
Abstract
Background
The gut microbiota has emerged as an important environmental factor associated with obesity, type 2 diabetes, and cardiovascular disease, through its interactions with dietary factors. Therefore, we analyzed the composition of the fecal microbiota and levels of biochemical markers related to metabolic disease according to dietary pattern in Korean adolescents.
Methods
We collected fecal samples from 112 student subjects aged 13–16 years with sufficient information available regarding clinical biomarkers and diet, and performed 16S rRNA targeted gene sequencing.
Results
Regarding bacterial composition according to taxonomic rank, we found that traditional dietary patterns enriched in plant-based and fermented foods were associated with higher proportions of Bacteroides (Bacteroidaceae) and Bifidobacterium (Bifidobacteriaceae-Actinobacteria) and a lower proportion of Prevotella (Prevotellaceae) relative to modified Western dietary patterns (a greater proportion of animal-based foods). Specifically, the proportion of Bacteroides (Bacteroidaceae) was associated with intake of plant-based nutrients such as fiber; however, that of Prevotella (Prevotellaceae) was negatively associated with these factors. Additionally, we observed that the increase of prevotella (Prevotellaceae) and decrease of Bacteroides (Bacteroidaceae) and Ruminococcaceae had a higher risk of obesity. We also found that the traditional dietary pattern was negatively associated with general and central adiposity and levels of clinical biomarkers, including AST, ALT, total cholesterol, triglyceride, hs-CRP, insulin, and HOMA-IR, whereas the positive associations were found for a modified Western dietary pattern.
Conclusions
These findings suggest that the gut microbiota composition differs markedly according to dietary intake and suggest a role for diet in promoting a gut microbiome associated with the pathogenesis of metabolic disease.
Similar content being viewed by others


Background
The increased prevalence of childhood obesity worldwide is associated with serious health risks, such as insulin resistance (IR), type two diabetes (T2D), and cardiovascular disease (CVD) in childhood and later life [1]. The etiology of obesity and its metabolic consequences are complex and involve environmental factors that are challenging to modify. It is therefore crucial to identify modifiable risk factors involved in the early development of metabolic disorders to facilitate prevention and treatment.
In recent years, the gut microbiota has emerged as an important environmental factor associated with host health. The gut is home to trillions of microbes, and plays a major role in energy metabolism and the immune system [2, 3]. Several studies in animal models and humans have suggested that the gut microbiota is linked to complex disease phenotypes such as obesity and insulin resistance [4, 5]. A balanced gut microbiota composition confers benefit to the host, whereas microbial dysbiosis is implicated in various diseases, including obesity, T2D, and CVD. These studies imply that the effects of environmental factors on the development of metabolic-related disorders are mediated in part by altered gut microbial composition and function.
Therefore, research into factors affecting the gut microbiota has become an area of growing scientific interest. The gut microbiota is influenced by various factors, including the microbial species acquired at birth, age, host genotype, geography, and diet [6–9]. Of these, diet is considered a key contributor to the diversity of the gut microbiota, explaining 57% of the total structural variation, while only 12% is related to genetic differences [10]. Therefore, many studies have focused on the relationship between the gut microbiota and dietary factors, such as dietary pattern (vegetarian and Western) [11, 12], specific foods (whole grain products, fruits, and vegetables) [13–17], and food constituents (dietary fiber, fat, and protein) [8, 18–20]. Interestingly, a recent study showed that European children who consumed a typical Western diet had a microbiota enriched in Firmicutes and Enterobacteriaceae, whereas rural African children, who consumed a diet low in fat and animal protein and rich in plant-based foods, had greater abundances of the genera Bacteroidetes and Prevotella [7]. Another study demonstrated that microbiota composition was strongly associated with long-term diet [8]. Bacteroides abundance was found to be associated with a diet enriched in animal products, whereas that of Prevotella was correlated with diets that contained more plant-based foods.
Dietary factors can vary widely according to ethnicity and geographical location; however, few studies have focused on Korean populations. Therefore, we performed an in-depth analysis of the association between the fecal microbiota and dietary factors in Korean adolescents. Because dietary intake is a complex exposure variable, we used a total diet approach to identity the overall dietary patterns of Korean adolescents and then examined their associations with the gut microbiota composition and levels of biochemical markers. Assessment of food intake as a whole using dietary pattern analysis yielded useful information regarding the link between diet and the gut microbiota that will facilitate formulation of dietary guidelines to prevent disease.
Methods
Study participants
For this study, data were obtained from the Korean Children-Adolescents Study (KoCAS) conducted by the Korea National Institute of Health (KNIH) [4, 21]. A total of 135 volunteers on collection of stool samples, aged 13–16 years, including those with severe obesity, were recruited from Seoul and Kyunggi Province in 2012. After excluding those who did not respond to the dietary survey (n = 19) and those for whom insufficient clinical biomarker information was available (n = 4), data from 112 subjects were analyzed. The study was approved by the Institutional Review Boards of Seoul-Paik Hospital (IIT-2012-092), Inje University, and the Korea Center for Disease Control (KCDC) and Prevention (2012-04EXP-06-R).
