
Abstract
Background
Sickle cell disease (SCD) is an inherited blood disorder that predominantly affects individuals in sub-Saharan Africa. However, research that elucidates links between SCD pathophysiology and nutritional status in African patients is lacking. This systematic review aimed to assess the landscape of studies in sub-Saharan Africa that focused on nutritional aspects of SCD, and highlights gaps in knowledge that could inform priority-setting for future research.
Methods
The study was conducted using the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines. Inclusion criteria comprised original, peer-reviewed research published between January 1995 and November 2020 involving individuals in Africa with any phenotypic variant of SCD and at least one nutritional status outcome. Nutritional status outcomes were defined as those that assessed dietary intakes, growth/anthropometry, or nutritional biomarkers. Databases used were Ovid Embase, Medline, Biosis and Web of Science.
Results
The search returned 526 articles, of which 76 were included in the final analyses. Most investigations (67%) were conducted in Nigeria. Studies were categorized into one of three main categories: descriptive studies of anthropometric characteristics (49%), descriptive studies of macro- or micronutrient status (41%), and interventional studies (11%). Findings consistently included growth impairment, especially among children and adolescents from sub-Saharan Africa. Studies assessing macro- and micronutrients generally had small sample sizes and were exploratory in nature. Only four randomized trials were identified, which measured the impact of lime juice, long-chain fatty acids supplementation, ready-to-use supplementary food (RUSF), and oral arginine on health outcomes.
Conclusions
The findings reveal a moderate number of descriptive studies, most with small sample sizes, that focused on various aspects of nutrition and SCD in African patients. There was a stark dearth of interventional studies that could be used to inform evidence-based changes in clinical practice. Findings from the investigations were generally consistent with data from other regional settings, describing a significant risk of growth faltering and malnutrition among individuals with SCD. There is an unmet need for clinical research to better understand the potential benefits of nutrition-related interventions for patients with SCD in sub-Saharan Africa to promote optimal growth and improve health outcomes.
Similar content being viewed by others


Background
Sickle cell disease (SCD) is the most common inherited blood disease worldwide, with the vast majority of cases occurring in sub-Saharan Africa [1]. The condition derives from a point mutation of the β-globin gene found on the short arm of chromosome 11 through which the hydrophilic amino acid glutamic acid is substituted with the hydrophobic amino acid valine at the sixth position [2, 3]. The result is a change in the structure and dynamics of hemoglobin such that certain conditions including deoxygenation and acidosis predispose to hemoglobin polymerization. When this occurs, erythrocytes assume a misshapen and rigid form that promotes pathological processes leading to intravascular inflammation and occlusion of small blood vessels [4]. Since these processes can take place anywhere in the body, the disease is highly complex and characterized by multiple potential life-threatening complications that include acute splenic sequestration, aplastic crises, acute chest syndrome, infection, heart failure, and stroke [5, 6]. The disease’s clinical hallmarks include acute painful crises and severe anemia [1]. In sub-Saharan Africa, it has been estimated that up to 90% of children born with SCD die before 5 years of age [7].
In high resource countries, mortality from SCD has decreased dramatically over the past five decades. The improvements in outcomes have been attributed in part to newborn screening and comprehensive care programs designed to prevent disease complications to the extent possible and to treat complications of disease when they occur [8]. Early detection of disease enables clinicians and families to institute measures to proactively prevent complications and facilitate timely treatment when needed. For example, the risk of fatal infection has been shown to be reduced through vaccination and administration of prophylactic antibiotics [9]. Furthermore, treatments with blood transfusion and hydroxyurea therapy has led to superior outcomes in the long term [10, 11]. Unfortunately, the availability of vaccines, medicines, and other interventions is highly variable in sub-Saharan Africa. Increasing access to proven preventative and treatment modalities is therefore an urgent priority [12]. At the same time, there is a need to identify new ways of maximizing the well-being of individuals with SCD in Africa and it is in this context that nutritional interventions could possibly play an important role.
There is evidence that the pathophysiology of SCD has substantial nutritional implications including higher energy and nutrients requirements, nutrient deficiencies, and growth abnormalities [13,14,15]. It is theorized that a main driver of disease complications is higher rates of metabolic expenditure in individuals with SCD resulting from increased hematopoiesis, increased cardiac output, chronic inflammation, and related processes [16, 17]. Since nutrition interventions could be a mechanism for addressing increased energy expenditure, attention to nutritional care is increasingly seen to be an important aspect of supportive management for patients with SCD [18, 19], especially in resource poor settings. However, evidence-based nutritional guidelines for patients with SCD in Africa are lacking and the extent of nutrition-focused research involving individuals in Africa with SCD is unclear. We undertook this systematic review to evaluate the existing literature focused on nutritional aspects of SCD in sub-Saharan Africa. Specifically, we sought to assess the number and nature of relevant studies, review their findings, and identify gaps in knowledge that could inform priority-setting for future research.
Methods
Eligibility criteria
We sought to include all studies involving original research that focused on the nutritional status of individuals with SCD in an African population. Studies involving nutritional status were defined as those that investigated topics of dietary intake, measurements of growth or anthropometry, or nutrition-related biomarkers. Studies that did not differentiate the cause of the anemia were excluded, as were studies that only included nutrition interventions as part of comprehensive care programmes (i.e., studies that did not clearly report nutrition-related outcomes). Studies involving both children and adults were included. The focus of this analysis was on studies involving individuals with various forms of SCD including HbSS, HbSC, and rarer genetic variants of disease; studies were excluded that only involved individuals with sickle cell trait. Case reports and review articles were also excluded.
Informational sources and search strategy
The databases employed for this search were Ovid Embase, Medline, Biosis, and Web of Science. The date range was January 1st, 1995, through November 30th, 2020, such that the reference list covered a period of approximately 25 years. We performed a Boolean search using specific Boolean operators and the following search terms: “Sickle cell disease” or “sickle cell anemia” or “hemoglobinopathy” AND Africa or specific African countries (all African countries were individually listed) AND various nutrition-related terms (i.e., nutrition, growth, macronutrient, micronutrient, vitamin, mineral, anthropometric, height, length, weight, head circumference, mid-upper arm circumference, MUAC, dietary intake, recommended dietary allowance, RDA, nutritional status) along with associated terms (both indexed and non-indexed) for nutrition, diet and growth, and specific vitamins and minerals. We also allowed for inclusion of articles that were identified through review of the bibliographies of papers that underwent full-text review. Investigations written in any language were included provided sufficient translation into English could be assured.
