
Abstract
Loktanella hongkongensis UST950701-009PT is a Gram-negative, non-motile and rod-shaped bacterium isolated from a marine biofilm in the subtropical seawater of Hong Kong. When growing as a monospecies biofilm on polystyrene surfaces, this bacterium is able to induce larval settlement and metamorphosis of a ubiquitous polychaete tubeworm Hydroides elegans. The inductive cues are low-molecular weight compounds bound to the exopolymeric matrix of the bacterial cells. In the present study we describe the features of L. hongkongensis strain DSM 17492T together with its genome sequence and annotation and novel aspects of its phenotype. The 3,198,444 bp long genome sequence encodes 3104 protein-coding genes and 57 RNA genes. The two unambiguously identified extrachromosomal replicons contain replication modules of the RepB and the Rhodobacteraceae-specific DnaA-like type, respectively.
Similar content being viewed by others


Introduction
Loktanella hongkongensis UST950701-00PT (= DSM 17492T = NRRL B-41039T = JCM 12479T) was isolated from a biofilm grown naturally on a glass coupon that had been submerged in the coastal seawater of Hong Kong for 7 days in July 1995 [1]. In the marine environment, bacteria in biofilms mediate the settlement and metamorphosis of the planktonic larvae of many benthic invertebrates. The cells of UST950701-00PT, when attached as a biofilm, were able to induce settlement and metamorphosis of the polychaete Hydroides elegans [2]. The chemical cues mediating the larval response were found to be low-molecular weight compounds associated with the exopolymeric matrix of the bacterial cells [3–5].
In this study we analyzed the genome sequence of L. hongkongensis DSM 17492T. We present a description of the genome sequencing, an annotation and a summary classification together with a set of features for strain, including novel aspects of its phenotype.
Organism information
Classification and features
Figure 1 shows the phylogenetic neighborhood of L. hongkongensis DSM 17492T in a 16S rRNA gene based tree. The sequence of the single 16S rRNA gene copy in the genome does not differ from the previously published 16S rRNA gene sequence (AY600300).

Phylogenetic tree highlighting the position of L. hongkongensis relative to the type strains of the other species within the genus Loktanella [6, 13]. The tree was inferred from 1353 aligned characters of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion as previously described [14]. Rooting was done initially using the midpoint method and then checked for its agreement with the current classification (Table 1). The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 350 ML bootstrap replicates (left) and from 1000 maximum-parsimony bootstrap replicates (right) if larger than 60 % [6]. Lineages with type strain genome sequencing projects registered in GOLD [7] are labeled with one asterisk, those also listed as ‘Complete and Published’ with two asterisks
The single genomic 16S rRNA gene sequence of L. hong-kongensis DSM 17492T was compared with the Greengenes database for determining the weighted relative frequencies of taxa and (truncated) keywords as previously described [6]. The most frequently occurring genera were Loktanella (46.2 %), Ketogulonicigenium (14.9 %), Methylarcula (10.3 %), Silicibacter (10.0 %) and Ruegeria (8.5 %) (65 hits in total). Regarding the five hits to sequences from representatives of the species, the average identity within high-scoring segment pairs was 99.6 %, whereas the average coverage by HSPs was 98.0 %. Regarding the 13 hits to sequences from other representatives of the genus, the average identity within HSPs was 95.6 %, whereas the average coverage by HSPs was 97.6 %. Among all other species, the one yielding the highest score was Loktanella vestfoldensis (NR_029021), which corresponded to an identity of 95.8 % and a HSP coverage of 99.4 %. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification). The highest-scoring environmental sequence was FJ869048 (Greengenes short name ‘ Roseobacter isolates Chesapeake Bay water 2 m depth isolate CB1079Rhodobacterales str. CB1079’), which showed an identity of 99.2 % and an HSP coverage of 99.9 %. The most frequently occurring keywords within the labels of all environmental samples which yielded hits were ‘lake’ (8.6 %), ‘tin’ (7.1 %), ‘qinghai’ (6.4 %), ‘microbi’ (3.2 %) and ‘sea’ (3.1 %) (185 hits in total). The most frequently occurring keywords within the labels of those environmental samples which yielded hits of a higher score than the highest scoring species were ‘sea’ (15.4 %), ‘water’ (7.7 %), ‘bloom, chl, concentr, contrast, diatom, dure, filter, non-bloom, spring, station, success, surfac, yel’ (5.1 %) and ‘bai, chesapeak, depth, roseobact’ (2.6 %) (3 hits in total). These keywords fit well to the isolation site of strain UST950107-009PT.

