin) will participate voluntarily and will give written informed consent. Participants will be informed that they can cancel their participation at any time without disclosing reasons for their cancellation and without negative consequences for their future medical care. This also applies to participants in the intervention group who already made use of ICU follow-up services.
The risks for participants arising from participation in the study are generally considered low. No negative effects of ICU follow-up clinics are reported [24]. Therefore, there are no specific risks related to the intervention and thus no rules for stopping the intervention.
Data collection, management and privacy issues
For study purposes, quantitative and qualitative primary data as well as clinical data will be collected. In order to keep the burden on the participants as low as possible, as much data as possible will be extracted directly from the hospital records (contact information, disease- and therapy-related characteristics, sociodemographic characteristics). More detailed information on sociodemographic characteristics will be obtained directly from the participants. Participants themselves will provide information on the primary outcome physical HRQOL at baseline and follow-up. At follow-up, further patient-reported outcomes are recorded as well as self-reported health care utilisation and physical functioning (e.g. Chair Rise Test). HRQOL of next of kin will also be recorded at follow-up. Personal information will only be collected on the consent form. These will be stored in the Trust Centre in locked cabinets without access by unauthorised persons.
All data will be handled according to the Medical Confidentiality Rules and German Federal Data Protection Act (BDSG), as well as the European General Data Protection Regulation (GDPR/DS-GVO) and all subordinate acts and ordinances. Except for contact information required for phone monitoring and home visits, all data will be recorded and stored pseudonymously. Person-identifying data will be handled by a trust centre and stored separately from study data and can only be linked via a separately stored pivot table on a PC without access to the internet. The chances that an illicit link can be made between the data are very small in light of the precautions we have taken. Therefore, the risk of an unauthorised de-pseudonymisation of the study participant’s data is considered extremely low (see recital 75 DS-GVO). In this context, we have drawn up a comprehensive data protection concept. All study related data and documents will be stored on protected servers of either the University of Regensburg or the University of Magdeburg. Only members of the study team will have access to the respective study files. Data exchange between the study team and the outcomes assessor will be via a secure platform where study data can only be accessed via double authentication (username/password for login and additional password to access the database). We have summarised the data flow schematically in Fig. 6.
Amendment to protocol
Due to the COVID-19 pandemic, the Bavarian authorities ordered restrictions on research activities at all study centres. Therefore, all activities related to recruitment, inclusion of participants, the work of the follow-up clinic as well as the follow-up survey had to be discontinued on 20 March 2020. As a result, we needed to plan for a restart of the RCT. Since the duration of the interruption was not foreseeable at this time, there was initially no other choice than to completely restart this pilot RCT afterwards. In a RCT, differences between the intervention group and the control group should be determined. However, if a part of all patients were treated before the pandemic and another part of the patients were treated during or even after the pandemic, these patients cannot be compared with each other anymore, as the care provided and the circumstances repeatedly changed. Furthermore, it was not foreseeable whether and how the intervention (ICU follow-up clinic) could have been carried out. After intensive consultations among the study team, with the hospital management and the funding body, it was decided to reduce the number of cases to 40 participants. This measure had to be taken mainly for research economic reasons. The limited duration of the project and limited financial resources did not allow a further extension of the project to recruit another 100 patients. We were able to resume activities on 15 June 2020 and believe that even with this reduced number of study participants, our overall aim to evaluate feasibility and effects of an ICU follow-up clinic can still be achieved. Study completion is expected in May 2021.
Discussion
To our knowledge, PINA is the first study evaluating the feasibility and preliminary effects of a complex intervention in form of an ICU follow-up clinic for ICU survivors in Germany. Based on the key elements of development and evaluation of complex interventions using the framework of the medical research council (MRC) [29], this study concerns mainly the phases development, feasibility and piloting.
We have invested significant resources in the development of the intervention which was done by a participatory process in which patients, next of kin and health care professionals were involved. To be more precise, focus groups with health care professionals and one-to-one interviews with experts from the PICS field were conducted. At the same time, face-to-face interviews with patients and with next of kin were conducted. The results of these qualitative research projects were then further elaborated into a final concept in several workshops.