Data management and selection process
Titles and abstracts were each screened by two independent reviewers. Any title or abstract that appeared to meet inclusion criteria or for which there was uncertainty prompted a full text review. Reviewing of full text articles were assigned to individual investigators. If eligibility of a full text article was unclear, it was resolved by discussing it with at least two other reviewers on the research team who were not earlier assigned the full text article using the inclusion criteria. To maximize consistency among reviewers, each reviewer initially reviewed 10 articles and the review team together discussed the initial dataset that had been extracted to ensure accuracy and completeness. The review process then proceeded according to the process described.
Data synthesis
Study data were extracted into standardized forms using Microsoft Excel (Microsoft, Redmond, USA) where they were organized for analyses. Depending on the nature of the articles that met inclusion criteria, we extracted information relating to geography, subject age group, sample size, comparison groups, biomarkers, and other relevant variables. Since the main purpose of this investigation was to broadly understand the types of nutrition-related studies that have been conducted involving individuals with SCD in Africa, we chose not to systematically judge the quality of evidence or risks of bias within individual studies. Rather, we discussed specific merits and limitations of individual studies where appropriate in the context of major themes that would emerge in the analyses. We planned for a quantitative categorization of the types of articles (e.g., descriptive versus interventional studies) and a narrative synthesis of data in table and text format to summarize and assess the results.
PRISMA
The study was conducted and reported according to PRISMA (Preferred Reporting Items for Systematic reviews and Meta-Analyses) guidelines [20, 21].
Results
Search results
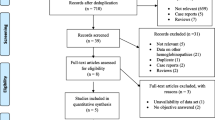
In total, 526 unique titles and abstracts were identified through the literature search. Of those, 347 did not meet inclusion criteria and 179 full-text articles were assessed. Seventy-six studies were deemed eligible and included in the final analyses (Fig. 1).

Literature search flowchart
Results overview
Most investigations (67%) were conducted in a single country (Nigeria). Each study was placed into one of three main categories according to the primary nature of the investigation: (a) descriptive study of anthropometric characteristics (37 articles; 49%); (b) descriptive study of macro- or micronutrient status (31 articles; 41%); and (c) interventional studies (8 articles; 11%). The studies are summarized in Tables 1, 2, 3 and described in greater detail below.
Descriptive studies of anthropometric characteristics
Nearly one-half of all studies identified focused on anthropometric characteristics. The studies typically assessed height, weight, and body mass index (BMI). Other measurements included head circumference, arm span, and various body composition parameters.
The majority (25/37; 68%) of studies were conducted in Nigeria. Other studies involved populations in Democratic Republic of Congo (DRC) [49, 50, 57], Ghana [33, 53], Tanzania [32], Egypt [23], Cameroon [54], and Algeria [37]. In addition, two multi-country studies involved patients in Cameroon, Ivory Coast, Gabon, Mali, and Senegal [43, 55]. The majority of reports focused on children and adolescents; only one study exclusively involved adults [24]. Most studies evaluated approximately 50–200 patients and a similar number of age- and gender-matched healthy controls; three large studies enrolled over 1000 SCD patients each [32, 43, 55]. Several studies used WHO growth standards for comparison rather than a non-sickle cell disease control group. Male and female subjects were generally equally represented in the study populations.
The three largest studies found significant growth defects compared to healthy controls. A multi-country study of more than 3500 SCD patients (aged 10–24 years) with nearly 1000 controls in Cameroon, Ivory Coast, Gabon, Mali, and Senegal was designed to evaluate determinants of vascular complications [43]. Anthropometric analyses showed that SCD patients were significantly shorter and had lower BMI than controls; weight was not reported. A caveat of the study was that the control group was significantly older (median age 24 vs 16 for patients) and more likely to be female (60% vs 54% for patients) than the SCD group. A follow-up study involving the same subject population revealed significantly higher rates of growth failure, defined as a height and/or weight and/or BMI below the 5th percentile using WHO growth reference [55]. Another large study followed a cohort of approximately 1000 SCD patients aged 6 months to 48 years over 5 years and found SCD to be significantly associated with stunting, underweight, and wasting, with the most pronounced effects associated with adolescent age and male gender [32]. Adult men were seven times more likely than adult women to be underweight and were significantly more likely to be stunted and wasted. Females demonstrated improved catch-up growth compared with males following growth deficits that were identified during adolescence.
The studies involving smaller sample sizes showed greater variation in the results, but some trends emerged. Several reports confirmed the finding that males were more likely to show growth defects than females [22, 27, 29, 45]. Multiple studies also noted that growth deficits became more pronounced with age. For example, a study that involved young patients aged 6–35 months showed no association with wasting, stunting, or underweight status [40]. A study of 233 children aged 2–17 years with SCD in Lagos, Nigeria found that the factor most significantly associated with both wasting and stunting was older age [45]. Additional studies similarly detected one or more growth deficits in adolescents but not in younger children [25, 28, 34, 39, 41, 46]. In several Nigerian studies, SCD patients were found to be underweight or to have low BMI, but showed no difference in height compared to controls [22, 25, 35, 42, 44, 52]; while other Nigerian studies showed differences in both weight and height [24, 26, 27, 29, 44, 51]. These variable findings may have resulted from the fact that the studies were not powered to detect significant differences in height specifically. Reports from DRC, Egypt, and Ghana found that children with SCD had a higher prevalence of stunting compared to controls, but did not always show differences in wasting or BMI [23, 33, 50]. SCD was also associated with delayed puberty [23, 31]. Three studies from Nigeria reported the presence of overweight and obesity among patients with SCD although in lower proportions (an average of less than 3% of the sample population) [36, 42, 45].
Descriptive studies of macro- or micronutrient status
The second most common group of studies identified involved assessment of biomarkers from serum samples for macronutrients or micronutrients. Most studies were conducted in Nigeria (21/31, 68%); other studies took place in DRC [78], Tanzania [32, 77, 79], Egypt [62], Kenya [40], Ghana [82], Uganda [69], and Malawi [64]. Nutritional parameters measured included proteins/amino acids, fatty acids, vitamins, and minerals. Most studies included less than 100 individuals with SCD. Both children and adults were studied, with male and female subjects generally equally represented.
Serum protein levels were investigated in one small study (13 children with SCD and 17 healthy controls) in Nigeria in which no significant differences were reported in the concentrations of total protein or albumin between SCD patients and controls [22]. However, serum prealbumin levels were significantly lower for the population of patients with SCD, which was hypothesized to result from poor nutrition or existing disease-related inflammation. The serum concentrations of all amino acids except alanine, glutamic acid, and proline were significantly reduced in SCD patients. A small study involving 23 participants in Tanzania measured the steady state nutrition status of SCD patients who later died (n = 11) compared with those who were alive at the end of the study period. Those who suffered mortality had a significantly lower BMI, plasma taurine levels and arginine bioavailability before succumbing [59].