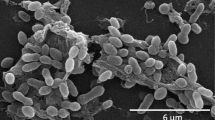
Phase-contrast micrograph of strain L. hongkongensis DSM 17492T
L. hongkongensis UST950107-009PT is Gram-negative and non-spore forming (Table 1). Cells are short rods and non-motile (Fig. 2). When grown on Marine Agar 2216 (Difco) at 30 ˚C in the absence of light, colonies are pink in color, convex with entire margin, and have smooth and shiny surface; brown diffusible pigment is produced. However, whitish colonies would emerge from every culture upon aging (3 days or beyond). The colonies of the white morphovar, with otherwise identical morphological properties, can be maintained as separate cultures (UST950701-009 W) without turning pink. L. hongkongensis UST950107-009PT cannot grow on nutrient agar or trypticase-soy agar (both from Oxoid).
The growth of L. hongkongensis UST950701-009PT is strictly aerobic and requires at least 2 % NaCl (up to 14 %). The ranges of temperature and pH where its growth can occur are 8–44 ˚C and 5.0–10.0, respectively. L. hongkongensis UST950107-009PT can utilize a wide range of mono-, di-, tri- and polysaccharides, and sugar alcohols. Citrate is not utilized. Catalase, oxidase and beta-galactosidase activities are positive whereas arginine dihydrolase, lysine decarboxylase, ornithine decarboxylase, urease, tryptophane deaminase and gelatinase are negative. L. hongkongensis UST950701-009PT does not produce bacteriochlorophyll a, indole, acetoin or H2S. It cannot hydrolysis casein or tween 80. Streptomycin, penicillin, chloramphenicol, amplicilin and tetracycline can inhibit the growth of L. hongkongensis UST950107-009PT but kanamycin cannot (all data from [1]).
The utilization of carbon compounds by L. hongkongensis DSM 17492T grown at 28 °C was also determined for this study using Generation-III microplates in an OmniLog phenotyping device (BIOLOG Inc., Hayward, CA, USA). The microplates were inoculated at 28 °C with dye IF-A and a cell suspension at a cell density of 95–96 % turbidity. Further additives were vitamin, micronutrient and sea-salt solutions [14]. The plates were sealed with parafilm to avoid a loss of fluid. The exported measurement data were further analyzed with the opm package for R [15, 16], using its functionality for statistically estimating parameters from the respiration curves such as the maximum height, and automatically translating these values into negative, ambiguous, and positive reactions. The reactions were recorded in three individual biological replicates. Positive results were received for the following substrates: positive control, pH 6, 1 % NaCl, 4 % NaCl, 8 % NaCl, D-galactose, 3-O-methyl-D-glucose, D-fucose, L-fucose, L-rhamnose, inosine, 1 % sodium lactate, myo-inositol, rifamycin SV, L-aspartic acid, L-glutamic acid, L-histidine, L-serine, D-glucuronic acid, glucuronamide, quinic acid, L-lactic acid, citric acid, α-keto-glutaric acid, D-malic acid, L-malic acid, nalidixic acid, acetic acid and sodium formate.
According to Generation-III plates the strain is negative for dextrin, D-maltose, D-trehalose, D-cellobiose, β-gentiobiose, sucrose, D-turanose, stachyose, pH 5, D-raffinose, α-D-lactose, D-melibiose, β-methyl-D-galactoside, D-salicin, N-acetyl-D-glucosamine, N-acetyl-β-D-mannosamine, N-acetyl-D-galactosamine, N-acetyl-neuraminic acid, D-glucose, D-mannose, D-fructose, fusidic acid, D-serine, D-sorbitol, D-mannitol, D-arabitol, glycerol, D-glucose-6-phosphate, D-fructose-6-phosphate, D-aspartic acid, D-serine, troleandomycin, minocycline, gelatin, glycyl-L-proline, L-alanine, L-arginine, L-pyroglutamic acid, lincomycin, guanidine hydrochloride, niaproof, pectin, D-galacturonic acid, L-galactonic acid-γ-lactone, D-gluconic acid, mucic acid, D-saccharic acid, vancomycin, tetrazolium violet, tetrazolium blue, p-hydroxyphenylacetic acid, methyl pyruvate, D-lactic acid methyl ester, bromo-succinic acid, lithium chloride, potassium tellurite, tween 40, γ-amino-n-butyric acid, α-hydroxy-butyric acid, β-hydroxybutyric acid, α-keto-butyric acid, acetoacetic acid, propionic acid, aztreonam, butyric acid and sodium bromate and the negative control.