Anthropometric and biochemical measurements
Professionally trained personnel performed the anthropometric examinations and blood collections using a standardized protocol. The levels of total cholesterol (TC), triglycerides (TG), and HDL cholesterol (HDL-C) were measured via enzymatic assays, and fasting serum glucose levels were measured using the hexokinase method (Autoanalyzer Model 7600 II; Hitachi, Tokyo, Japan). The levels of high-sensitivity C-reactive protein (hs-crp) were measured by latex agglutination turbidimetry and an analyzer (Model 7600; Hitachi, Tokyo, Japan). Fasting serum insulin levels were measured using an enhanced chemiluminescence immunoassay analyzer (E170; Roche, Germany). Insulin resistance (IR) was estimated from fasting serum measurements using the homeostasis model assessment of insulin resistance (HOMA-IR): insulin (μIU/mL) × glucose (mmol/L) ÷ 22.5.
Isolation of fecal DNA and pyrosequencing of 16S rRNA
The stool samples were self-collected in sterile boxes containing dry ice, transported to the study center with dry ice within 12 h, and stored at −70 °C in the laboratory freezer until DNA extraction. DNA was extracted using a QIAamp DNA Stool kit (Qiagen, Valencia, CA), and the 16S rRNA gene fragments were amplified. The following barcoded primers were designed to target the hypervariable regions (V1 to V3) within the 16S rRNA gene as described previously [4]. The quality of the PCR product was assessed using a Gel Doc system (Bio–Rad, Hercules, CA, USA), and the amplified products were purified using a QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA). Short DNA fragments were removed using an Ampure Beads Kit (Agencourt Bioanalyzer, MA, USA). The quality and size of the products were assessed using a Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA). The 16S rRNA gene amplicons were sequenced using the GS Junior Sequencing System (Roche, Branford, CT, USA) according to the manufacturer’s protocol. The raw reads were filtered to remove those with a low quality score (average score <25) or reads less than 300 bp. Potential chimeric sequences were detected using the Bellerophon method [22].
Determination of operational taxonomic units and taxonomic classification
To estimate the taxonomic composition of each sample, the numbers of operational taxonomic units (OTUs) in the filtered reads were calculated. As reported previously [4], the number of OTUs was determined by clustering the sequences from each sample using a 97% sequence identity cutoff [23, 24] using the QIIME software (v. 1.8.0). Taxonomic abundance was assessed using RDP Classifier v. 1.1 with a confidence threshold of 0.8 derived from the pre-processed reads for each sample. The microbial compositions were normalized using the value calculated from the count of taxonomy abundance divided by the number of pre-processed reads of each sample. To normalize the sequence reads, we calculated the extent of coverage using the number of singleton phylotypes and the number of sequences which is a percentage value indicative of how complete sample coverage was. Also, we generated rarefaction curves using a re-sampling without replacement approach to calculate the observed number of OTUs on each iteration. We plotted the curves allowed for comparison of sampling intensity and rarefaction curves appeared parallel. A total of 1,185,358 high-quality sequences (reads) (range, 2,065–42,522 reads) from 135 samples were obtained after filtering out primer sequences and low-quality and chimeric sequences.
Assessment of dietary intake
Typical dietary intake for each subject was estimated based on 3-day food diaries maintained for three consecutive days (2 weekdays and 1 weekend day), which were completed by 112 adolescent subjects. Dietary questionnaires certified by Inje University Paik Hospital and Hallym University Sacred Heart Hospital were checked by researchers trained to determine whether records contained sufficient information. Nutrient intakes were determined from food intakes using the Computer-Aided Nutritional Analysis for Professionals software v. 3.0 (CAN-pro 3.0, Korean Nutrition Society, Seoul, Korea).
Dietary pattern analysis
For a factor analysis [25] to generate dietary patterns, foods from the data of the 2012 KoCAS were classified into 31 food groups based on those used in the Korean Nutrient Database [26] and designed to reflect the food habit characteristics of adolescents (Additional file 1: Supplementary Table S1). The factors were rotated using Varimax rotation to achieve a simpler structure with greater interpretability. We considered components with an eigenvalue by scree test > 2 as significant to identify more meaningful factors. To identify the characteristics of the factors, the collection of food groups with an absolute value factor loading of at least 0.2 was used [27]. A cluster analysis was performed using two factor scores for the subjects calculated by factor analysis, and subjects were grouped into two clusters of dietary patterns through the k-means clustering method.
Definition of metabolic syndrome
Metabolic syndrome was defined as the International Diabetes Federation (IDF) in children and adolescents [28]. According to this criteria, subjects with abdominal obesity (waist circumference above 90th percentile for age, sex and ethnicity), and presenting two or more other clinical features (elevated TG, glucose, blood pressure and low HDL-C), were considered as having metabolic syndrome.