The implementation of the complex intervention in the feasibility and piloting phase aims at testing procedures, estimating retention and determining the sample size. In spite of the sample size being too small for a full effectiveness evaluation, we will determine effectiveness preliminarily, thus anticipating the full-scale evaluation phase in a large pragmatic trial in the future. The implementation will be monitored by process evaluation to provide insights into unexpected or unanticipated consequences or to show why the intervention works. Depending on the evaluation and feasibility outcomes, the next steps will be either to modify the intervention and repeat the piloting phase or to proceed to implement the intervention on a large scale and test it in a pragmatic real-world RCT.
Limitations
Even though the randomisation process may have been conducted correctly using appropriate randomisation strategies, a balanced distribution of known and unknown confounding factors (such as sociodemographic or clinical factors) between intervention and control group might not be achieved. Therefore, we will compare baseline characteristics of participants in the intervention and the control group. The follow-up period may be too short, and the effects of the intervention might appear only after the follow-up period of 6 months. A minimal spillover effect, e.g. due to the information about the study, cannot be completely ruled out.
Strengths
The study introduces the first ICU follow-up clinic in Germany and thus tries to improve the aftercare of ICU survivors. The greatest advantage of the ICU follow-up clinic is its participatory development, which increases the likelihood of the intervention being effective. In addition, all intervention treatments will use the standard health care system processes, allowing the ICU follow-up clinic to be well integrated into the existing health care system in the future.
Trial status and dissemination
The first participant was enrolled on 2 December 2019. Participants’ recruitment started at the time of submission of the study protocol and was completed during the review process. Results will be presented at national and international conferences and reported in peer-reviewed journals.
Availability of data and materials
Data can be requested from 1 year after study completion from the principal investigator.
References
Iwashyna TJ, Cooke CR, Wunsch H, Kahn JM. Population burden of long-term survivorship after severe sepsis in older Americans. J Am Geriatr Soc. 2012;60(6):1070–7.
Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000-2012. JAMA. 2014;311(13):1308–16.
Needham DM, Davidson J, Cohen H, Hopkins RO, Weinert C, Wunsch H, et al. Improving long-term outcomes after discharge from intensive care unit: report from a stakeholders' conference. Crit Care Med. 2012;40(2):502–9.
Wolters AE, Slooter AJ, van der Kooi AW, van Dijk D. Cognitive impairment after intensive care unit admission: a systematic review. Intensive Care Med. 2013;39(3):376–86.
Rabiee A, Nikayin S, Hashem MD, Huang M, Dinglas VD, Bienvenu OJ, et al. Depressive symptoms after critical illness: a systematic review and meta-analysis. Crit Care Med. 2016;44(9):1744–53.
Needham DM, Dinglas VD, Morris PE, Jackson JC, Hough CL, Mendez-Tellez PA, et al. Physical and cognitive performance of patients with acute lung injury 1 year after initial trophic versus full enteral feeding. EDEN trial follow-up. Am J Respir Crit Care Med. 2013;188(5):567–76.
Jackson JC, Pandharipande PP, Girard TD, Brummel NE, Thompson JL, Hughes CG, et al. Depression, post-traumatic stress disorder, and functional disability in survivors of critical illness in the BRAIN-ICU study: a longitudinal cohort study. Lancet Respir Med. 2014;2(5):369–79.
Marra A, Pandharipande PP, Girard TD, Patel MB, Hughes CG, Jackson JC, et al. Co-occurrence of post-intensive care syndrome problems among 406 survivors of critical illness. Crit Care Med. 2018;46(9):1393–401.
Pandharipande PP, Girard TD, Jackson JC, Morandi A, Thompson JL, Pun BT, et al. Long-term cognitive impairment after critical illness. N Engl J Med. 2013;369(14):1306–16.
Nikayin S, Rabiee A, Hashem MD, Huang M, Bienvenu OJ, Turnbull AE, et al. Anxiety symptoms in survivors of critical illness: a systematic review and meta-analysis. Gen Hosp Psychiatry. 2016;43:23–9.
