
Abstract
Obesity is one of the most serious global health problems, with an incidence that increases yearly and coincides with the development of cancer. Adipose tissue macrophages (ATMs) are particularly important in this context and contribute to linking obesity-related inflammation and tumor progression. However, the functions of ATMs on the progression of obesity-associated cancer remain unclear. In this review, we describe the origins, phenotypes, and functions of ATMs. Subsequently, we summarize the potential mechanisms on the reprogramming of ATMs in the obesity-associated microenvironment, including the direct exchange of dysfunctional metabolites, inordinate cytokines and other signaling mediators, transfer of extracellular vesicle cargo, and variations in the gut microbiota and its metabolites. A better understanding of the properties and functions of ATMs under conditions of obesity will lead to the development of new therapeutic interventions for obesity-related cancer.
Similar content being viewed by others


Background
As an increasingly serious global epidemic, increasing evidence from clinical and preclinical studies suggests that obesity is a preventable risk factor for cancer morbidity and mortality, and results in approximately 20% of adult cancer-associated deaths [1,2,3]. In particular, obesity-induced chronic inflammation plays a promoting role in cancer initiation and progression via its ability to form a permissive microenvironment for neoplastic transformation [4]. For example, in the PC-3 M-Luc-C6 prostate cancer mouse model, high-fat diet-induced obesity activated the macrophage inhibitory cytokine-1 signaling pathway by metabolically increasing adipose lipolysis and free fatty acid release, which further elevated the expression of IL-8 and IL-6 and subsequently advanced prostate cancer progression [5]. Although some studies have focused on obesity, the impact of obesity on cancer initiation and progression remains unclear. Elucidating the core pathological characteristics of adipose tissue may provide crucial insights for understanding the crosstalk between obesity and cancer.
Adipose tissue exists extensively in various anatomical locations, and there are different adipose tissue depots, including subcutaneous and visceral white adipose tissue, brown adipose tissue, inter- and intra-muscular adipose tissue, marrow adipose tissue, and dermal adipose tissue [6]. Adipose tissue is thought to be composed of adipocytes and diverse nonadipocyte cells, including pericytes, endothelial cells, monocytes, macrophages, and adipose-derived stromal/stem cells [7, 8]. Adipose tissue macrophages (ATMs) are a hybrid of macrophages composed of tissue-resident macrophages and myeloid monocytes. Crown-like structures (CLSs) are a representative configuration of macrophages in adipose tissues, in which macrophages surround and phagocytose a dead or dying adipocyte [9]. Notably, the number and density of CLSs are positively correlated with a high body mass index, large adipocyte size, postmenopausal status, and insulin resistance in obese subjects. Compared with lean mice, CLS counts were 5.2-fold greater in obese mice, and CLSs were closer to the tissue center in obese mice [10]. In patients with breast cancer, the pathological elevation of CLSs in mammary adipose tissue was associated with inferior prognosis and aggressive behavior [11]. Therefore, understanding the role of ATMs is essential for obesity-associated cancer research.
Notably, the macrophage phenotype can vary among different cancer types and intratumor regions [12]. First, the reprogramming of macrophages from one phenotype into another partly explains the diversity of macrophages [13]. In response to different environmental cues, ATMs change their transcriptional programs to ‘polarize’ from a homeostatic state into an inflammatory state or vice versa [14, 15]. Subsequently, the crosstalk between macrophages and other cells, such as fibroblasts [16], endothelial cells [17], and adipocytes [18], contributes to the alteration of ATM phenotypes. For example, exosomes derived from adipose-derived stromal/stem cells promote macrophage polarization to an alternatively polarized phenotype, thereby facilitating angiogenesis [19]. In contrast, macrophages, especially classically polarized macrophages, secrete proinflammatory cytokines, such as vascular endothelial growth factor α, which activate endothelial cells and facilitate proliferation and blood vessel development [20, 21]. Therefore, understanding multifunctional macrophages and the intersection between macrophages and other cells is essential for obesity-associated cancer research.