Statistical analysis
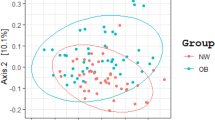
Statistical analysis was performed using the SAS software package (version 9.2; SAS Institute, Inc., Cary, NC, USA), and values are presented as means ± standard deviations (SD) for continuous variables or as raw numbers and percentages for categorical variables. Variables with non-normal distributions were log-transformed before analysis. Microbial alpha diversity of each sample using the Shannon Index with OTUs, H′ = − ∑ S i = 1 (pi ln(p i )) was measured and Beta diversity was measured by the difference in organism composition between samples using a Bray-Curtis distance \( BCij=\frac{Si+ Sj-2 Cij}{Si+ Sj} \). Principal component analysis (PCA) from measured beta diversities was performed in microbiota. Mean values were compared between dietary pattern groups using the general linear model adjusted for age and gender. Chi-squared and Fisher’s exact tests were used to compare prevalence data, and Spearman’s correlation coefficients were used to evaluate the relationship between the abundance of gut microbial taxa and diet. The association between fecal microbiota and metabolic disease were analyzed using a logistic regression model to estimate the odds ratios (ORs) and 95% confidence intervals (CI) after adjusting for age and sex. Differences in the relative abundance of components of the fecal microbiota between the dietary pattern groups were analyzed using the nonparametric Wilcoxon rank-sum test and correction was performed by FDR multiple test. Heat maps and box plots were generated using the R package. P values <0.05 and adjusted P-values < 0.05 were considered to indicate statistical significance.
Results
Dietary pattern
Two major factors were extracted by factor analysis using 31 food groups (Table 1). Factor 1, which had the highest eigenvalue, was characterized as plant-based and fermented foods because it showed higher factor-loading values for whole grains, sweet potatoes, sweets, vegetables, shellfish, seaweeds, and Oriental sources, whereas the contributions of flour, bread, instant products, poultry, eggs, milks, fats, carbonated beverages, and seasonings were negative. Factor 2 was characterized as animal-based foods, with intake of potatoes, nuts & seeds, red meats, fish, oils, fats, and seasonings, but noodles, cookies, fast food, legumes, fruits, milk, and yoghurts had negative contributions. White rice, which is the staple in the Korean diet, had high loadings in both the traditional and modified Western diets.
To categorize the subjects according to dietary pattern, we performed a cluster analysis based on the two factors and derived two clusters that comprised 73.2 and 26.8% of the total subjects, respectively. Factor scores of the two clusters are presented in Table 2. Cluster 1 had characteristics of a traditional (Korean) dietary pattern, with the highest score for Factor 1. Cluster 2 had a high score for Factor 2, representing a modified Western dietary pattern.
Nutrient intakes
The nutrient intakes of the two dietary pattern groups are presented in Additional file 1: Supplementary Table S2. Cluster 1 had significantly higher intakes of carbohydrate, plant protein, fiber, vitamin B6, folate, vitamin C, phosphorus, potassium, calcium, iron (especially plant iron), and sodium than Cluster 2. Additionally, fat intake in Cluster 1 was significantly lower than that in Cluster 2. The percentage of energy from carbohydrate was higher in Cluster 1, whereas that from fat was higher in Cluster 2.
Clinical biomarkers according to dietary pattern group
Table 3 summarizes the clinical and biochemical characteristics of the subjects stratified according to dietary pattern group. There were no significant differences in age, sex, height, glucose, and HDL-cholesterol (HDL-C) between the dietary pattern groups. However, Cluster 2 had significantly higher levels of general and central adiposity—including BMI, waist circumference, fat percent, and fat mass—and clinical biomarkers such as liver enzymes (AST and ALT), TC and TG levels, hs-crp, insulin and insulin-resistance surrogate markers (HOMA-IR) compared with Cluster 1. The prevalence of obesity and metabolic syndrome (MS) was also higher in Cluster 2 compared with Cluster 1 (86.7 and 39.0% for obesity (P = <0.0001), 17.3 and 36.7% for MS (P = 0.0299), respectively).