Fan E, Dowdy DW, Colantuoni E, Mendez-Tellez PA, Sevransky JE, Shanholtz C, et al. Physical complications in acute lung injury survivors: a two-year longitudinal prospective study. Crit Care Med. 2014;42(4):849–59.
Harvey MA, Davidson JE. Postintensive care syndrome: right care, right now...and later. Crit Care Med. 2016;44(2):381–5.
Davydow DS, Gifford JM, Desai SV, Needham DM, Bienvenu OJ. Posttraumatic stress disorder in general intensive care unit survivors: a systematic review. Gen Hosp Psychiatry. 2008;30(5):421–34.
Parker AM, Sricharoenchai T, Raparla S, Schneck KW, Bienvenu OJ, Needham DM. Posttraumatic stress disorder in critical illness survivors: a metaanalysis. Crit Care Med. 2015;43(5):1121–9.
Herridge MS, Moss M, Hough CL, Hopkins RO, Rice TW, Bienvenu OJ, et al. Recovery and outcomes after the acute respiratory distress syndrome (ARDS) in patients and their family caregivers. Intensive Care Med. 2016;42(5):725–38.
Brandstetter S, Dodoo-Schittko F, Brandl M, Blecha S, Bein T, Apfelbacher C, et al. Ambulatory and stationary healthcare use in survivors of ARDS during the first year after discharge from ICU: findings from the DACAPO cohort. Ann Intensive Care. 2019;9(1):70.
Kosilek RP, Baumeister SE, Ittermann T, Gründling M, Brunkhorst FM, Felix SB, et al. The association of intensive care with utilization and costs of outpatient healthcare services and quality of life. PLoS One. 2019;14(9):e0222671.
Cuthbertson BH, Roughton S, Jenkinson D, MacLennan G, Vale L. Quality of life in the five years after intensive care: a cohort study. Crit Care. 2010;14(1):R6.
Griffith D, Salisbury LG, Lee R, Lone N, Merriweather JL, Walsh T, et al. Determinants of health-related quality of life after intensive care: importance of patient demographics, previous comorbidity, and severity of illness. Crit Care Med. 2018;46(4):594–601.
Salisbury C, Johnson L, Purdy S, Valderas JM, Montgomery AA. Epidemiology and impact of multimorbidity in primary care: a retrospective cohort study. Br J Gen Pract. 2011;61(582):e12–21.
Tan T, Brett SJ, Stokes T. Rehabilitation after critical illness: summary of NICE guidance. Bmj. 2009;338:b822.
Jonasdottir RJ, Klinke ME, Jonsdottir H. Integrative review of nurse-led follow-up after discharge from the ICU. J Clin Nurs. 2016;25(1-2):20–37.
Schmidt K, Worrack S, Von Korff M, Davydow D, Brunkhorst F, Ehlert U, et al. Effect of a primary care management intervention on mental health-related quality of life among survivors of sepsis: a randomized clinical trial. JAMA. 2016;315(24):2703–11.
Schofield-Robinson OJ, Lewis SR, Smith AF, McPeake J, Alderson P. Follow-up services for improving long-term outcomes in intensive care unit (ICU) survivors. Cochrane Database Syst Rev. 2018;11:CD012701.
Sevin CM, Jackson JC. Post-ICU Clinics Should Be Staffed by ICU Clinicians. Crit Care Med. 2019;47(2):268–72.
Lasiter S, Oles SK, Mundell J, London S, Khan B. Critical care follow-up clinics: a scoping review of interventions and outcomes. Clin Nurse Specialist CNS. 2016;30(4):227–37.
Griffiths JA, Barber VS, Cuthbertson BH, Young JD. A national survey of intensive care follow-up clinics. Anaesthesia. 2006;61(10):950–5.
Sevin CM, Bloom SL, Jackson JC, Wang L, Ely EW, Stollings JL. Comprehensive care of ICU survivors: Development and implementation of an ICU recovery center. J Crit Care. 2018;46:141–8.
Craig P, Dieppe P, Macintyre S, Michie S, Nazareth I, Petticrew M. Developing and evaluating complex interventions: the new Medical Research Council guidance. Bmj. 2008;337:a1655.