Proportions of fatty acid and the state of metabolism were evaluated in four related studies of young SCD patients in Nigeria [26, 29, 60, 61]. These reports found perturbed pathways of fatty acid elongation and desaturation in children with SCD. Specifically, arachidonic acid, eicosapentanoic acid (EPA), and decosahexanoic acid (DHA) were significantly reduced, whereas saturated (palmitic acid) and monounsaturated (oleic acid) were significantly elevated in patients compared to controls. Another study in a population of 26 SCD patients aged 11–43 in Enugu, Nigeria, confirmed the finding that EPA and DHA fatty acids are reduced in SCD patients [63]. The authors of these studies hypothesized that reduced polyunsaturated fatty acids in the phospholipids of the cell membrane of SCD patients could lead to their being more rigid, thereby contributing to disease symptoms. A study of 30 children with SCD in Egypt found that patients also had significantly lower cholesterol, triglycerides, and LDL (but not HDL) in blood plasma compared to healthy controls [62].
Acknowledging that interpretation of plasma concentrations of vitamins and minerals can be problematic in patients with ongoing inflammation, analysis of serum vitamin levels in SCD patients generally indicated lower concentrations of vitamin A [63, 65] vitamin C [65, 66] and vitamin E [62,63,64,65]. One study of 14 SCD patients in Kenya found no association of HbSS phenotype and low concentrations vitamin A [40]. Three reports of a related study population in Ilese, Nigeria, investigated vitamin D status of young SCD patients [48, 67, 68]. When compared to healthy controls, mean 25-hydroxyvitamin D levels were significantly lower in SCD patients and suboptimal vitamin D levels were seen in greater than 10% of patients. However, no SCD patients with severe vitamin D deficiency (defined as < 20 ng/ml) were observed. A limitation of the latter two studies was the lack of a healthy comparator group; each used vitamin D deficiency cut-off values for a healthy population in other published studies as reference.
Selected minerals were evaluated in eight small studies of SCD patients and compared to healthy controls. Serum iron concentration was reduced in patients compared to controls in all studies that evaluated it [74,75,76, 82]. Serum or plasma zinc was also generally reduced in SCD patients [7, 71, 72, 74, 75, 78, 83], although zinc was elevated in one population of 59 Nigerian adult SCD patients [76]. Measures of other minerals showed mixed results. Magnesium levels were either reduced [71, 75], elevated [72] or unchanged [73, 75, 76] in SCD patients compared to healthy controls. Similarly, copper was reduced [71], elevated [72, 81, 82] or unchanged [75, 76] in SCD patients. Other minerals measured in only a few studies included manganese, chromium, selenium, potassium, rubidium, cadmium, and calcium.
Interventional studies
A very small number of clinical studies involving nutritional interventions in SCD patients in African countries were identified. There were four randomized trials [88,89,90,91]. The first was a study of 125 SCD patients in Nigeria (involving children aged 1 year and above) that tested the effect of lime juice on SCD parameters. All patients in the treatment group (n = 65) as well as controls (n = 60) were given folic acid, vitamin B complex, and proguanil, with the treatment group also given twice-daily oral lime juice with weight-based dosing ranging from 5 to 15 mL. Each child was assessed monthly for 6 months. The group receiving lime juice was reported to have significantly fewer pain episodes, febrile illnesses, and hospital admission rates. There was no change in transfusion rate, organomegaly, or jaundice. The positive effect was postulated to result from vitamin C, amino acids (in particular, phenylalanine), and flavonoids contained in the juice, but no direct evidence for this was provided [88].
A second study of 128 SCD patients aged 2–24 years in Sudan investigated the impact of 1 year of treatment with oral omega-3 capsules containing EPA and DHA fatty acids (using age- and weight-dependent dosing) compared to placebo. The hypothesis was that omega-3 fatty acids could reduce red blood cell aggregation, adherence, and inflammation that occur during sickle cell disease-mediated vaso-occlusive crises. The treatment group had significantly fewer clinical vaso-occlusive events, as well as reduced rates of severe anemia and need for blood transfusions. The study was not powered to detect changes in other outcomes such as stroke, sequestration crisis, or vascular necrosis [89].
A third trial utilized a double-blind, random order design. Ready-to-use-supplementary food was studied in 119 children with SCD in Tanzania [90]. Two different formulations of the supplements were compared: a commercially available (Nutriset, France) ready-to-use-supplementary food (RUSF) fortified with vitamins and minerals according to recommended daily allowances and an “enhanced” version of the same RUSF (providing 500 kcal/day) that was additionally fortified with arginine and citrulline. Arginine is the substrate for endothelial nitric oxide synthase, a natural vasodilator, and has been implicated in pathophysiology of SCD complications. In the cross-over study design, children received each treatment for 4 months, with 4-month washout periods following the intervention. Ready-to-use-supplementary food led to small weight gains, an increased arginine bioavailability ratio, and improved measures of endothelial function compared to baseline; addition of arginine and citrulline to the supplement did not provide additional benefits [90].
The final randomized study identified involved the regular administration of oral arginine therapy to 35 hospitalized patients with SCD in Nigeria and compared the effects with 33 control subjects. Plasma arginine levels increased by 125% in the arginine arm compared with 29% in the control arm [91]. Arginine treatment was associated with quicker discharge and reduced pain events. The rate of adverse events was non-significant between the two treatment arms, however there was a trend towards increased vomiting in the patients treated with arginine. A previous study outside of Africa also found positive clinical effects associated with the use of arginine [92].
One of the non-randomized interventional studies identified investigated the use of vitamin D supplementation. A small treatment arm was nested in a Nigerian study comparing blood levels of vitamin D and pro-inflammatory cytokines [48]. The hypothesis was that low vitamin D levels might lead to a pro-inflammatory environment that exacerbates SCD symptoms. Twelve children with SCD who were determined to have low vitamin D levels were given 3 months of oral vitamin D supplementation (2000 U). At the end of treatment, mean serum 25-hydroxyvitamin D levels were significantly increased compared to baseline, levels of several proinflammatory cytokines were significantly decreased, and the levels of anti-inflammatory cytokine IL-11 were significantly increased.