The phenotype of the strain was described as well as the assimilation of a wide range of sugars was tested by Lau et al. [1] with the API50CH system, which is based on the detection of biochemical reactions. Using the API50CH system positive reactions were found for more than 20 carbon sources. None of these results could be confirmed by the OmniLog measurement. L. hongkongensis was positive for only five sugars, as well as for a number of carboxylic acids (e.g. malate and citrate) and amino acids. This observation agrees with the finding of Van Trappen et al. [6], who determined the phenotype of three Loktanella strains using API20NE, except for the difference that no positive reaction was found for the carbon sources given in [6]. Positive reactions found in the OmniLog measurements but not in growth experiments might be due to the higher sensitivity of the former [17].
Chemotaxonomy
The predominant fatty acids of L. hongkongensis UST950107-009PT are C18:1 Ω7C (84.5 %), C16:0 (5.8 %), C18:0 (3.5 %), C10:0 3-OH (2.0 %) and C12:0 3-OH (1.9 %), making up to 97.7 % of the total [1]. The remaining fatty acids are C12:1 3-OH, C17:O, C18:1 Ω7C 11-methyl, summed feature 3 (comprising C16:1 Ω7c and C15 iso 2-OH), and an unknown peak with an expected chain length equivalent to 11.799.
Genome sequencing and annotation
Genome project history
The genome was sequenced within the project “Ecology, Physiology and Molecular Biology of the Roseobacter clade: Towards a Systems Biology Understanding of a Globally Important Clade of Marine Bacteria”. The strain was chosen for genome sequencing according to the Genomic Encyclopedia of Bacteria and Archaea criteria [29]. For the same reason it was previously also chosen as part of the “Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes project” [51, 52], a follow-up of the GEBA project [30], which aims at increasing the sequencing coverage of key reference microbial genomes. Two draft sequences were produced independently from the same source of DNA and finally joined. According project information can found in the Genomes OnLine Database [31]. The Whole Genome Shotgun sequence is deposited in Genbank and the Integrated Microbial Genomes database (IMG) [32]. A summary of the project information is shown in Table 2.
Growth conditions and genomic DNA preparation
A culture of strain DSM 17492T was grown aerobically in DSMZ medium 514 [33] at 28 °C. Genomic DNA was isolated using Jetflex Genomic DNA Purification Kit (GENOMED 600100) following the standard protocol provided by the manufacturer but modified by an incubation time of 60 min, incubation on ice over night on a shaker, the use of additional 50 μl proteinase K, and the addition of 100 μl protein precipitation buffer. DNA is available from the DSMZ through the DNA Network [34].
Genome sequencing and assembly
The genome was sequenced using a combination of two libraries (Table 2). Illumina sequencing was performed on a GA IIx platform with 150 cycles. The paired-end library contained inserts of an average of 500 bp in length. The first run on Illumina GAII platform delivered 1.0 million reads. A second Illumina run was performed on a Miseq platform to gain a higher sequencing depth. To achieve longer reads, the library was sequenced in one direction for 300 cycles, providing another 2.1 million reads. After error correction and clipping by fastq-mcf [35] and quake [36], the data was assembled using velvet [37]. A total of 2,403,257 reads with a mean length of 126 bp passed the filter step and were assembled in 54 contigs. To gain information on the contig arrangement an additional 454 run was performed. The paired-end jumping library of 3 kb insert size was sequenced on a 1/8 lane. Pyrosequencing resulted in 158,608 reads with an average length of 337 bp. A total of 41 scaffolds was obtained from Newbler assembler (Roche Diagnostics).