Hence, we review these primary factors mediating the functionality of ATMs, including metabolism, inflammatory factors and extracellular vesicles. We also investigate the effects of these factors on obesity-related cancer progression mainly in tumor types with a high composition of adipose tissue (e.g., breast cancer).
Origin, feature, and function of ATMs
Evidence has demonstrated that macrophages are derived from three different pathways, including embryonic or adult hematopoietic stem cell (HSC) progenitor cells, as well as monocytes [22]. Tissue-resident macrophages originate from embryonic precursors or adult HSCs, with the relative contributions of these populations varying by tissue. Likewise, monocyte-derived macrophages account for a certain proportion of several tissues under inflammatory conditions. Importantly, macrophages in adipose tissues have a mixed origin [23]. In general, most ATMs are thought to be derived from the embryonic origin (also named tissue-resident macrophages) in homeostatic conditions, when there is an extensive accumulation of monocytes that differentiate into macrophages later in obesity-related adipose tissues [24]. In solid tumors, the proportions of monocyte-derived macrophages and tissue-resident macrophages vary by tumor type. In cervical and mammary tumors, recent evidence has shown that tumor-infiltrating macrophages usually originate from monocytes [25]; however, other studies revealed that tissue-resident macrophages account for 50% of the macrophage populations in pancreatic ductal adenocarcinoma [26]. Moreover, the proportion also changes with tumor development. Tissue-resident macrophages dominate in the early stage of tumorigenesis; however, during tumor growth, tissue-resident macrophages were gradually replaced by monocyte-derived macrophages in the mouse and human non-small cell lung carcinoma microenvironment [27]. Hence, several novel approaches, such as single-cell RNA sequencing (scRNA-seq), should be applied to explore the origin and impact of ATMs in human cancers.
Abundant evidence has revealed significant heterogeneities among ATMs, and the features of different ATMs have been described by scRNA-seq [28,29,30]. In visceral adipose tissue, ATMs are further classified into two monocyte and three macrophage subtypes. The two monocyte subsets, Mon1 and Mon2, differ by the expression of Retnla, Fn1 (Mon1), Plac8, and Clec4e (Mon2). One macrophage subtype (Mac1) was characterized by a high expression of Retnla, Cd163, Lyve1, and Cd209f, while the other macrophage subtype (Mac2) mainly overexpresses Cd9 and Nceh1 [28]. In addition, a new type (Mac3) of macrophages was identified, lipid-associated macrophages (LAMs). They are differentiated from monocytes and express Trem2, Lipa, Lpl, Ctsb, Ctsl, Fabp4, Fabp5, Lgals1, Lgals3, Cd9 and Cd36, which are closely related to lipid metabolism and phagocytosis [28]. In subcutaneous fatty tissue, three distinct ATM subsets were identified, lymphocyte antigen 6C (Ly6C+) and two different Ly6C– (CD9+Ly6C– and CD9−Ly6C–) subgroups [31].
Generally, embryonic-derived macrophages in tumor tissues function in tissue remodeling and wound healing [26], while macrophages derived from HSCs exert an immunosuppressive effect [32]. Apart from this, the ATM subtype has very different functions. Among them, LAMs mainly accumulate in tumor-adipose junctional regions and lung metastatic lesions in mammary tumors, which contribute to tumor growth and metastasis [30, 33]. Interestingly, obesity led to a significant increase in lipid metabolism-related genes in ATMs expressing Lpl, Trem2, and Cd9 but a significant decrease in macrophage-specific genes [34, 35]. Thus, LAMs may be a link between obesity and malignant behaviors. Additionally, Ly6C+ ATMs in subcutaneous fatty tissue are mainly distributed outside the CLS and enable it to accelerate adipogenesis, while CD9+ ATMs are located within the CLS, which are rich in lipids and proinflammatory. Thus, diverse ATMs exert multitudinous effects on tumor growth and development.
