
Abstract
Background
Multiple endocrine neoplasia type 1 (MEN1) is a hereditary tumor syndrome characterized by endocrine tumors with mainly a parathyroid, pancreatic, or anterior pituitary origin. Low-grade fibromyxoid sarcoma (LGFMS) is a rare low-grade soft tissue tumor. There is one known report of a patient with MEN1 complicated by LGFMS, which is very rare. Our report represents the second documented case, providing valuable insights.
Case presentation
A 31-year-old man with the chief complaint of a cough underwent chest contrast-enhanced computed tomography, which revealed a giant hypoabsorptive tumor with a maximum diameter of 23 cm in the left thoracic cavity. The patient was diagnosed with MEN1, as he also possessed a pancreatic neuroendocrine tumor and parathyroid tumor, and because his father had been found to have MEN1. To control hypercalcemia, surgery for the parathyroid tumor was initially performed, followed by surgical resection of the giant thoracic tumor for diagnosis and treatment. Histopathological examination findings of the tumor resulted in a diagnosis of LGFMS.
Conclusion
We experienced a very rare MEN1 with LGFMS. Although endocrine tumors generally occur more frequently in MEN1, non-endocrine tumors such as the present case should also be noted, reinforcing the importance of systemic imaging scrutiny in addition to early diagnosis and long-term follow-up of MEN1 patients.
Similar content being viewed by others


Background
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant inherited tumor syndrome characterized by endocrine tumors, mainly with a parathyroid, pancreatic, or anterior pituitary origin. Furthermore, MEN1 is known to be associated with non-endocrine tumors of the central nervous system, skin, smooth muscle, and other mesenchymal tumors, most of which are benign [1]. A low-grade fibromyxoid sarcoma (LGFMS) is a rare type of low-grade soft tissue tumor, with a mixture of fibrous and myxomatous components that accounts for only 0.6% of all soft tissue sarcomas [2], most often occurring within skeletal muscles and rarely in the thoracic cavity [3]. There is only one known report of MEN1 with LGFMS [4], which is very rare. Our report represents the second documented case, providing valuable insights.
Case presentation
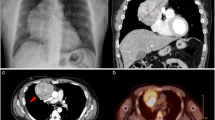
The patient was a 31-year-old man with a chief complaint of cough, while his medical history and comorbidities included diabetes mellitus, hyperparathyroidism, urinary tract stones, chronic kidney disease, pancreatic glucagonoma, and duodenal gastrinoma. Physical examination findings revealed obesity with a BMI of 34 and blood testing indicated mildly elevated inflammatory response, hypercalcemia, mild renal dysfunction, and a high glycohemoglobin level. Chest X-ray showed decreased permeability in the left middle and lower lung fields (Fig. 1A). Chest contrast-enhanced computed tomography (CT) was performed, which revealed a giant hypoabsorptive tumor with a maximum diameter of 23 cm in the left thoracic cavity compressing the left lower lobe and heart (Fig. 2A), while F-18 fluorodeoxyglucose (FDG) positron emission tomography (PET) showed heterogeneous FDG uptake consistent with the same area (SUVmax = 3.3) (Fig. 2B). Based on these imaging findings, the giant tumor in the left thoracic cavity was suspected to be a chronic expanding hematoma, solitary fibrous tumor, or neurogenic tumor. As for other complications, the patient possessed a pancreatic neuroendocrine tumor and parathyroid tumor along with hyperparathyroidism. In addition, a family history showed that his father suffered from MEN1. Although genetic testing could not be performed due to a lack of patient consent, MEN1 gene mutations are not essential for a diagnosis of MEN1 [5], and based on the clinical findings and familial information, a diagnosis of MEN1 in the present case was determined.