Fecal microbial composition according to dietary pattern group
As reported previously [4], we calculated the number of OTUs to examine the diversity of the gut microbiota based on an average of 8,846 reads which covered each sample. The mean number of OTUs was 356 ± 140 (range, 105–876) and the number of OTUs between normal and obese individuals (Mann–Whitney U test, p = 0.072) had no significant difference. Also, there was no obvious difference between these samples when the alpha-diversity (Shannon Index) values between normal and obese samples were compared (OTUs, average normal 6.94 ± 0.49, obese 6.98 ± 0.59). In addition, PCA result of beta diversity did not show differential pattern between the normal and obese samples (data not shown). Table 4 and Fig. 1 show the average fecal microbiota compositions of the dietary pattern groups according to taxonomic rank (P < 0.05). Notably, there was a marked difference in the abundance of the genus Bifidobacterium between Cluster 1 and Cluster 2 (P = 0.011). The proportion of Bifidobacterium in Cluster 1 was 0.26%, compared with 0.18% in Cluster 2. This trend was also evident for the family Bifidobacteriaceae and the phylum Actinobacteria (0.26 and 0.18%, and 0.35 and 0.25% respectively). We also found significant differences in the proportions of the genera Bacteroides, Prevotella, Clostridium XlVa, and Roseburia (P = 0.046, 0.015, 0.026, and 0.046, respectively) and the families Bacteroidaceae, Prevotellaceae, and Ruminococcaceae (P = 0.046, 0.047, and 0.047, respectively) according to dietary pattern group. Cluster 1 had a higher proportion of Bacteroides (Bacteroidaceae), Clostridium XlVa, and Roseburia, but a lower proportion of Prevotella (Prevotellaceae) compared with Cluster 2. However, there were no significant difference in above-mentioned genera and families by the dietary patterns after adjustment of FDR.

Association of clusters with abundant microbiota in family and genus levels. The Wilcoxon rank-sum test was used to assess the association of clusters with microbiota (P < 0.05). Boxes represent the interquartile range (IQR) between the first and third quartiles with a line at the median
Risk of obesity and metabolic syndrome
We evaluated the association between fecal microbiota and metabolic disease/or disorders using multivariate-adjusted ORs (Table 5). Because of the absence of normal weight subjects with MS, the normal weight group without MS was used as the reference and compared with obesity group with and without MS in this study. Lower proportion of Bacteroides (Bacteroidaceae) or higher proportion of Prevotella (Prevotellaceae) had significantly higher risk effect on obesity and the risk tended to increase when obesity subjects had with MS. We also found that lower proportion of Ruminococcaceae was associated with a higher risk of obesity and had a tendency slightly to increase risk in obesity with MS although there was no significant effect.
Associations of microbiota composition with dietary intake
Fig. 2 shows a heat map of Spearman’s correlations between dietary intake and microbial taxa. We considered only five genera and four families that exhibited a significant difference in abundance according to dietary pattern group. After adjustment age, sex, and BMI, the high proportion of Bifidobacteriaceae and Bifidobacterium were associated with high intakes of whole grain and low white rice consumption. The prevotellaceae and prevotella were negatively associated with potatoes, whereas Bacteroidaceae and Bacteroides showed the opposite association. The microbiota composition positively associated with legumes were Bacteroides (Bacteroidaceae), Roseburia, and Clostridium XlVa. In addition, Clostridium XlVa was associated with high intakes of legumes and fermented foods, such as yoghurt, cheese, and oriental sauce, and low intake of milk, poultry, and Flour. Roseburia was inversely associated with seaweeds and instant products.

Spearman’s correlations between dietary intakes and microbial taxa. Columns correspond to bacterial taxa showing a significant difference in abundance according to dietary pattern group; rows correspond to a age, sex BMI-adjusted intakes of food items and b age, sex, energy and BMI-adjusted nutrients intakes (* adj P < 0.05) Columns and rows are cluster by hierarchical method
We next examined the association between microbiota composition and nutrient intake. Bacteroides (Bacteroidaceae) was positively associated with iron, especially plant iron. We also found several positive associations, e.g., folate with Bifidobacterium (Bifidobacteriaceae) and niacin with Roseburia.
Discussion
There has been increasing interest in analyzing dietary factors associated with chronic disease. The analysis of dietary patterns has advantages over nutrient-based or food-group-based dietary assessment approaches because people consume foods in the form of meals, which are combinations of various foods and nutrients. Therefore, we analyzed dietary patterns in Korean adolescents using a combination of factor and cluster methods and compared the fecal microbiota and host phenotype across these dietary pattern groups.
In this study, we identified two major dietary patterns: traditional (Korean) and modified Western diets. The traditional diets were characterized as comprising predominantly plant-based and fermented foods because they included higher proportions of oriental sauce, sweet potatoes, vegetables, seaweeds, and whole grains, which are naturally high in fiber and undergo minimal processing. In contrast, the modified Western diets were characterized as comprising animal-based foods with intake of red meats, fishes, oils, and fats. Although the two dietary patterns were statistically independent as determined by the orthogonal rotation procedure, it would be possible for one individual to have high or low scores for both dietary patterns simultaneously [29]. In our study, white rice had high loadings in both the traditional and modified Western diets, as rice is the staple in the Korean diet.