Polarization of ATMs
Macrophages have great plasticity, as evidenced by diverse polarization patterns, which further form multiple functional phenotypes in response to various environmental cues. Two distinct polarization types for macrophages have been clarified: the classically polarized (M1) macrophage and the alternatively polarized (M2) macrophage [36]. Macrophages are activated toward the M1 phenotype in response to interferon-gamma (IFN-γ) or bacterial moieties such as lipopolysaccharide (LPS). In contrast, macrophages are polarized toward the M2 subtype upon stimulation with IL-4 [37]. The main features of M1 macrophages are the secretion of proinflammatory cytokines, increased cytotoxic activity against bacteria and viruses, and facilitation of antitumor immunity [38]. However, M2 macrophages have an anti-inflammatory effect and are detected in allergies, parasitic infections, tissue remodeling, and tumor development [39]. Additionally, M2 macrophages exhibit the generation of ornithine and polyamines via the arginase pathway, increased generation of scavenging, galactose, mannose receptors, and secretion of anti-inflammatory cytokines (including IL-4 and IL-10) as well as chemokines [chemokine (C-C motif) ligand (CCL)17, CCL22 and CCL24] [12]. M2 macrophages also promote angiogenesis and attract Th2 and T regulatory cells to inevitably have immunosuppressive functions [40].
Crucially, ATMs can be activated into a switching phenotype, which secretes both proinflammatory and anti-inflammatory cytokines and is also found in cancers, such as malignant mesothelioma [41]. These macrophages may be self-reprogramming or undergoing a polarization shift, leading to mutual immunosuppression by the alternative phenotype [42]. Macrophages in intermediate or overlapping states have been detected in vivo under pathophysiological conditions with diverse and temporally changing activating signals. For instance, CD11c+ ATMs derived from obese mice also show an overlapping profile, with the increased transcription of both M1- and M2-associated genes [14]. These observations demonstrate the different polarized phenotypes of macrophages and show that the typical M1 and M2 phenotypes are extremes of macrophage functional states [43].
Regulatory mechanisms of macrophage polarization in adipose tissue and cancer progression
Apart from M1/M2 macrophage phenotypes, evidence demonstrates that there is a more complex scenario dictating macrophage functional states in adipose tissues [37]. Increasing evidence indicates that macrophage polarization has a vital effect on the progression of obesity and obesity-associated cancer [44, 45]. However, the main mechanisms that result in macrophage polarization in adipose tissues and obesity-related cancers are still ambiguous. Thus, the following sections discuss the integrated mechanisms of obesity-associated macrophage polarization in either adipose tissues or both adipose tissues and tumor physiology: metabolic dysregulation; secreted molecules, including chemokines, cytokines, adipokines, and other inflammatory factors; extracellular vesicles (EVs); and gut microbes.
Metabolic dysregulation
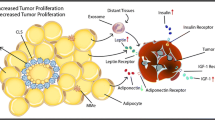
Obesity-induced pathological changes have an important impact on cell metabolism. Macrophage polarization may be stimulated by various metabolic intermediates caused by obesity. With different stimuli, macrophages display different metabolic preferences that impact their differentiation, polarization, mobilization and the establishment of effective antitumor response capabilities. For example, LPS- and/or IFN-γ-stimulated M1 macrophages exhibit elevated glucose uptake and aerobic glycolysis, while IL-4-stimulated M2 macrophages are more prone to oxidative phosphorylation and fatty acid oxidation [46]. However, the metabolic preference for ATMs is not clear. Several major metabolites that stimulate macrophage activation and function in adipose tissues have been identified, as detailed below (Fig. 1a).