Chest X-ray images. A Preoperative image showing decreased permeability in the left middle and lower lung fields. B Postoperative image showing that the decreased transparency in the left lung field has disappeared and the expansion is good

A Chest contrast-enhanced computed tomography image showing giant hypoabsorptive tumor with a maximum diameter of 23 cm in the left thoracic cavity, and compressing the left lower lobe and heart. B F-18 fluorodeoxyglucose (FDG) positron emission tomography image showing heterogeneous FDG uptake (SUVmax = 3.3). Yellow triangles in A indicate the tumor
To control hypercalcemia, resection of the parathyroid tumor was initially performed. Three weeks later, surgical resection of the giant thoracic tumor was performed for diagnosis and treatment, during which stand-by extracorporeal membrane oxygenation (ECMO) was prepared for intraoperative circulatory instability. First, the 5th intercostal space was opened and a huge tumor covered with a white capsule was observed in the thoracic cavity, leading to suspicion of a solitary fibrous or neurogenic tumor. Because of the large size, it was difficult to obtain a clear view, thus the 6th rib was removed and an additional chest opening was made in the 7th intercostal space. The tumor had relatively sparse inflammatory adhesions to the chest wall, while adhesions near the vertebral body on the caudal side of the tumor were strong, indicating a pleural tumor arising from this site. There was no grossly apparent bony invasion, and the pleura was dissected to the vicinity of the vertebral body, and the tumor was removed. Post-extraction left lower lobe re-expansion was good and there was no evidence of reperfusion injury (see Additional file 1).
The excised tumor was 25 cm in its greatest diameter and weighed 3 kg. Macroscopic findings showed a yellowish-white mass surrounded by a capsule, along with internal septal structures and mucus (Fig. 3A, B). Hematoxylin–eosin imaging indicated a spindle-shaped tumor cell proliferation, with a mixture of fibrous and mucus components (Fig. 4A, B). Immunostaining was negative for epithelial, vascular, lymphatic, and neural markers, and positive for alpha-smooth muscle actin (αSMA), a smooth muscle marker. The Ki-67 proliferation index was 1% and positive findings for mucin 4, a marker noted in LGFMS cases and related mesenchymal tumors [6], were noted (Fig. 4C). These findings led to a diagnosis of LGFMS, and the resection margins were negative. Postoperative lung expansion was good (Fig. 1B) and the patient was discharged without complications. At three years after surgery, there was no evidence of disease, and we will continue to follow the patient every 6 months, including any local recurrence.

Macroscopic appearance of tumor. A A yellowish-white mass surrounded by a capsule, B with internal septal structures and mucus

Histological imaging. Hematoxylin–eosin images (× 200) showing spindle-shaped tumor cell proliferation, with a mixture of A fibrous and B mucus components. C Mucin 4 was positive with immunostaining (×200)
Discussion
MEN1 is an inherited tumor syndrome characterized by endocrine tumors, mainly with a parathyroid, pancreatic, or anterior pituitary origin, with a wide variety of tumors known to occur in affected individuals throughout life. However, it is difficult to predict which types are likely to develop because of no correlation between genotype and phenotype [1]. The effects of MEN1 on affected patients depends on complications associated with malignancy, and early detection and treatment lead to improved prognosis. Therefore, an asymptomatic diagnosis based on genetic testing findings and long-term follow-up examinations are very important [7,8,9,10]. However, the awareness of MEN1 in Asia including Japan is lower than in Europe and the United States, and genetic testing for such asymptomatic diagnosis is not widely performed [11]. The present patient was estranged from his father who had a history of MEN1, thus he did not undergo regular examinations and the disease was discovered in association with a large lesion occupying the thoracic cavity. As he had previously been diagnosed with and treated for relatively young onset diabetes and multiple urinary tract stones associated with hypercalcemia, it is highly likely that MEN1 could have been diagnosed and treated earlier had detailed systemic examinations been performed at that time. As for the future, conducting long-term surveillance in accordance with the guidelines [5] is important.