Many studies have attempted to identify an association between dietary patterns and the composition of the gut microbiota. Here, we examined bacterial composition at the genus, family, and phylum levels according to dietary pattern group. We found that adolescents who consumed traditional diets showed a higher proportion of Bacteroides (Bacteroidaceae), Clostridium XlVa, Roseburia, Bifidobacterium (Bifidobacteriaceae–Actinobacteria) and Ruminococcaceae and a lower proportion of Prevotella (Prevotellaceae), whereas inverse associations were found for those who consumed modified Western diets. In our study, traditional diets comprised predominantly plant-based and fermented foods, which are low in fat and rich in carbohydrate, plant protein, vitamin, mineral, and fiber; in contrast, modified Western diets comprised greater proportions of animal-based foods and were high in fat and low in fiber. In support, previous studies have reported that vegetarian diets were associated with Bacteroides [11], and fat-restricted diets, together with higher carbohydrate intake, were linked to increased proportions of Bacteroides and Bifidobacterium [30]. However, Cani et al. reported a reduction in Clostridium cluster XIVa and lower Bifidobacterium and Bacteroides levels [31] and Kim et al. found the enrichment of Ruminococcaceae [32] in mice fed a high-fat diet.
Meanwhile, Wu et al. [8] reported that diets high in animal protein and fats, similar to a Westernized diet, were associated with a Bacteroides enterotype. By contrast, the Prevotella enterotype was associated with diets high in carbohydrates that contained more plant-based foods. Similarly, De Filippo et al. [7] found that children in rural Africa showed a higher abundance of Prevotella, whereas European children had a higher abundance of Bacteroides. The authors speculated that the abundance of Prevotella in rural African children was a consequence of their higher fiber intake because the traditional rural African diet is primarily vegetarian, being low in fat and animal protein and rich in starch, fiber, and polysaccharides. In contrast, the Western diet of the European children was high in animal protein, sugar, starch, and fat, and low in fiber. We assume that the inconsistencies with our findings are due to the complex relationships among genetic, geographical, environmental, technical, and/or clinical factors. The diet of people living in Korea, even those who consuming modified Western diets, is usually rather low in fat and provides a high amount of carbohydrate and fiber compared with the diet of people living in Western countries. Several studies showed that the microbiota composition clustered according to country in spite of a cultural area similar to the Western diet and quantity of Prevotella was greater in people from geographical regions where plant-based dietary pattern dominated [6, 33]. Furthermore, some reports suggested that the genus Prevotella was underrepresented in Americans probably due to discriminatory taxon, which is the enrichment in Prevotella in African children compared with European children, in Africans compared with African Americans, in the Hadza hunter-gatherers (from Tanzania) compared with Italian people, and in Succinivibrio and Treponema in several African populations [6, 34]. Therefore, more integrated approaches are needed to enhance our understanding of these complex associations.
It has been reported that the gut microbial communities of Koreans individuals differed from those of US, Japanese, and Chinese subject, but tended to vary less between individual Koreans, suggesting that diet type affects the gut microbiota [35]. They also found the core gut microbiota in Korean individuals and many of these are related to butyrate-producing bacteria. Koreans have high intakes of carbohydrate and fiber [36, 37], which are related to production of short-chain fatty acids (SCFA) such as acetate, propionate, and butyrate. SCFA, especially butyrate, have been suggested as important in maintaining gut health. Most subjects in our study were in the traditional diet group (73.2%), which was associated with a greater proportion of butyrate-producing bacteria, including Bacteroides, Clostridium XlVa, and Roseburia. Although we could not evaluate the subsequent effects of the gut microbiota related to dietary pattern and fecal metabolite in the current study, we believe that this should be taken into consideration in future in-depth studies.
Several studies have reported an association of major dietary patterns with obesity and metabolic disease. In this study, we found that subjects who consumed a modified Western diet had a higher prevalence of obesity and metabolic syndrome than those who consumed a traditional diet. In support of this, previous cross-sectional studies reported that Western dietary patterns were positively associated with obesity [38, 39]. Two large prospective cohort studies, the Nurses’ Health Study and the Health Professionals Follow-up Study, also reported that adoption of a Western dietary pattern was associated with greater weight gain [40], whereas reduction of weight gain was linked to diets rich in plant-based foods [41]. Furthermore, these studies reported that a Western diet was significantly related to metabolic diseases such as CVD [42, 43] and T2D [44, 45] and to all-cause mortality [43]. The Western dietary pattern was associated with increased risks of these diseases, whereas a prudent dietary pattern, diet high in unprocessed foods and fiber and low in salt, fat, and sugar, was associated with beneficial effects on host health [42–45]. In our study, we found that several important components of metabolic disorder were associated with dietary pattern. Subjects who consumed a modified Western diet had higher levels of liver enzymes, TC and TG, hs-CRP, insulin, and HOMA-IR than those who consumed a traditional diet, and these values were associated with increased risk of CVD and T2D. Also, we investigated that high consumption of whole grain and fiber or low consumption of fats is positively associated with Bacteroides, but not Prevotella (data not shown). However, the association disappeared after adjustment for BMI, because most obese adolescents consumed modified Western diet characterized with low intake of fiber and high intake of fat in the current study. This indicates the effects of fiber and fat intakes on gut microbiota composition including Bacteroides and Prevotella, which is associated with the pathogenesis of metabolic disease.