Discussion
To our knowledge this is the first review of nutrition-related studies involving individuals living with SCD in sub-Saharan Africa. While a moderate number of studies were identified, most were descriptive in nature and small in terms of numbers of subjects. Approximately two-thirds of studies took place in a single country (Nigeria). In addition, there were very few interventional trials designed to measure the impact of an isolated nutritional intervention and only four randomized studies. The findings of this review suggest an outstanding need for nutrition-focused research relating to the care of individuals with SCD in Africa, with a particular emphasis on research with practical implications for clinical management in order to improve patient outcomes.
The findings of studies identified through this review are generally consistent with nutrition-related investigations involving SCD patients in other parts of the world. More than 50 years ago, poor growth was first reported in patients with SCD, and that observation has since been repeated in multiple studies involving SCD populations in Jamaica, Brazil, India, and North America [13, 15, 93,94,95,96,97,98,99]. Many of these studies specifically note that the growth faltering occurred in patients that were receiving recommended daily protein and calorie intakes. The pathophysiology of growth problems in SCD patients has come into sharper focus in recent decades. A leading view is that the increased rate of red cell turnover, a primary feature of SCD patients, underlies a hypermetabolic state. The biochemical and physiological factors that contribute to hypermetabolism include increased protein turnover, increased myocardial activity, and production of proinflammatory cytokines [100,101,102,103,104]. The supposition is that the energy and nutrient requirements normally recommended are not adequate in patients with SCD given their increased energy expenditures and other unusual metabolic demands, which compete directly with energy needs required to sustain adequate growth.
Evidence derived from robust interventional studies is important to support recommendations for specific nutrition-related practices for patients with sickle cell disease. Only four randomized trials were identified. The studies were small, each involving less than 150 individuals with SCD. Positive clinical benefits were found with the use of lime juice, long-chained fatty acid supplementation, RUSF, and oral arginine; ideally these findings would be confirmed in larger follow-up investigations. It is worth noting the paucity of robust interventional trials designed to test the effect of macronutrient supplementation in individuals with SCD despite the evidence, as described above, that nutrition deficits in this population are likely to be caused at least in part from the increased energy demands that result from altered metabolism.
Guidelines for clinical management of patients with SCD published by internationally recognized organizations do not provide special guidance for nutritional care [105, 106]. Given that the risk of poor growth in SCD patients is increasingly reported, and the fact that there is plausible pathophysiologic drivers of nutritional disturbances in SCD patients, there appears to be a substantial gap in research in this area to inform much needed evidence-based recommendations.
Limitations of this systematic review include the fact that nearly half of studies identified were largely anthropometry-based descriptive studies. Few studies involving nutrition interventions in sub-Saharan Africa were identified, only several had robust methodologies, and none have been validated in repeated studies. In addition, the studies involving analyses of vitamin and mineral levels in SCD patients in sub-Saharan Africa overall involved small numbers of patients and generally were unable to link findings with meaningful clinical correlations in ways that might influence nutritional care practices. Another limitation is that most investigations identified took place in the single country of Nigeria (at the same time, acknowledging that Nigeria is home to the largest population of SCD patients globally).
Conclusion
Despite the reality that most SCD patients globally live in sub-Saharan Africa, and the fact that nutritional disturbances in SCD patients are increasingly well described, there has been limited research focused on ways that nutritional care might help to improve clinical outcomes in this patient population. A systematic review of the literature revealed studies that consistently reported stunted growth and malnutrition in African SCD patients during childhood and adolescence, but failed to identify robust, validated studies that could be used to inform clinical management. Our study suggests an outstanding need to determine if and how supportive nutritional care can reduce disease severity and improve health outcomes for individuals with SCD in sub-Saharan Africa. As such, priority research in this area in the future may include systematic assessment of the drivers of nutritional status in SCD patients, studies that directly advance the understanding of macro- and micronutrient deficiencies associated with clinically significant physiologic effects, and investigations that evaluate the impact of nutritional interventions to inform evidence-based nutrition guidance.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- SCD:
-
Sickle cell disease
- EPA:
-
Eicosapentanoic acid
- DHA:
-
Decosahexanoic acid
- PRISMA:
-
Preferred reporting items for systematic reviews and meta-analysis
References
Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390:311–23. https://doi.org/10.1016/S0140-6736(17)30193-9.
Pauling L, Itano H. Sickle cell anemia a molecular disease. Science (80- ). 1949;110:543–8.
Ingram VM. Abnormal human haemoglobins. I. the comparison of normal human and sickle-cell haemoglobins by fingerprinting. Biochim Biophys Acta. 1958;28:539–45. https://doi.org/10.1016/0006-3002(58)90516-x.
Bunn H, Forget B. Hemoglobin: molecular, genetic and clinical aspects. Philadelphia, PA, USA: WB Saunders; 1986.
Mbanya N. Sickle cell disease in subsaharan Africa. Vox Sang. 2015;109:63–4.
Otoikhian CSO, Okoror LE. Sickle cell disease african killer: biologists alternative. Int J Pharma Med Biol Sci. 2012;1:232–45.
Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. 2011;41:S398–405. https://doi.org/10.1016/j.amepre.2011.09.013.
Consensus conference. Newborn screening for sickle cell disease and other hemoglobinopathies. JAMA. 1987;258:1205–9 Available: http://www.ncbi.nlm.nih.gov/pubmed/3626004.
Lanzkron S, Carroll CP, Haywood C. Mortality rates and age at death from sickle cell disease: U.S., 1979–2005. Public Health Rep. 2013;128:110–6. https://doi.org/10.1177/003335491312800206.
Tshilolo L, Tomlinson G, Williams TN, Santos B, Olupot-Olupot P, Lane A, et al. Hydroxyurea for children with sickle cell Anemia in sub-Saharan Africa. N Engl J Med. 2019;380:121–31. https://doi.org/10.1056/NEJMoa1813598.
Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5–11. https://doi.org/10.1056/NEJM199807023390102.
McGann PT. Time to invest in sickle cell Anemia as a Global Health priority. Pediatrics. 2016;137:e20160348. https://doi.org/10.1542/peds.2016-0348.
Al-Saqladi A-WM, Cipolotti R, Fijnvandraat K, Brabin B. Growth and nutritional status of children with homozygous sickle cell disease. Ann Trop Paediatr. 2008;28:165–89. https://doi.org/10.1179/146532808X335624.
Bello-Manga H, DeBaun MR, Kassim AA. Epidemiology and treatment of relative anemia in children with sickle cell disease in sub-Saharan Africa. Expert Rev Hematol. 2016:1031–42. https://doi.org/10.1080/17474086.2016.1240612.