LGFMS is a low-grade soft tissue tumor with a mixture of fibrous and myxomatous components that occurs mainly within the skeletal muscles of the limbs and trunk, and rarely in the thoracic cavity. Therefore, it is difficult to identify LGFMS preoperatively in thoracic tumor cases [3]. Imaging findings such as low to moderate absorption in CT scanning [12] are typical, and helpful for diagnosis. While FDG-PET typically shows relatively faint FDG uptake [13], cases without FDG uptake have been reported [2]. Findings obtained with preoperative imaging in this case were consistent with LGFMS, though a differential diagnosis was not considered and, unexpectedly, LGFMS was demonstrated by the histopathological results. Although MEN1 is known to be associated with a variety of tumors, only a single case of LGFMS with MEN1 has been reported [4]. LGFMS is not known as a common complicating tumor, making preoperative differentiation in the present case difficult. In addition, non-endocrine tumors associated with MEN1 are often benign [1] and intraoperative findings, which showed the mass to be covered by a capsule, presented no evidence of surrounding invasion or extensive adhesions, leading us to strongly suspect that the tumor was benign. Treatment for LGFMS is based on complete resection, which has been shown to be associated with a good long-term prognosis [14,15,16]. On the other hand, there are cases of local recurrence and postoperative irradiation may be considered when the resection margin cannot be secured [17]. The resection margins in this case were negative, and postoperative irradiation was not performed.
Various non-endocrine tumors associated with MEN1 have been reported, including facial angiofibroma [18], lipoma [18], meningioma [19], leiomyoma [20], and breast cancer [21], but only one case of MEN1 with LGFMS has been reported. As for the only reported case of LGFMS with MEN1 [4], that patient had no family history of MEN1, thus it was most likely a sporadic occurrence, indicating that the present is the first reported case of LGFMS with familial MEN1. The authors of that previous case report raised a question regarding the lack of correlation between genotype and phenotype in LGFMS, because their patient had sporadic MEN1 and a novel genetic mutation was detected. While the present patient was diagnosed with familial MEN1 based on complications associated with an endocrine tumor and family history, he did not wish to undergo genetic testing, thus the possible presence of MEN1 gene mutations or mutation patterns was not examined. This is a limitation in our case report. However, although this case was a familial MEN1, the father of our patient had no history of LGFMS, supporting no correlation between genotype and phenotype even in LGFMS cases.
Conclusion
We experienced a very rare MEN1 with LGFMS. Although endocrine tumors generally occur more frequently in MEN1, non-endocrine tumors such as the present case should also be noted, reinforcing the importance of systemic imaging scrutiny in addition to early diagnosis and long-term follow-up of MEN1 patients.
Availability of data and materials
Not applicable.
Abbreviations
- LGFMS:
-
Low-grade fibromyxoid sarcoma
- MEN 1:
-
Multiple endocrine neoplasia type 1
- CT:
-
Computed tomography
- FDG:
-
Fluorodeoxyglucose
- PET:
-
Positron emission tomography
- ECMO:
-
Extracorporeal membrane oxygenation
- αSMA:
-
Alpha-smooth muscle actin
References
Kamilaris CDC, Stratakis CA. Multiple endocrine neoplasia type 1 (MEN1): an update and the significance of early genetic and clinical diagnosis. Front Endocrinol (Lausanne). 2019;10:339.
Maretty-Nielsen K, Baerentzen S, Keller J, Dyrop HB, Safwat A. Low-grade fibromyxoid sarcoma: incidence, treatment strategy of metastases, and clinical significance of the FUS gene. Sarcoma. 2013;2013: 256280.
Jakowski JD, Wakely PE Jr. Primary intrathoracic low-grade fibromyxoid sarcoma. Hum Pathol. 2008;39:623–8.
Radman M, Milicevic T. A novel mutation of the MEN1 gene in a patient with multiple endocrine neoplasia type 1 and recurrent fibromyxoid sarcoma—a case report. BMC Med Genet. 2020;21:190.
Giusti F, Marini F, Brandi ML, et al. Multiple endocrine neoplasia type 1. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews(®) copyright © 1993–2023. Seattle: University of Washington; 1993.