Chan A-W, Tetzlaff JM, Altman DG, Dickersin K, Moher D. SPIRIT 2013: new guidance for content of clinical trial protocols. Lancet. 2013;381(9861):91–2.
Chan A-W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, et al. SPIRIT 2013 Statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7.
Hertzog MA. Considerations in determining sample size for pilot studies. Res Nurs Health. 2008;31(2):180–91.
Teare MD, Dimairo M, Shephard N, Hayman A, Whitehead A, Walters SJ. Sample size requirements to estimate key design parameters from external pilot randomised controlled trials: a simulation study. Trials. 2014;15(1):264.
Schulz KF, Grimes DA. Allocation concealment in randomised trials: defending against deciphering. Lancet. 2002;359(9306):614–8.
Brandl M, Apfelbacher C, Weiß A, Brandstetter S, Baumeister SE. Incidence estimation in Post-ICU populations: challenges and possible solutions when using claims data; 2020.
ICUsteps. Intensive Care. A guide for patients and relatives. 2010 Available from: http://icusteps.org/assets/files/booklet/languages/Intensiv_Info_A5_Web.pdf.
German Society of Skilled Nursing and Functional Services e.V. (DGF). Die Zeit der Intensivstation. Available from: https://www.dgf-online.de/die-zeit-der-intensivstation/. Accessed 22 Jan 2019.
Borson S, Scanlan JM, Chen P, Ganguli M. The Mini-Cog as a screen for dementia: validation in a population-based sample. J Am Geriatr Soc. 2003;51(10):1451–4.
Kroenke K, Spitzer RL. The PHQ-9: a new depression diagnostic and severity measure. Psychiatr Ann. 2002;32(9):509–15.
Spitzer RL, Kroenke K, Williams JB, Löwe B. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166(10):1092–7.
Cooper R, Kuh D, Cooper C, Gale CR, Lawlor DA, Matthews F, et al. Objective measures of physical capability and subsequent health: a systematic review. Age Ageing. 2010;40(1):14–23.
Bobos P, Nazari G, Lu S, MacDermid JC. Measurement properties of the hand grip strength assessment. A systematic review with meta-analysis. Arch Phys Med Rehabil. 2019:101(3):553–65.
Greiner W. A European EQ-5D VAS valuation set. The measurement and valuation of health status using EQ-5D: a European perspective. Dordrecht: Springer; 2003. p. 103–42.
Moore GF, Audrey S, Barker M, Bond L, Bonell C, Hardeman W, et al. Process evaluation of complex interventions: Medical Research Council guidance. BMJ. 2015;350:h1258.
Oakley A, Strange V, Bonell C, Allen E, Stephenson J. Process evaluation in randomised controlled trials of complex interventions. BMJ. 2006;332(7538):413–6.
Griffiths J, Hatch RA, Bishop J, Morgan K, Jenkinson C, Cuthbertson BH, et al. An exploration of social and economic outcome and associated health-related quality of life after critical illness in general intensive care unit survivors: a 12-month follow-up study. Crit Care (London, England). 2013;17(3):R100.
Bein T, Weber-Carstens S, Apfelbacher C, Brandstetter S, Blecha S, Dodoo-Schittko F, Karagiannidis C. The quality of acute intensive care and the incidence of critical events have an impact on health-related quality of life in survivors of the acute respiratory distress syndrome–a nationwide prospective multicenter observational study. GMS German Med Sci. 2020;18.
Ware JE Jr, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care. 1996:220–33.
Bullinger M, Kirchberger I. Der SF-36 Fragebogen zum Gesundheitszustand (SF-36). In: Handbuch für die deutschsprachige Fragebogenversion Göttingen, Hogrefe; 1998.
Brandstetter S, Dodoo-Schittko F, Blecha S, Sebök P, Thomann-Hackner K, Quintel M, et al. Influence of quality of care and individual patient characteristics on quality of life and return to work in survivors of the acute respiratory distress syndrome: protocol for a prospective, observational, multi-centre patient cohort study (DACAPO). BMC Health Serv Res. 2015;15(1):563.
Resnick B, Nahm ES. Reliability and validity testing of the revised 12-item Short-Form Health Survey in older adults. J Nurs Meas. 2001;9(2):151–61.