Platt OS, Rosenstock W, Espeland MA. Influence of sickle Hemoglobinopathies on growth and development. N Engl J Med. 1984;311:7–12. https://doi.org/10.1056/NEJM198407053110102.
Akohoue SA, Shankar S, Milne GL, Morrow J, Chen KY, Ajayi WU, et al. Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatr Res. 2007;61:233–8. https://doi.org/10.1203/pdr.0b013e31802d7754.
Hyacinth HI, Gee BE, Hibbert JM. The role of nutrition in sickle cell disease. Nutr Metab Insights. 2010;3:57–67. https://doi.org/10.4137/nmi.s5048.
Canadian Haemoglobinopathy Association. Consensus Statement on the Care of Patients with Sickle Cell Disease in Canada. Ottawa; 2014.
National Health Service. Sickle Cell Disease in Childhood: Standards and Guidelines for Clinical Care. 2010.
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2714672&tool=pmcentrez&rendertype=abstract.
Shamseer L, Moher D, Clarke M, Ghersi D, Liberati A, Petticrew M, et al. Preferred reporting items for systematic review and meta-analysis protocols (prisma-p) 2015: Elaboration and explanation. BMJ 2015;349. doi:https://doi.org/10.1136/bmj.g7647.
VanderJagt DJ, Kanellis GJ, Isichei C, Pastuszyn A, Glew RH. Serum and urinary amino acid levels in sickle cell disease. J Trop Pediatr. 1997;43:220–5. https://doi.org/10.1093/tropej/43.4.220.
Soliman AT, ElZalabany M, Amer M, Ansari BM. Growth and pubertal development in transfusion-dependent children and adolescents with thalassaemia major and sickle cell disease: a comparative study. J Trop Pediatr. 1999;45:23–30. https://doi.org/10.1093/tropej/45.1.23.
Aderibigbe A, Omotoso AB, Awobusuyi JO, Akande TM. Arterial blood pressure in adult Nigerian sickle cell anaemia patients. West Afr J Med. 1999;18:114–8.
VanderJagt DJ, Okolo SN, Rabasa AI, Glew RH. Bioelectrical impedance analysis of the body composition of Nigerian children with sickle cell disease. J Trop Pediatr. 2000;46:67–72. https://doi.org/10.1093/tropej/46.2.67.
Vandegt DJ, Huang YS, Chuang LT, Bonnett C, Glew RH. Phase angle and n-3 polyunsaturated fatty acids in sickle cell disease. Arch Dis Child. 2002;87:252–4. https://doi.org/10.1136/adc.87.3.252.
VanderJagt DJ, Bonnett C, Okolo SN, Glew RH. Assessment of the bone status of Nigerian children and adolescents with sickle cell disease using calcaneal ultrasound and serum markers of bone metabolism. Calcif Tissue Int. 2002;71:133–40. https://doi.org/10.1007/s00223-001-1107-x.
Oredugba F, Savage K. Anthropometric finding in Nigerian children with sickle cell disease. Pediatr Dent. 2002;24:321–5.
Glew RH, Casados J, Huang YS, Chuang LT, VanderJagt DJ. Correlation of the fatty acid composition and fluid property of the cholesteryl esters in the serum of Nigerian children with sickle cell disease and healthy controls. Prostaglandins Leukot Essent Fat Acids. 2003;68:61–8. https://doi.org/10.1016/S0952-3278(02)00275-2.
Vanderjagt DJ, Trujillo MR, Jalo I, Bode-Thomas F, Glew RH, Agaba P. Pulmonary function correlates with body composition in Nigerian children and young adults with sickle cell disease. J Trop Pediatr. 2008;54:87–93. https://doi.org/10.1093/tropej/fmm070.
Aina OF, Fadaka K, Temiye E, Renner JK. Sexual maturation and psychiatric morbidity among persons with sickle cell Anaemia in a Nigerian teaching hospital. Int J Psychiatry Med. 2010;40:31–43. https://doi.org/10.2190/pm.40.1.c.
Cox SE, Makani J, Fulford AJ, Komba AN, Soka D, Williams TN, et al. Nutritional status, hospitalization and mortality among patients with sickle cell anemia in Tanzania. Haematologica. 2011;96:948–53. https://doi.org/10.3324/haematol.2010.028167.
Osei-Yeboah C, Rodrigues O, Enweronu-Laryea C. Nutritional status of children with sickle cell disease at Korle Bu teaching hospital, Accra. Ghana West Afr J Med. 2011;30:262–7.
Iwalokun BA, Iwalokun SO, Hodonu SO, Aina AO, Agomo PU. Serum levels of leptin in Nigerian patients with sickle cell anaemia. BMC Blood Disord. 2011;11. https://doi.org/10.1186/1471-2326-11-2.
Animasahun BA, Temiye EO, Ogunkunle OO, Izuora AN, Njokanma OF. The influence of socioeconomic status on the hemoglobin level and anthropometry of sickle cell anemia patients in steady state at the Lago; university teaching hospital. Niger J Clin Pract. 2011;14:422–7. https://doi.org/10.4103/1119-3077.91748.
Akodu SO, Diaku-Akinwumi IN, Njokanma OF. Obesity-does it occur in Nigerian children with sickle cell anemia. Pediatr Hematol Oncol. 2012;29:358–64. https://doi.org/10.3109/08880018.2012.680682.
Tebbani F, Rouabah L, Grifi F, Boudiba N, Rouabah A, Necib Y. Descriptive approach for sickle cell disease in eastern of Algeria. Int J Pharm Sci Rev Res. 2014;25:97–101.
Akingbola TS, Tayo BO, Salako B, Layden JE, Hsu LL, Cooper RS, et al. Comparison of patients from Nigeria and the USA highlights modifiable risk factors for sickle cell anemia complications. Hemoglobin. 2014;38:236–43. https://doi.org/10.3109/03630269.2014.927363.
Akodu SO, Diaku-Akinwumi IN, Kehinde OA, Njokanma OF. Evaluation of arm span and sitting height as proxy for height in children with sickle cell Anemia in Lagos, Nigeria. J Am Coll Nutr. 2014;33:437–41. https://doi.org/10.1080/07315724.2013.875356.
Tsang BL, Sullivan KM, Ruth LJ, Williams TN, Suchdev PS. Nutritional status of young children with inherited blood disorders in Western Kenya. Am J Trop Med Hyg. 2014;90:955–62. https://doi.org/10.4269/ajtmh.13-0496.
Eke CB, Chukwu BF, Ikefuna AN, Ezenwosu OU, Emodi IJ. Bioelectric impedance analysis of body composition of children and adolescents with sickle cell anemia in Enugu, Nigeria. Pediatr Hematol Oncol. 2015;32:258–68. https://doi.org/10.3109/08880018.2015.1010111.