Many studies have attempted to identify an association between the composition of the gut microbiota and metabolic disease. In this study, we found that increased in prevotella (Prevotellaceae) and decreased in Bacteroides (Bacteroidaceae) and Ruminococcaceae had a higher risk of obesity. Theses tendency increased when they had with MS in genera and families of prevotella (Prevotellaceae) and Bacteroides (Bacteroidaceae). However, the evidence for an association between gut microbiota and metabolic disease is scarce. Recent studies suggested a potential role for diet in promoting a gut microbiome associated with the pathogenesis of metabolic disease. Koeth et al. [46, 47] found that microbial metabolism of choline, phosphatidylcholine and L-carnitine resulted in production of trimethylamine (TMA), which is further metabolized to the proatherogenic species, trimethylamin-N-oside (TMAO). Together, these findings suggested that individuals adhering to an omnivorous diet have higher fasting TMAO concentrations and produce more TMAO than do vegans or vegetarians after ingestion of carnitine through a microbiota-dependent mechanism. Furthermore, a correlation analysis of the fecal microbiota and plasma TMAO levels indicated that subjects with a Prevotella enterotype had significantly higher plasma TMAO concentrations than did Bacteroides enterotype subjects [47]. Interestingly, our study showed that subjects who consumed a modified Western diet had a greater abundance of Prevotella. Additionally, we recently reported that an increased Prevotella population was associated with increased TC, TG, and hs-crp levels, but negatively associated with HDL-C [4].
Due to the fact that QIAamp DNA Stool kit based on enzymatic DNA-extraction has the lower diversity of fecal microbiota comparing other DNA extraction kits [48] and is a insufficient method to lyse cell wall efficiently in detecting genes of various Gram-positive bacteria [49], caution must be used when interpreting these findings as regards to the general adolescent fecal microbiota. Additionally, short-term dietary data on 3-day food diaries maintained for three consecutive days (2 weekdays and 1 weekend day) were obtained from self-reported questionnaires which are not representative of the long-term dietary pattern of the subjects and may have resulted in an estimation bias. Finally, our study was cross-sectional in design. In this context, further studies with different methods using a prospective design and measurements before and after interventions are needed to determine core microbiota, the effects of dietary patterns, and the causal relationships among microbiota, dietary patterns, metabolic markers, and disease.
Nevertheless, we found that the proportions of the genera Bacteroides, Prevotella, Clostridium XlVa, Roseburia, and Bifidobacterium were markedly different in subjects who consumed traditional compared with those who consumed modified Western diets. Furthermore, a traditional (Korean) dietary pattern was associated with reduced risk of metabolic disease, whereas opposite associations were found for a modified Western dietary pattern. These findings suggest that dietary habits affect both the gut microbiota composition and host health.
Conclusions
We found that traditional dietary patterns were associated with higher proportions of Bacteroides (Bacteroidaceae) and Bifidobacterium (Bifidobacteriaceae-Actinobacteria) and a lower proportion of Prevotella (Prevotellaceae) relative to modified Western dietary patterns. Specifically, the proportion of Bacteroides (Bacteroidaceae) was associated with intake of plant-based nutrients such as fiber; however, that of Prevotella (Prevotellaceae) was negatively associated with these factors. Also, we found that prevotella (Prevotellaceae) increased and Bacteroides (Bacteroidaceae) and Ruminococcaceae decreased in higher risk group of obesity. The risk had a tendency to increase in obesity subjects with MS in genera of prevotella and Bacteroides and families of Prevotellaceae and Bacteroidaceae. We also found that the traditional dietary pattern was negatively associated with general and central adiposity and levels of clinical biomarkers, including AST, ALT, total cholesterol, triglyceride, hs-CRP, insulin, and HOMA-IR, whereas the positive associations were found for a modified Western dietary pattern. These findings suggest that dietary habits affect both the gut microbiota composition and host health.
Abbreviations
- ALT:
-
Alalnine aminotransferase
- AST:
-
Aspartate aminotransferase
- BMI:
-
Body mass index
- CVD:
-
Cardiovascular disease
- HDL-C:
-
High-density lipoprotein-cholesterol
- HOMA-IR:
-
Homeostasis model assessment of insulin resistance
- hs-crp:
-
High-sensitivity C-reactive protein
- IR:
-
Insulin resistance
- MS:
-
Metabolic syndrome
- OTUs:
-
Operational taxonomic units
- T2D:
-
Type 2 diabetes
- TC:
-
Total cholesterol
- TG:
-
Triglyceride
- TMAO:
-
Trimethylamin-N-oside
References
Poyrazoglu S, Bas F, Darendeliler F. Metabolic syndrome in young people. Curr Opin Endocrinol Diabetes Obes. 2015;21:56–63.