Both draft assemblies (Illumina and 454 sequences) were fractionated into artificial Sanger reads of 1000 nt in length plus 75 bp overlap on each site. These artificial reads served as an input for the phred/phrap/consed package [38]. By manual editing the number of contigs was reduced to 13. Using minimus2 [39], the resulting sequence was mapped to an existing permanent draft version of the genome published on IMG-ER by the DOE Joint Genome Institute, which was sequenced as described earlier [53]. The source DNA of both samples was obtained from the same origin DSM 17492T. The combined sequences provided a 132 × coverage of the genome.
Genome annotation
Genes were identified using Prodigal [40] as part of the JGI genome annotation pipeline. The predicted CDSs were translated and used to search the National Center for Biotechnology Information nonredundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Identification of RNA genes were carried out by using HMMER 3.0rc1 [41] (rRNAs) and tRNAscan-SE 1.23 [42] (tRNAs). Other non-coding genes were predicted using INFERNAL 1.0.2 [43] Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review platform [44] CRISPR elements were detected using CRT [45] and PILER-CR [46].
Genome properties
The genome statistics are provided in Table 3 and Fig. 3. The genome of strain DSM 17492T has a total length of 3,198,444 bp and a G + C content of 68.3 %. Of the 3161 genes predicted, 3104 were identified protein-coding genes, and 57 RNAs. The majority of the protein-coding genes were assigned a putative function (83.9 %) while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical map of the largest scaffold. From bottom to the top: Genes on forward strand (colored by COG categories), Genes on reverse strand (colored by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content (black), GC skew (purple/olive)
Insights from the genome sequence
Genome sequencing of L. hongkongensis DSM 17492T reveals the presence of two plasmids with sizes of about 85 kb and 103 kb (Table 5). These plasmids contain characteristic replication modules of the RepB and DnaA-like type comprising a replicase as well as the parAB partitioning operon. The respective replicases that mediate the initiation of replication are designated according to the established plasmid classification scheme [47]. The different numbering of the replicases (RepB-I, DnaA-like I) corresponds to specific plasmid compatibility groups that are required for a stable coexistence of the replicons within the same cell. Type-IV secretion systems for conjugative plasmid transfer [48, 49] and postsegregational killing systems, consisting of a typical operon with two small genes encoding a stable toxin and an unstable antitoxin [50], are missing on both plasmids. The presence of a RepA-I plasmid replicase (lokhon_02202) in close proximity to a complete rRNA operon on the chromosomal 1,0 MB contig 684.8 is conspicuous. The parAB partitioning operon is located 15 genes downstream of repA-I indicating that the replication module has been subjected to several recombination events with the chromosome and is probably not functional any more. However, genome finishing would be required to document the presence of a single chromosomal replicon in L. hongkongensis DSM 17492T.
Conclusion
The marine Roseobacter group is widely distributed in the marine environment. In this study we analyzed the genome sequence of L. hongkongensis UST950701-009PT, which was isolated from a marine biofilm, and summarized known and newly revealed aspects of its phenotype. Genome analysis of this type strain demonstrated at least two extrachromosomal elements with replication systems specific or at least characteristic for the family Rhodobacteraceae .
Abbreviations
- DOE:
-
Department of Energy
- GEBA:
-
Genomic encyclopedia of Bacteria and Archaea
- HSP:
-
High-scoring segment pair
- IMG:
-
Integrated microbial genomes
- JGI:
-
Joint Genome Institute
- KMG:
-
1000 microbial genomes
- WGS:
-
Whole genome shotgun
References
Lau SCK, Tsoi MMY, Li X, Plakhotnikova I, Wu M, Wong PK, et al. Loktanella hongkongensis sp. nov., a novel member of the α-Proteobacteria originating from marine biofilms in Hong Kong waters. Int J Syst Evol Microbiol. 2004;54:2281–4.
Lau SCK, Qian PY. Phlorotannins and related compounds as larval settlement inhibitors of the tube-building polychaete Hydroides elegans. Mar Ecol Prog Ser. 1997;159:219–27.
Lau SCK, Qian PY. Larval settlement and metamorphosis in the serpulid polychaete Hydroides elegans (Haswell) in response to cues from bacterial films. Mar Biol. 2001;138:321–8.