Bismarck Eke C, Onyeka Edelu B, Nnaemeka Ikefuna A, Josephine Emodi I, Chidozie IB. Obesity in preschool-aged children with sickle cell Anemia: emerging nutritional challenge in a resource limited setting. Pediatr Hematol Oncol. 2015;32:390–8. https://doi.org/10.3109/08880018.2015.1030716.
Ranque B, Menet A, Boutouyrie P, Diop IB, Kingue S, Diarra M, et al. Arterial stiffness impairment in sickle cell disease associated with chronic vascular complications. Circulation. 2016;134:923–33. https://doi.org/10.1161/CIRCULATIONAHA.115.021015.
Odetunde OI, Chinawa JM, Achigbu KI, Achigbu EO. Body Mass Index and other anthropometric variables in children with sickle cell anaemia. Pak J Med Sci. 2016;32:341–6. https://doi.org/10.12669/pjms.322.9046.
Esezobor CI, Akintan P, Akinsulie A, Temiye E, Adeyemo T. Wasting and stunting are still prevalent in children with sickle cell anaemia in Lagos, Nideria. Ital J Pediatr. 2016;42:45. https://doi.org/10.1186/s13052-016-0257-4.
Senbanjo IO, Oshikoya KA, Salisu M, Diaku-Akinwumi IN. Head circumference of children with sickle cell disease in Lagos, Nigeria. Pan Afr Med J. 2016;25. https://doi.org/10.11604/pamj.2016.25.4.8030.
Oluwole OB, Noll RB, Winger DG, Akinyanju O, Novelli EM. Cognitive functioning in children from Nigeria with sickle cell anemia. Pediatr Blood Cancer. 2016;63:1990–7. https://doi.org/10.1002/pbc.26126.
Adegoke SA, Figueiredo MS, Adekile AD, Braga JAP. Comparative study of the growth and nutritional status of Brazilian and Nigerian school-aged children with sickle cell disease. Int Health. 2017;9:327–34. https://doi.org/10.1093/inthealth/ihx035.
Mikobi TM, Lukusa Tshilobo P, Aloni MN, Akilimali PZ, Mvumbi-Lelo G, Mbuyi-Muamba JM. Clinical phenotypes and the biological parameters of Congolese patients suffering from sickle cell anemia: a first report from Central Africa. J Clin Lab Anal. 2017;1. https://doi.org/10.1002/jcla.22140.
Lukusa Kazadi A, Ngiyulu RM, Gini-Ehungu JL, Mbuyi-Muamba JM, Aloni MN. Factors associated with growth retardation in children suffering from sickle cell Anemia: first report from Central Africa. Anemia. 2017;1. https://doi.org/10.1155/2017/7916348.
Sokunbi OJ, Ekure EN, Temiye EO, Anyanwu R, Okoromah CAN. Pulmonary hypertension among 5 to 18 year old children with sickle cell anaemia in Nigeria. PLoS One. 2017;12. https://doi.org/10.1371/journal.pone.0184287.
Onukwuli VO, Ikefuna AN, Nwokocha AR, Emodi IJ, Eke CB. Relationship between zinc levels and anthropometric indices among school-aged female children with sickle cell anemia in Enugu, Nigeria. Niger J Clin Pract. 2017;20:1461–7. https://doi.org/10.4103/njcp.njcp_104_17.
Osei TB, Apprey C, Mills-Robertson FC, Ohemeng AN. Nutritional status of children with sickle cell disease: a study at the Komfo Anokye teaching Hospital of Ghana. Nutr Food Sci. 2019;49:232–9. https://doi.org/10.1108/NFS-03-2018-0100.
Um SSN, Seungue J, Alima AY, Mbono R, Mbassi H, Chelo D, et al. A cross sectional study of growth of children with sickle cell disease, aged 2 to 5 years in Yaoundé, Cameroon. Pan Afr Med J. 2019;34. https://doi.org/10.11604/pamj.2019.34.85.16432.
Alexandre-Heymann L, Dubert M, Diallo DA, Diop S, Tolo A, Belinga S, et al. Prevalence and correlates of growth failure in young African patients with sickle cell disease. Br J Haematol. 2019;184. https://doi.org/10.1111/bjh.15638.
Arigliani M, Castriotta L, Zubair R, Dogara LG, Zuiani C, Raywood E, et al. Differences in lung function between children with sickle cell anaemia from West Africa and Europe. Thorax. 2019;74:1154–60. https://doi.org/10.1136/thoraxjnl-2019-213717.
Arigliani M, Kitenge R, Castriotta L, Ndjule P, Barbato V, Cogo P, et al. Lung function in children with sickle cell disease from Central Africa. Thorax. 2019;74:604–6. https://doi.org/10.1136/thoraxjnl-2018-212720.
Ukoha OM, Emodi IJ, Ikefuna AN, Obidike EO, Izuka MO, Eke CB. Comparative study of nutritional status of children and adolescents with sickle cell anemia in Enugu, Southeast Nigeria. Niger J Clin Pract. 2020;23:1079–86. https://doi.org/10.4103/njcp.njcp_476_19.
Cox SE, Makani J, Komba AN, Soka D, Newton CR, Kirkham FJ, et al. Global arginine bioavailability in Tanzanian sickle cell anaemia patients at steady-state: a nested case control study of deaths versus survivors. Br J Haematol. 2011;155:522–4. https://doi.org/10.1111/j.1365-2141.2011.08715.x.
Enomoto TM, Isichei C, Vanderjagt DJ, Fry DE, Glew RH. Decreased polyunsaturated fatty acids in sickle cell anaemia. J Trop Pediatr. 1998;44:28–34. https://doi.org/10.1093/tropej/44.1.28.
Glew RH, Casados JK, Huang YS, Chuang LT, VanderJagt DJ. The fatty acid composition of the serum phospholipids of children with sickle cell disease in Nigeria. Prostaglandins Leukot Essent Fat Acids. 2002;67:217–22. https://doi.org/10.1054/plef.2002.0423.
Hamdy M, Mosallam D, Rabie W. Vitamin E and selenium in Egyptian thalassemia and sickle cell disease patients: Are they deficient? Haematologica. 2014;99:741.
Ren H, Ghebremeskel K, Okpala I, Lee A, Ibegbulam O, Crawford M. Patients with sickle cell disease have reduced blood antioxidant protection. Int J Vitam Nutr Res. 2008;78:139–47. https://doi.org/10.1024/0300-9831.78.3.139.