Doyle LA, Möller E, Dal Cin P, Fletcher CD, Mertens F, Hornick JL. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am J Surg Pathol. 2011;35:733–41.
Dean PG, van Heerden JA, Farley DR, Thompson GB, Grant CS, Harmsen WS, et al. Are patients with multiple endocrine neoplasia type I prone to premature death? World J Surg. 2000;24:1437–41.
Doherty GM, Olson JA, Frisella MM, Lairmore TC, Wells SA Jr, Norton JA. Lethality of multiple endocrine neoplasia type I. World J Surg. 1998;22:581–6 (discussion 586–587).
Geerdink EA, Van der Luijt RB, Lips CJ. Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening? Eur J Endocrinol. 2003;149:577–82.
Goudet P, Murat A, Binquet C, Cardot-Bauters C, Costa A, Ruszniewski P, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d’Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg. 2010;34:249–55.
Sakurai A, Suzuki S, Kosugi S, Okamoto T, Uchino S, Miya A, et al. Multiple endocrine neoplasia type 1 in Japan: establishment and analysis of a multicentre database. Clin Endocrinol (Oxf). 2012;76:533–9.
Fujii S, Kawawa Y, Horiguchi S, Kamata N, Kinoshita T, Ogawa T. Low-grade fibromyxoid sarcoma of the small bowel mesentery: computed tomography and magnetic resonance imaging findings. Radiat Med. 2008;26:244–7.
Williams HT, Gossage JR Jr, Allred TJ, Kallab AM, Pancholy A, Anstadt MP. F-18 FDG positron emission tomography imaging of rare soft tissue sarcomas: low-grade fibromyxoid sarcoma and malignant hemangiopericytoma. Clin Nucl Med. 2004;29:581–4.
Folpe AL, Lane KL, Paull G, Weiss SW. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol. 2000;24:1353–60.
Evans HL. Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol. 2011;35:1450–62.
Chamberlain F, Engelmann B, Al-Muderis O, Messiou C, Thway K, Miah A, et al. Low-grade fibromyxoid sarcoma: treatment outcomes and efficacy of chemotherapy. In Vivo. 2020;34:239–45.
Delaney TF, Kepka L, Goldberg SI, Hornicek FJ, Gebhardt MC, Yoon SS, et al. Radiation therapy for control of soft-tissue sarcomas resected with positive margins. Int J Radiat Oncol Biol Phys. 2007;67:1460–9.
Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97:2990–3011.
Asgharian B, Turner ML, Gibril F, Entsuah LK, Serrano J, Jensen RT. Cutaneous tumors in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas: prospective study of frequency and development of criteria with high sensitivity and specificity for MEN1. J Clin Endocrinol Metab. 2004;89:5328–36.
McKeeby JL, Li X, Zhuang Z, Vortmeyer AO, Huang S, Pirner M, et al. Multiple leiomyomas of the esophagus, lung, and uterus in multiple endocrine neoplasia type 1. Am J Pathol. 2001;159:1121–7.
Dreijerink KM, Goudet P, Burgess JR, Valk GD. Breast-cancer predisposition in multiple endocrine neoplasia type 1. N Engl J Med. 2014;371:583–4.
Acknowledgements
We would like to thank Dr. Brian Quinn for the English language editing.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
HI: conceptualization, data duration, writing—original draft, writing—review and editing, visualization. SF: conceptualization, data curation, writing—review and editing. ST: data curation, writing—review and editing, visualization. EM: data curation, writing—review and editing, visualization. YS: writing—review and editing, supervision. All of the authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent for publication of the details of this case was obtained from the patient.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Surgical video.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ishida, H., Funaki, S., Taniguchi, S. et al. Familial multiple endocrine neoplasia type 1 with intrathoracic low-grade fibromyxoid sarcoma. surg case rep 10, 16 (2024). https://doi.org/10.1186/s40792-024-01809-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40792-024-01809-w