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;10:15718–23.
Goodman AL, Gordon JI. Our unindicted coconspirators: human metabolism from a microbial perspective. Cell Metab. 2010;12:111–6.
Hu HJ, Park SG, Jang HB, Choi MG, Park KH, Kang JH, et al. Obesity alters the microbial community profile in Korean adolescents. PLoS One. 2015;10:e0134333.
Diamant M, Blaak EE, de Vos WM. Do nutrient-gut-microbiota interactions play a role in human obesity, insulin resistance and type 2 diabetes? Obes Rev. 2011;12:272–81.
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominquez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2009;457:480–4.
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural africa. Proc Natl Acad Sci U S A. 2010;104:14691–6.
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–8.
Koleva PT, Bridgman SL, Kozyrskyj AL. The infant gut microbiome: evidence for obesity risk and dietary intervention. Nutrients. 2015;7:2237–60.
Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 2010;4:232–41.
Liszt K, Zwielehner J, Handschur M, Hippe B, Thaler R, Haslberger AG. Characterization of bacteria, clostridia and bacteroides in faeces of begetarinas using qPCR and OCR-DGGE fingerprinting. Ann Nutr Metab. 2009;54:253–7.
Zimmer J, Lange B, Frick JS, Sauer H, Zimmermann K, Schwiertz A, et al. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur J Clin Nutr. 2012;505:559–63.
Carvalho-Wells AL, Helmolz K, Nodet C, Molzer C, Leonard C, McKevith B, et al. Determination of the in vivo previotic potential of a maize-based whole grain breakfast cereal: a human feeding study. Br J Nutr. 2010;104:1353–6.
Costabile A, Klinder A, Fava F, Napolitano A, Fogliano V, Leonard C, et al. Whole-grain wheat breakfast cereal has a previotic effect on the human gut microbiota: a double-blind, placebo-controlled, crossover study. Br J Nutr. 2008;99:110–20.
Vendrame S, Guglielmetti S, Riso P, Arioli S, Klimis-Zacas D, Porrini M. Six-week consumption of a wild blueberry powder drink increases bifidobacteria in the human gut. J Agric Food Chem. 2011;59:12815–20.
Fernando WM, Hill JE, Zello GA, Tyler RT, Dahl WJ, Van Kessel AG. Diets supplemented with chickpea or its main oligosaccharide component raffinose modify faecal microbial composition in healthy adults. Benef Microbes. 2010;1:197–207.
Han K, Bose S, Wang JH, Kim BS, Kim MJ, Kim EJ, et al. Contrasting effects of fresh and fermented kimchi consumption on gut microbiota compostion and gene expression related to metabolic syndrome in oveses korean women. Mol Nutr Food Res. 2015;59:1004–8.
Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2010;5:220–30.
Martinez I, Kim J, Duffy PR, Schlegel VL, Walter J. Resistant starches types two and four habe differential effects on the composition of the fecal microbiota in human subjects. PLoS One. 2010;5:e15046.
Russell WR, Gratz SW, Duncan SH, Holtrop G, Ince J, Scobbie L, et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr. 2011;93:1062–72.
Lee A, Jang HB, Ra M, Choi Y, Lee HJ, Park JY, et al. Prediction of future risk of insulin resistance and metabolic syndrome based on Korean boy’s metabolite profiling. Obes Res Clin Pract. 2015;9:336–45.
Huber T, Faulkner G, Hugenholtz P. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics. 2004;20:2317–9.
Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the health “core microbiome” of oral microbial communities. BMC Microbiol. 2009;9:259.
Chiu CM, Huang WC, Weng SL, Tseng HC, Liang C, Wang WC, et al. Systematic analysis of the association between gut flora and obesity through high-throughput sequencing and bioinformatics approaches. Biomed Res Int. 2014;2014:906168.
Gorsuch RI. Factor analysis. Philadelphia: Lawrence Erlbaum Associates; 1983.
The Korean Nutrition Society. Recommended Dietary Allowances for Koreans. 7th revision. Seoul: Korean Nutrition Society; 2000 (in Korean).
Hu FB, Rimm EB, Smith-Warner SA, Feskanich D, Stampfer MJ, Ascherio A, et al. Reproducibility and validity of dietary patterns assessed with a food-frequency questionnaire. Am J Clin Nutr. 1999;69:246–9.
Zimmet P, Alberti G, Kaufman F, Tajima N, Silink M, Arslaina S, et al. The metabolic syndrome in children and adolescents. Lancet. 2007;369:2059–61.
Hu FB. Dietary pattern analysis: a new direction in nutritional epidemiology. Curr Opin Lipidol. 2002;13:3–9.