Shukla P, Graham SM, Borgstein A, Nhlane A, Harper G, Brabin BJ. Sickle cell disease and vitamin E deficiency in children in developing countries. Trans R Soc Trop Med Hyg. 2000;94:109. https://doi.org/10.1016/S0035-9203(00)90460-7.
Jiya NM, Matazu IK, Bilbis LS, Shehu RA. Serum levels of antioxidant vitamins in foetal haemoglobin (HbF) persistent sickle cell anaemia children in Sokoto, Nigeria. Ann Afr Med. 2005;4:168–71.
Cox SE, L’Esperance V, Makani J, Soka D, Hill CM, Kirkham FJ. Nocturnal haemoglobin oxygen saturation variability is associated with vitamin C deficiency in Tanzanian children with sickle cell anaemia. Acta Paediatr. 2011;100:594–7. https://doi.org/10.1111/j.1651-2227.2010.02078.x.
Adegoke SA, Smith OS, Adekile AD, Figueiredo MS. Relationship between serum 25-hydroxyvitamin D and inflammatory cytokines in paediatric sickle cell disease. Cytokine. 2017;96:87–93. https://doi.org/10.1016/j.cyto.2017.03.010.
Adegoke SA, Oyelami OA, Adekile A, Figueiredo MS. Influence of serum 25-hydroxyvitamin D on the rate of pain episodes in Nigerian children with sickle cell anaemia. Paediatr Int Child Health. 2017;37:217–21. https://doi.org/10.1080/20469047.2017.1295012.
Siegert T, Cusick S, Ware R, Opoka R, John C. Micronutrient levels in a pediatric Ugandan cohort with sickle cell anemia. Am J Trop Med Hyg. 2018;99:158–9.
Anyanwu JN, Williams O, Sautter CL, Kasirye P, Hume H, Opoka RO, et al. Novel use of Hydroxyurea in an African region with malaria: protocol for a randomized controlled clinical trial. JMIR Res Protoc. 2016;5:e110. https://doi.org/10.2196/resprot.5599.
Ajayi G. Zinc, magnesium, and copper concentrations in serum, erythrocytes, and urine in sickle cell homozygotes and heterozygote Nigerian women. Trace Elem Electrocytes. 1997;14:69–71.
Akenami FO, Aken’Ova YA, Osifo BO. Serum zinc, copper and magnesium in sickle cell disease at Ibadan, south western Nigeria. Afr J Med Med Sci. 1999;28:137–9.
Oladipo OO, Temiye EO, Ezeaka VC, Obomanu P. Serum magnesium, phosphate and calcium in Nigerian children with sickle cell disease. West Afr J Med. 2005;24:120–3. https://doi.org/10.4314/wajm.v24i2.28180.
Ojo JO, Oluwole AF, Osoniyi RO, Durosinmi MA, Aboderin AO. Determination of trace elements status of Nigerians with sickle cell anaemia using INAA and PIXE. Afr J Med Med Sci. 2006;35:461–7.
Arinola OG, Olaniyi JA, Akiibinu MO. Evaluation of antioxidant levels and trace element status in Nigerian sickle cell disease patients with Plasmodium parasitaemia. Pak J Nutr. 2008;7:766–9. https://doi.org/10.3923/pjn.2008.766.769.
Olaniyi JA, Arinola OG. Nitric oxide and trace metals in relation to haemoglobin F concentration in Nigerian sickle cell disease patients. Turkish J Med Sci. 2010;40:109–13. https://doi.org/10.3906/sag-0901-30.
Cox SE, L’Esperance V, Makani J, Soka D, Prentice AM, Hill CM, et al. Sickle cell anemia: Iron availability and nocturnal oximetry. J Clin Sleep Med. 2012;8:541–5. https://doi.org/10.5664/jcsm.2152.
Sungu JK, Mukuku O, Mutombo AM, Mawaw P, Aloni MN, Luboya ON. Trace elements in children suffering from sickle cell anemia: a case–control study. J Clin Lab Anal. 2018;32. https://doi.org/10.1002/jcla.22160.
Lee N, Makani J, Tluway F, Makubi A, Armitage AE, Pasricha SR, et al. Decreased Hepcidin levels are associated with low steady-state hemoglobin in children with sickle cell disease in Tanzania. EBioMedicine. 2018;34:158–64. https://doi.org/10.1016/j.ebiom.2018.07.024.
Ajibola KA, Adedokun KA, Oduola T, Oparinde DP, Ayelagbe OG, Ojokuku HO. Assessment of iron status and interplay between lipid peroxidation and antioxidant capacity in common hemoglobin variants in Osun state, southwestern Nigeria. Kaohsiung J Med Sci. 2019;35. https://doi.org/10.1002/kjm2.12062.
Emokpae MA, Fatimehin EB, Obazelu PA. Serum levels of copper, zinc and disease severity scores in sickle cell disease patients in Benin City, Nigeria. Afr Health Sci. 2019;19:2798–805. https://doi.org/10.4314/ahs.v19i3.56.
Antwi-Boasiako C, Dankwah GB, Aryee R, Hayfron-Benjamin C, Doku A, N’guessan BB, et al. Serum iron levels and copper-to-zinc ratio in sickle cell disease. Medicina (B Aires). 2019;55. https://doi.org/10.3390/medicina55050180.
Kudirat AA, Shehu UA, Kolade E, Ibrahim M. Serum zinc level during and after acute painful episodes in children with sickle cell anemia at the aminu Kano teaching hospital, Kano, Northern Nigeria. Niger J Clin Pract. 2019;22:16–23. https://doi.org/10.4103/njcp.njcp_169_18.
Erhabor O, Ogar K, Erhabor T, Dangana A. Some haematological parameters, copper and selenium level among children of African descent with sickle cell disease in specialist hospital Sokoto, Nigeria. Hum Antibodies. 2019;27:143–54. https://doi.org/10.3233/HAB-180360.
Afolabi IS, Osikoya IO, Fajimi OD, Usoro PI, Ogunleye DO, Bisi-Adeniyi T, et al. Solenostemon monostachyus, Ipomoea involucrata and Carica papaya seed oil versus glutathione, or Vernonia amygdalina: Methanolic extracts of novel plants for the management of sickle cell anemia disease. BMC Complement Altern Med. 2012;12:262. https://doi.org/10.1186/1472-6882-12-262.
Imaga NA, Chukwu CE, Blankson A, Gbenle GO. Biochemical assessment of Ciklavit®, a nutraceutical used in sickle cell anaemia management. J Herb Med. 2013;3:137–48. https://doi.org/10.1016/j.hermed.2013.05.003.