Harder T, Lau SCK, Dahms HU, Qian PY. Isolation of bacterial metabolites as natural inducers for larval settlement in the marine polychaete Hydroides elegans (Haswell). J Chem Ecol. 2002;28:2029–43.
Lau SCK, Harder T, Qian PY. Larval settlement induction in the serpulid polychaete Hydroides elegans (Haswell): role of bacterial extracellular polymers. Biofouling. 2003;19:197–204.
Van Trappen S, Mergaert J, Swings J. Loktanella salsilacus gen. nov., sp. nov., Loktanella fryxellensis sp. nov. and Loktanella vestfoldensis sp. nov., new members of the Roseobacter group, isolated from microbial mats in Antarctic lakes. Int J Syst Evol Microbiol. 2004;54:1263–9.
Ivanova EP, Zhukova NV, Lysenko AM, Gorshkova NM, Sergeev AF, Mikhailov VV, et al. Loktanella agnita sp. nov. and Loktanella rosea sp. nov., from the north-west Pacific Ocean. Int J Syst Evol Microbiol. 2005;55:2203–7.
Hosoya S, Yokota A. Loktanella atrilutea sp. nov., isolated from seawater in Japan. Int J Syst Evol Microbiol. 2007;57:1966–9.
Tsubouchi T, Shimane Y, Mori Y, Miyazaki M, Tame A, Uematsu K, et al. Loktanella cinnabarina sp. nov., isolated from a deep subseafloor sediment, and emended description of the genus Loktanella. Int J Syst Evol Microbiol. 2013;63:1390–5.
Weon HY, Kim BY, Yoo SH, Kim JS, Kwon SW, Go SJ, et al. Loktanella koreensis sp. nov., isolated from sea sand in Korea. Int J Syst Evol Microbiol. 2006;56:2199–202.
Yoon JH, Jung YT, Lee JS. Loktanella litorea sp. nov., isolated from seawater. Int J Syst Evol Microbiol. 2013;63:175–80.
Yoon JH, Kang SJ, Lee SY, Oh TK. Loktanella maricola sp. nov., isolated from seawater of the East Sea in Korea. Int J Syst Evol Microbiol. 2007;57:1799–802.
Moon YG, Seo SH, Lee SD, Heo MS. Loktanella pyoseonensis sp. nov., isolated from beach sand, and emended description of the genus Loktanella. Int J Syst Evol Microbiol. 2010;60:785–9.
Buddruhs N, Pradella S, Göker M, Päuker O, Michael V, Pukall R, et al. Molecular and phenotypic analyses reveal the non-identity of the Phaeobacter gallaeciensis type strain deposits CIP 105210T and DSM 17395. Int J Syst Evol Microbiol. 2013;63:4340–9.
Vaas LAI, Sikorski J, Michael V, Göker M, Klenk HP. Visualization and curve-parameter estimation strategies for efficient exploration of phenotype microarray kinetics. PLoS One. 2012;7:e34846.
Vaas LAI, Sikorski J, Hofner B, Fiebig A, Buddruhs N, Klenk HP, et al. opm: an R package for analysing OmniLog phenotype microarray data. Bioinformatics. 2013;29:1823–4.
Vaas LAI, Marheine M, Sikorski J, Göker M, Schumacher M. Impacts of pr-10a overexpression at the molecular and the phenotypic level. Int J Mol Sci. 2013;14:15141–66.
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–7.
Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, et al. Clarifying concepts and terms in biodiversity informatics. PLoS Biol. 2013;9:e1001088.
Woese CR, Kandler O, Weelis ML. Towards a natural system of organisms. Proposal for the domains Archaea and Bacteria. Proc Natl Acad Sci U S A. 1990;87:4576–9.
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl nov. In: Brenner DJ, Krieg NR, Stanley JT, Garrity GM, editors. Bergey’s manual of systematic bacteriology. Second edition, Volume 2 (The Proteobacteria part B The Gammaproteobacteria). New York: Springer; 2005. p. 1.
Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. nov. In: Brenner DJ, Krieg NR, Stanley JT, Garrity GM, editors. Bergey’s manual of sytematic bacteriology, Second edition, Volume 2 (The Proteobacteria part C The Alpha-, Beta-, Delta-, and Epsilonproteobacteria). New York: Springer; 2005. p. 1.