Polarization of ATMs is induced by metabolites and inflammatory factors derived from tumor adipose microenvironment (TAME). a Function and polarization of macrophages in TAME are regulated by metabolites, derived from glycolysis, lipid metabolism, and amino acid metabolism. Lactate from aerobic glycolysis promotes the alternative polarization of macrophages. Free fatty acids (FFAs) activate the toll-like receptor (TLR) 4 and increase the expression of proinflammatory genes dependent on nuclear factor kappa B (NF-κB). Branched-chain amino acids (BCAAs), another metabolite from triacylglycerol lipolysis, participate in inducing immune-suppressing macrophages. b Polarization of ATMs is also regulated by inflammatory factors. For example, leptin activated the JAK2/pSTAT3 pathway to function as a proinflammatory factor. Leptin-activated phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway in macrophages to increase the production of lipid droplets and induce the classical polarization of macrophages. In contrast, M2 stimuli such as IL-6, IL-10, and CCL2, trigger alternative macrophage polarization. CCL2 promotes the production of IL-10 and increases the generation of M2-associated markers. c Tumor macroenvironment has a fundamental effect on tumor growth and metastasis. Tim4+ ATMs have a high level of activation of JAK/STAT signaling and promoted the progression and metastasis of ovarian cancer. d Obesity results in the activation of NLRC4 inflammasome in macrophages to increase their infiltration and the generation of IL-1β. In turn, IL-1β contributes to elevated angiogenesis. e Though integrating with CD8+ T cells with PS overexpression, Tim4+ macrophages suppress their proliferation and make them away from tumor targets. Oleate, one of the long-chain unsaturated fatty acids, amplifies the immunosuppressive effects of TAMs by inducing the polarization of macrophages into the CD206+ suppressive subtype by hyper-phosphorylating mTORC2. ATMs adipose tissue macrophages, BCAAs branched-chain amino acids, CCL2 chemokine (C-C motif) ligand 2, IL-1β interleukin-1beta, IL-10 interleukin-10, IL-6 interleukin-6, mTORC2 mTOR complex 2, NF-κB nuclear factor kappa B, NLRC4 NOD-like receptor (NLR) family CARD-containing protein 4, PS phosphatidylserine, TAM tumor-associated macrophage, TLR toll-like receptor, TNF-α Tumor necrosis factor-α, TNFR TNF-α receptor
Glycolysis
Glycolysis is the decomposition of glucose into pyruvate under anaerobic or hypoxic conditions [47]. Importantly, hypoxia is a major characteristic of hypertrophic adipose tissue or the tumor microenvironment (TME). Hypoxia-driven glycolysis enables the recruitment of macrophages and activates their functions via diverse mechanisms. First, lactate is one of the main metabolites during glycolysis and has a crucial effect on ATMs. Malignant cells secrete high amounts of lactate into the extracellular environment due to the significant consumption of glucose [48]. Likewise, lactate derived from endothelial cells was taken up by macrophages in a monocarboxylate transporter 1-dependent manner [49]. Lactate in the microenvironment mainly contributes to the alternative polarization of macrophages and their functions [50]. Mechanistically, lactate binds to histone lysine residues to promote histone lactylation and then induces a switch to an M2-like phenotype in the late phase [51]. In addition, by directly binding to mitochondrial antiviral-signaling protein, lactate inhibited interferon-mediated pathways and alternatively promoted macrophage polarization indirectly [52]. Lactate can also activate G protein-coupled receptor 132 (Gpr132) on macrophages to facilitate macrophage alternative polarization, which further promotes the migration and invasion of breast cancer cells; accordingly, the deletion of Gpr132 reduces M2 macrophages and impairs breast cancer lung metastasis in mice [53]. Thus, high levels of lactate in hypertrophic adipose tissue stimulate protumorous functions and the alternative activation of macrophages. Furthermore, as a proinflammatory metabolite, succinate stabilizes hypoxia-inducible factor 1α (HIF-1α) by suppressing the activity of prolyl hydroxylase to increase glycolysis [54]. Metabolomic analysis of LPS-activated macrophages revealed an increase in aerobic glycolysis and a decrease in tricarboxylic acid cycle (TCA cycle) intermediates, which were directly related to succinate expression [54]. Therefore, succinate may be an important regulator in macrophage polarization by enhancing aerobic glycolysis.