Kaddam L, Fadl-Elmula I, Eisawi OA, Abdelrazig HA, Saeed AM. Acacia Senegal (gum Arabic) supplementation modulate lipid profile and ameliorated dyslipidemia among sickle cell anemia patients. J Lipids. 2019. https://doi.org/10.1155/2019/3129461.
Adegoke SA, Shehu UA, Mohammed LO, Sanusi Y, Oyelami OA. Influence of lime juice on the severity of sickle cell Anemia. J Altern Complement Med. 2013;19:588–92. https://doi.org/10.1089/acm.2012.0567.
Daak AA, Ghebremeskel K, Hassan Z, Attallah B, Azan HH, Elbashir MI, et al. Effect of omega-3 (n-3) fatty acid supplementation in patients with sickle cell anemia: randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2013;97:37–44. https://doi.org/10.3945/ajcn.112.036319.
Cox SE, Ellins EA, Marealle AI, Newton CR, Soka D, Sasi P, et al. Ready-to-use food supplement, with or without arginine and citrulline, with daily chloroquine in Tanzanian children with sickle-cell disease: a double-blind, random order crossover trial. Lancet Haematol. 2018;5:E147–60. https://doi.org/10.1016/S2352-3026(18)30020-6.
Onalo R, Cooper P, Cilliers A, Nnebe-Agumadu U, Oniyangi O, Oladimeji D, et al. Oral arginine therapy as a novel adjuvant in the nanagement of acute pain in children with sickle cell anemia in Nigeria: a randomized placebo-controlled trial. Blood. 2019;134:613. https://doi.org/10.1182/blood-2019-122510.
Morris CR, Kuypers FA, Lavrisha L, Ansari M, Sweeters N, Stewart M, et al. A randomized, placebo-controlled trial of arginine therapy for the treatment of children with sickle cell disease hospitalized with vaso-occlusive pain episodes. Haematologica. 2013;98:1375–82. https://doi.org/10.3324/haematol.2013.086637.
Scott R, Ferguson A, Jenkins M, Clark H. Studies in sickle-cell anemia. VIII. Further observations on the clinical manifestations of sickle-cell anemia in children. AMA Am J Dis Child. 1955;90:682–91 Available: http://www.ncbi.nlm.nih.gov/pubmed/13268049.
Whitten C. Growth status of children with sickle-cell Anemia. Arch Pediatr Adolesc Med. 1961;102:355. https://doi.org/10.1001/archpedi.1961.02080010357009.
Heyman MB, Vichinsky E, Katz R, Gaffield B, Hurst D, Castillo R, et al. Growth retardation in sickle-cell disease treated by nutritional support. Lancet. 1985;1:903–6. https://doi.org/10.1016/s0140-6736(85)91677-0.
Ashcroft MT, Serjeant GR, Desai P. Heights, weights, and skeletal age of jamaican adolescents with sickle cell Anaemia. Arch Dis Child. 1972. https://doi.org/10.1136/adc.47.254.519.
Nikhar H, Shinde G, Meshram S. An anthropometric and hematological comparison of sickle cell disease children from rural and urban areas. Indian J Hum Genet. 2012;18:40. https://doi.org/10.4103/0971-6866.96643.
Mitchell MJ, Carpenter GJO, Crosby LE, Bishop CT, Hines J, Noll J. Growth status in children and adolescents with sickle cell disease. Pediatr Hematol Oncol. 2009. https://doi.org/10.1080/08880010902896882.
Silva CM, Viana MB. Growth deficits in children with sickle cell disease. Arch Med Res. 2002. https://doi.org/10.1016/S0188-4409(01)00360-5.
Badaloo A, Jackson AA, Jahoor F. Whole body protein turnover and resting metabolite rate in homozygous sickle cell disease. Clin Sci. 1989;77:93–7. https://doi.org/10.1042/cs0770093.
Barden EM, Zemel BS, Kawchak DA, Goran MI, Ohene-Frempong K, Stallings VA. Total and resting energy expenditure in children with sickle cell disease. J Pediatr. 2000;36:73–9. https://doi.org/10.1016/S0022-3476(00)90053-2.
Hibbert JM, Creary MS, Gee BE, Buchanan ID, Quarshie A, Hsu LL. Erythropoiesis and myocardial energy requirements contribute to the hypermetabolism of childhood sickle cell anemia. J Pediatr Gastroenterol Nutr. 2006;43:680–7. https://doi.org/10.1097/01.mpg.0000228120.44606.d6.
Hibbert JM, Hsu LL, Bhathena SJ, Irune I, Sarfo B, Creary MS, et al. Proinflammatory cytokines and the Hypermetabolism of children with sickle cell disease. Exp Biol Med. 2005;230:68–74. https://doi.org/10.1177/153537020523000109.
Umeakunne K, Hibbert JM. Nutrition in sickle cell disease: recent insights. Nutr Diet Suppl. 2019;11:9–17. https://doi.org/10.2147/nds.s168257.
Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. J Am Med Assoc. 2014;312:1033–48. https://doi.org/10.1001/jama.2014.10517.
National Institutes of Health. Evidence-based management of sickle cell disease. Expert Panel Report, 2014.
Acknowledgements
We thank Mary Isgro, Emily Mannix and Sherri Schwaninger for database searches and collection of articles.
Funding
No financial support was required to conduct this study and there was no sponsor of the study. Non-financial sources of support were the provision of information specialist services by Novartis Institutes for BioMedical Research to assist with the mechanics of the database searches and acquisition of full text articles.
Author information
Authors and Affiliations
Contributions
EBN was responsible for coordinating the entire project and is the guarantor of the review. EBN, CJ, JS, and MSA developed the project. EBN, CJ, RS, AKD and JS reviewed the abstracts and articles included in this study and carried out the data collection. EBN, CJ, JS, and MSA participated in the analyses. EBN and CJ wrote the first version of the manuscript. AJ, AKD, EM, AO, RS, JS, SAA, ABB, HIH and MSA reviewed and edited the manuscript during its production. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Rajiv Shah is an employee of Novartis Global Health and Corporate Responsibility. Jonathan Spector is an employee at the Novartis Institute for Tropical Diseases. Catherine Jones was an employee at the Novartis Institute for Tropical Diseases at the time she contributed to this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nartey, E.B., Spector, J., Adu-Afarwuah, S. et al. Nutritional perspectives on sickle cell disease in Africa: a systematic review. BMC Nutr 7, 9 (2021). https://doi.org/10.1186/s40795-021-00410-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40795-021-00410-w