Editor L. Validation list No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2006;56:1–6.
Garrity GM, Bell JA, Lilburn T. Order III. Rhodobacterales ord. nov. In: Brenner DJ, Krieg NR, Stanley JT, Garrity GM, editors. Bergey’s manual of sytematic bacteriology, Second edition, Volume 2 (The Proteobacteria part C The Alpha-, Beta-, Delta-, and Epsilonproteobacteria). New York: Springer; 2005. p. 161.
Garrity GM, Bell JA, Lilburn T. Family I. Rhodobacteraceae fam. nov. In: Brenner DJ, Krieg NR, Stanley JT, Garrity GM, editors. Bergey’s manual of sytematic bacteriology, Second edition, Volume 2 (The Proteobacteria part C The Alpha-, Beta-, Delta-, and Epsilonproteobacteria). New York: Springer; 2005. p. 161.
Lee SD. Loktanella tamlensis sp. nov., isolated from seawater. Int J Syst Evol Microbiol. 2012;62:586–90.
BAuA. TRBA 466: classification of bacteria and archaea in risk groups. Berlin: BAuA; 2010. p. 93.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9.
Göker M, Klenk H-P. Phylogeny-driven target selection for large-scale genome-sequencing (and other) projects. Stand Genomic Sci. 2013;8:360–74.
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, et al. A phylogeny-driven Genomic Encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–60.
Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, et al. The Genomes OnLine Database (GOLD) v. 4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40:D571–9.
Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparitive analysis system. Nucleic Acids Res. 2012;40:D115–22.
List of growth media used at the DSMZ. http://www.dsmz.de/.
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, et al. The DNA bank network: the start from a German initiative. Biopreserv Biobank. 2011;9:51–5.
ea-utils: Command-line tools for processing biological sequencing data. http://code.google.com/p/ea-utils/.
Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 2010;11:R116.
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9.
Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202.
Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119.
Finn DR, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–7.
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–8.
Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, et al. CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209.
Edgar RC, Myers EW. PILER: identification and classification of genomic repeats. Bioinformatics. 2005;21:i152–8.
Petersen J, Brinkmann H, Berger M, Brinkhoff T, Päuker O, Pradella S. Origin and evolution of a novel DnaA-like plasmid replication type in Rhodobacterales. Mol Biol Evol. 2011;28:1229–40.
Cascales E, Christie PJ. The versatile bacterial type IV secretion systems. Nat Rev Microbiol. 2003;1:137–49.
Petersen J, Frank O, Göker M, Pradella S. Extrachromosomal, extraordinary and essential-the plasmids of the Roseobacter clade. Appl Microbiol Biotechnol. 2013;97:2805–15.
Zielenkiewicz U, Ceglowski P. Mechanisms of plasmid stable maintenance with special focus on plasmid addiction systems. Acta Biochim Pol. 2001;48:1003–23.
Kyrpides NC, Hugenholtz P, Eisen JA, Woyke T, Göker M, Parker CT, et al. Genomic Encyclopedia of Bacteria and Archaea: sequencing a myriad of type strains. PLoS Biol. 2014;12:e1001920.
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol. 2010;33:175–82.
Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS One. 2012;7:e48837.
Acknowledgements
The authors gratefully acknowledge the help of Iljana Schröder, DSMZ, for growing cells of DSM 17492T and of Evelyne Brambilla, DSMZ, for DNA extraction and quality control. The work was performed under the auspices of the German Research Foundation (DFG) Transregio-SFB 51 Roseobacter grant and as part of the KMG-2 project funded by the U.S. Department of Energy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare they have no competing interests.
Authors’ contributions
SCKL, TR, PYQ and MG wrote the manuscript. AF provided phenotypic data. JP, TR and MG analysed the annotated genome. HPK provided genomic DNA. JH, MH, NNI, VM, TW and NCK sequenced, assembled and annotated the genome. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lau, S.C., Riedel, T., Fiebig, A. et al. Genome sequence of the pink–pigmented marine bacterium Loktanella hongkongensis type strain (UST950701–009PT), a representative of the Roseobacter group. Stand in Genomic Sci 10, 51 (2015). https://doi.org/10.1186/s40793-015-0050-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-015-0050-9