Bohannon RW, Maljanian R, Lee N, Ahlquist M. Measurement properties of the short form (SF)-12 applied to patients with stroke. Int J Rehabil Res. 2004;27(2):151–4.
Bullinger M, Kirchberger I. SF-36: Fragebogen zum Gesundheitszustand. Handanweisung: Hogrefe, Verlag für Psychologie; 1998.
Mahoney FI, Barthel DW. Functional evaluation: the Barthel Index: a simple index of independence useful in scoring improvement in the rehabilitation of the chronically ill. Md State Med J. 1965;14:61–5.
Lübke N, Meinck M, Von WR-K. The Barthel Index in geriatrics. A context analysis for the Hamburg Classification Manual. Z Gerontol Geriatr. 2004;37(4):316–26.
Fuchs J, Busch M, Gößwald A, Hölling H, Kuhnert R, Scheidt-Nave C. Physical and cognitive capabilities among persons aged 65–79 years in Germany; 2013.
Beaton DE, O'Driscoll SW, Richards RR. Grip strength testing using the BTE work simulator and the Jamar dynamometer: A comparative study. J Hand Surg. 1995;20(2):293–8.
Dodds RM, Syddall HE, Cooper R, Benzeval M, Deary IJ, Dennison EM, et al. Grip strength across the life course: normative data from twelve British studies. PLoS One. 2014;9(12):e113637.
Weisæth L. Torture of a Norwegian ship's crew: The torture, stress reactions and psychiatric after-effects. Acta Psychiatr Scand. 1989;80:63–72.
Stoll C, Kapfhammer H, Rothenhäusler H, Haller M, Briegel J, Schmidt M, et al. Sensitivity and specificity of a screening test to document traumatic experiences and to diagnose post-traumatic stress disorder in ARDS patients after intensive care treatment. Intensive Care Med. 1999;25(7):697–704.
Löwe B, Spitzer R, Zipfel S, Herzog W. PHQ-D Gesundheitsfragebogen für Patienten (German Version of the Patient Health Questionnaire). Karlsruhe: Pfizer; 2002.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Diseases. 4th ed. Washington, DC; 2000.
White IR, Horton NJ, Carpenter J, Pocock SJ. Strategy for intention to treat analysis in randomised trials with missing outcome data. BMJ. 2011;342:d40.
Acknowledgements
The authors would like to thank Annette Weiß who supported the PINA study as a valuable colleague during the first year of study. We would like to thank Stefanie March and Christoph Stallmann (Institute of Social Medicine and Health Systems Research, Otto-von-Guericke University Magdeburg) for providing consulting advice.
Funding
The study is funded by the Innovation Fund of the Federal Joint Committee (Innovationsfond G-BA), which is involved neither in the study design, nor in the collection, analysis and interpretation of data, nor the writing of the article, nor the decision to submit the results for publication. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
CA and SB had the idea for the study and obtained funding for the study. CA, SB, MR, CF and MVM developed the research design and methods. PD, MR and CA developed the data protection concept. CF, KS and MVM provided input for the development of study materials (both intervention and study manual). MR, PD and CA wrote the article with input from SB, CF, VB and MVM. All authors reviewed the article for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The institutional Ethics Committee of the University of Regensburg approved the study protocol on 26 September 2019 (19-1522-101). Every participant will actively agree to be part of the study and informed consent will be obtained according the Helsinki Declaration.
Consent for publication
All authors carefully read the manuscript and approved it for publication.
Competing interests
The following authors declare that they have no competing interests: CA, CB, MR, SB, PD, CF, VB, MVM, KS.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:.
SPIRIT 2013 Checklist: Recommended items to address in a clinical trial protocol and related documents
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rohr, M., Brandstetter, S., Bernardi, C. et al. Piloting an ICU follow-up clinic to improve health-related quality of life in ICU survivors after a prolonged intensive care stay (PINA): study protocol for a pilot randomised controlled trial. Pilot Feasibility Stud 7, 90 (2021). https://doi.org/10.1186/s40814-021-00796-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40814-021-00796-1