Lipid metabolism
Adipose tissue is composed of large lipid droplets, which are a necessary form of energy storage and regulate lipid metabolism. Macrophages in hypertrophic adipose tissue facilitate lipid storage to form specific macrophage foam cells [55]. These macrophages have elevated inflammatory potential, which in turn exacerbates a positive inflammatory loop in adipose tissue [56]. Under obesity-induced lipid metabolic disorders, ATMs are capable of promoting lysosome biogenesis to accumulate lipid droplets [57]. However, the origin of lipids in ATMs remains unknown. Recent study has revealed that obese adipocytes enable the release of a large amount of lipid-filled exosomes, which provide lipids for macrophages [58]. Then, adipocyte-derived lipid-filled exosomes and relevant factors were sufficient to induce the differentiation of bone marrow precursors into adipose tissue macrophage-like cells in vitro[58]. Hence, the increased infiltration of lipid-overloaded macrophages was discovered in obese adipose tissues. Additionally, Tim4+ Lyve1+ ATMs exhibit a high level of lipid uptake and metabolism, with high lysosomal activity and cholesterol efflux [59]. Furthermore, lipid metabolites such as triacylglycerol and free fatty acids (FFAs) contribute to plastic changes in macrophages via the activation of stress and inflammatory pathways. First, triacylglycerol substrates are internalized by M2-polarized macrophages via scavenger receptor cluster of differentiation 36 (CD36), subsequently undergoing lipolysis by lysosomal acid lipases [60, 61]. Likewise, alternatively polarized macrophages can take up oxidated fatty acids and increase mitochondrial biogenesis [62]. Additionally, FFAs derived from entrapped adipocytes could activate the toll-like receptor (TLR) 4 on the plasma membrane of macrophages, upregulating the expression of proinflammatory genes dependent on nuclear factor kappa B (NF-κB) [63]. Finally, the dominant effectors involved in fatty acid oxidation are transcriptionally activated by peroxisome proliferator-activated receptor-gamma (PPARγ) and its downstream effector PPARγ-coactivator-1β [62, 64]. PPARγ plays an important role in alternative or protumorigenic polarization of macrophages both in vivo and in vitro [62]. Additionally, PPARγ-coactivator-1β is pivotal for macrophage polarization toward the M2 phenotype [64].
Amino acids
Adipose tissue has also emerged as a key regulator of amino acid metabolism [65]. Multiple amino acids derived from adipose tissues stimulate the polarization and function of macrophages. Originally, glutamate was found to be increased in plasma in obese individuals [66]. Glutamine replenishes intermediate catabolites for the TCA cycle and provides nitrogen and carbon for protein, amino acid, nucleotide, and lipid synthesis [67]. Glutamine stimulates the generation of inflammatory cytokines in macrophages [68]. The potential mechanism is glutamate-produced TCA cycle anaplerosis, which has a key impact on macrophage activation [69]. Integrated high-throughput transcriptional-metabolic profiling and analytical studies indicated that M2-polarized macrophages exhibit enhanced glutamine catabolism and activate the hexosamine biosynthesis pathway [69]. Correspondingly, both M2 polarization and the expression of M2-specific markers are inhibited by the deprivation of glutamine or inhibition of N-glycosylation. Nevertheless, glutamine is not required for the activation of classically polarized macrophages induced by LPS [69].
In addition, tryptophan is also one of the major amino acids that stimulate macrophage polarization. The increased production of tryptophan catabolites, including kynurenine, is associated with the pathophysiology of obesity [70]. It is worth noting that tryptophan catabolism in macrophages suppresses the activity of the adaptive immune system [47]. LPS, IFN-γ, and activation of TLRs regulate the expression of indoleamine 2,3-dioxygenase, a rate-limiting enzyme in tryptophan catabolism, and promote flux along the kynurenine pathway. Kynurenine, an agonist of the aryl hydrocarbon receptor, can be released by macrophages and strongly regulates the response of effector T cells and T regulatory cells to both inflammation and antigens [71].