Fava F, Gitau R, Griffin BA, Gibson GR, Tuohy KM, Lovegrove JA. The type and quantity of dietary fat and carbohydrate later faecal microbiome and short-chain fatty acid excretion in a metabolic syndrome “at-risk” population. Int J Obes (Lond). 2013;37:216–23.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72.
Kim KA, Gu W, Lee IA, Joh EH, Kim DH. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One. 2012;7:e47713.
Grzeskowiak K, Collado MC, Mangani C, Maleta K, Laitinen K, Ashorn P, et al. Distinct gut microbiota in southeastern African and northern european infants. J Pediatr Gastroenterol Nutr. 2012;54:812–6.
Graf D, Di Cagno R, Fak F, Flint HJ, Nyman M, Saarela M, et al. Contribution of diet to the composition of the human gut microbiota. Microb Ecol Health Dis. 2015;26:26164.
Nam Y, Jung M, Roh SW, Kim M, Bae J. Comparative analysis of Koran human gut microbiota by barcoded pyrosequencing. PLoS One. 2011;6:e22109.
EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Scientific opinion on dietary reference values for carbohydrate and dietary fibre. EFSA J. 2010;8:1462.
Lee Y, Lee HJ, Lee HS, Jang YA, Kim C. Analytical dietary fiber database for the National Health and Nutrition Survey in Korea. J Food Compos Anal. 2008;21:S35–42.
Slattery ML, Edwards SL, Boucher KM, Anderson K, Caan BJ. Lifestyle and colon cancer: an assessement of factors associated with risk. Am J Epidemiol. 1999;150:869–77.
Esmaillzadeh A, Azadbakht L. Major dietary patterns in relation to general obesity and central adiposity among Iranian women. J Nutr. 2008;138:358–63.
Schulze MB, Fung TT, Manson JE, Willett WC, Hu FB. Dietary patterns and changes in body weight in women. Obesity (Silver Spring). 2006;14:1444–53.
Mozaffarian D, Hao T, Rimm EB, Willett WC, Hu FB. Changes in diet and lifestyle and long-term weight gain in women and men. N Engl J Med. 2011;364:2392–404.
Hu FB, Rimm EB, Stampfer MJ, Ascherio A, Spiegelman D, Willett WC. Prospective study of major dietary patterns and risk of cornoay heart diseas in men. Am J Clin Nutr. 2002;72:912–21.
Heidemann C, Schulze MB, Franco OH, van Dam RM, Mantzoros CS, Hu FB. Dietary patterns and risk of mortality from cardioascular diseas, cancer, and allcauses in a prospective cohort of women. Circulation. 2008;118:230–7.
Van Dam RM, Rimm EB, Willett WC, Stampfer MJ, Hu FB. Dietary patterns and risk for type 2 diabetes mellitus in U.S. men. Ann Intern Med. 2002;136:201–9.
Fung TT, Schulze M, Manson JE, Willett WC, Hu FB. Dietar patterns, meat intake, and the risk of type 2 dieabetes in women. Arch Intern Med. 2004;164:2235–40.
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63.
Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–85.
Claassen S, du Toit E, Kaba M, Moodley C, Zar HJ, Nicol MP. A comparison of the efficiency of five different commercial DNA extraction kits for extraction of DNA from faecal samples. J Microbiol Methods. 2013;94:103–10.
Ferrand J, Patron K, Legrand-Frossi C, Frippiat JP, Merlin C, Alauzet C, et al. Comparison of seven methods for extraction of bacterial DNA from fecal and cecal samples of mice. J Microbiol Methods. 2014;105:180–5.
Funding
We thank all of the participating schools, children, and parents. This work was supported by intramural grants from the Korea National Institute of Health and Korea Center for Disease Control and Prevention (project numbers: 2016-NG64002-00 and 2012-E64001-00).
Availability of data and materials
The data supporting this study will be shared through corresponding author for any interested party with appropriate rationale.
Authors’ contributions
HBJ and HJL: conceived and designed the study; HBJ: analyzed and drafted the article; HJL: revised the article; MKC and JHK: provided phenotypic information; HBJ, HJL and SIP: interpreted the data. All authors read and approved the final manuscript.
Competing interests
The authors declare no conflict of interest.
Consent to publication
Not applicable.
Ethics approval and consent to participate
The study was approved by the Institutional Review Boards of Seoul-Paik Hospital (IIT-2012-092), Inje University, and the Korea Center for Disease Control (KCDC) and Prevention (2012-04EXP-06-R). Informed consent was obtained from the subject’s parents.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
Food grouping used in the dietary pattern analysis. Table S2. Nutrient intakes of subjects across dietary pattern groups. (DOCX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jang, H.B., Choi, MK., Kang, J.H. et al. Association of dietary patterns with the fecal microbiota in Korean adolescents. BMC Nutr 3, 20 (2017). https://doi.org/10.1186/s40795-016-0125-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40795-016-0125-z