Ultimately, large amounts of branched-chain amino acids (BCAAs) are metabolized by adipose tissue [65], and elevations in the concentrations of circulating BCAAs are significantly associated with obesity [72]. BCAAs, including leucine, valine, and isoleucine, also have an impact on macrophage activation. Specifically, BCAAs decrease the expression of IL-6 in macrophages and then attenuate proinflammatory function [73]. Branched-chain aminotransferase (BCAT) is the enzyme that initiates BCAA catabolism. As the main isoform of BCAT, BCAT1 is highly expressed in macrophages and stimulates oxygen consumption, glycolysis, immune responsive gene 1 (IRG1) levels, and the synthesis of itaconate [74]. Long-term treatment with ERG240, a leucine analog, inhibits the activity of BCAT1 and decreases the production of IL-1β in mice. This finding indicates that ERG240 could elevate BCAA levels and reduce IL-1β by suppressing the IRG1/itaconate axis [74]. Similarly, miRNA-93 reduces the production of IRG1-itaconic acid, thereby inhibiting the M2-like polarization of macrophages [75]. In db/db mice, long-term high BCAA supplementation further increases the levels of branched-chain alpha-keto acid (BCKA, a metabolite of BCAA) and results in macrophage hyperactivation [76]. Elevated BCKAs, not BCAAs, promote the generation of cytokines in primary macrophages [76]. BCKAs derived from glioblastoma can be absorbed and re-animated to BCAAs by tumor-associated macrophages (TAMs) [77]. Then, the phagocytic activity of macrophages decreases. This study indicates that tumor-derived BCKAs may play a direct role in the polarization of TAMs [77]. These studies demonstrate that BCAAs participate in the immunosuppressive function of macrophages, while BCKAs mainly play a role in the formation of the inflammatory microenvironment. Therefore, glutamine, tryptophan, and BCAAs alter macrophage functions in various ways.
Abnormally secreted molecules
Due to the accumulation of metabolites in the cell and the formation of hypoxic areas, the secretion of cytokines and adipocytokines in adipose tissue emerges to become dysfunctional. Under such conditions, the diverse products derived from adipose tissue may also influence the function and polarization of macrophages. In this section, we summarize the alterations of these factors resulting from adipose tissue dysfunction and how they affect macrophage polarization (Fig. 1b).
Cytokines and chemokines
In obese conditions, the hypertrophic expansion of adipose tissue has many common features with the growth of solid tumors. Hypoxia in obese adipose tissue as well as tumorous tissues induces the expression of the transcription factor HIF-1α to stimulate the high expression of proinflammatory cytokines, including IL-6, tumor necrosis factor-α (TNF-α) and CCL2 [78, 79]. IL-6 is involved in various biological activities, including immune regulation, hematopoiesis, and tumorigenesis [80]. In the mammary adipose tissue of humans and mice with obesity, IL-6 can be secreted by macrophages with a proinflammatory phenotype in an NADPH oxidase 2-dependent manner. Furthermore, IL-6 stimulates macrophage polarization toward the M2 phenotype and excessive proliferation via the upregulation of IL-4 receptor α (IL-4Rα) [81]. IL-6 facilitates the stem-like properties of breast cancer cells by interacting with glycoprotein 130 [82]. Second, TNF-α released from tumor and stromal cells acts as a critical inflammatory factor in the obesity-induced microenvironment. A recent study showed that macrophages derived from bone marrow from IL-1β−/− mice underwent M1 polarization, while TNF-α deletion inhibited M1 macrophage polarization [83]. In mice with knockout of TNF-α receptors (TNFR1 and TNFR2) (RKO), adipose tissue in high-fat/sucrose-diet-fed RKO mice exhibited elevated infiltration of macrophages; however, compared with those in wild-type mice, the macrophage phenotype markers in RKO mice were characterized by anti-inflammatory M2 over proinflammatory M1 markers [84]. Thus, TNF-α has a vital effect on the classically polarized phenotype of macrophages to promote inflammatory reactions. The CCL2/C-C chemokine receptor 2 (CCR2) pathway derived from hypertrophic adipocytes is enabled to recruit more macrophages in obese adipose tissue [61]. Subsequently, CCL2 upregulates the mannose receptor (CD206) in stimulating CD11b+ cells, and CD206 is a classical marker of M2-polarized macrophages [85]. Likewise, CCL2 promotes the generation of IL-10 induced by LPS in macrophages, while the blockade of CCL2 leads to an increased level of M1-associated genes and reduces the production of M2-associated markers in human macrophages [86]. Therefore, CCL2 not only recruits macrophages to the microenvironment but also shapes the M2-like polarization of macrophages. Moreover, IL-33 and IL-4 promote the alternative polarization of macrophages through metabolic reprogramming in a transcription factor GATA3-dependent manner [87]. Specifically, IL-4 induces de novo adipogenesis by activating the transcription of sterol regulatory element-binding protein 1. During the process of lipogenesis, a large amount of NADPH is consumed, and the levels of reactive oxygen species are elevated, which acts as a secondary messenger to signal adequate de novo lipogenesis and promote alternative polarization of macrophages [88].
Adipokines
Adipokine disorder emerges in obese individuals, commonly manifesting as upregulated secretion of leptin and resistin but decreased secretion of adiponectin. Leptin is now at the center of the association between obesity and cancer because it is generated in proportion to fat mass [89]. Leptin leads to the elevated secretion of IL-6 and IL-1β, demonstrating that leptin seems to facilitate the generation of proinflammatory factors by macrophages [90]. Likewise, leptin injection induced the activation of macrophages in TNF-α- and chemokine (C-X-C motif) ligand (CXCL) 1-dependent manners [91]. Mechanistically, leptin activates the JAK2/pSTAT3 pathway to upregulate the expression of chemokines to function as a proinflammatory factor [92]. Leptin also indirectly regulates macrophages through mast cell signaling. Recently, a study showed that leptin-deficient mast cells induce a swift from M1 to M2 for macrophages due to impaired cell signaling and changes in the balance between proinflammatory and anti-inflammatory cytokines [93]. From metabolic aspects, leptin-stimulated macrophages exhibit an increased production of lipid droplets through activation of the phosphatidylinositol 3-kinase (PI3K)/mammalian target of the rapamycin (mTOR) pathway [94]. Taken together, these findings demonstrate that leptin can trigger the macrophage M1 phenotype and proinflammatory cytokine production. In contrast, adiponectin is mainly produced by adipose tissue but has an anti-inflammatory impact by suppressing the generation of TNF-α and IL-6 [95]. Furthermore, adiponectin promotes the production of M2 markers but has a reverse effect on the expression of M1 markers in macrophages derived from human monocytes and stromal vascular fraction cells derived from human adipose tissue [96]. These observations demonstrate that adiponectin facilitates anti-inflammatory M2 polarization of macrophages.
Resistin is another predominant adipokine. Resistin has been shown to regulate glucose metabolism and exert proinflammatory effects. Consistently, its secretion level in the circulatory system is also elevated in obese models [97]. Recently, resistin was observed to facilitate the formation of foam cells from macrophages by increasing lipid uptake [98], which was induced by the upregulation of scavenger receptors (SR-1 and CD36) [99]. In addition, resistin upregulates the generation of proinflammatory cytokines, including TNF-α and IL-12, in macrophages through an NF-κB-dependent pathway [100]. Therefore, resistin may provide novel insights into the crosstalk among obesity, macrophages, and cancer. Nicotinamide phosphoribosyltransferase (NAMPT, also known as visfatin), a major enzyme of the NAD salvage pathway, was also discovered to mediate the polarization of macrophages [101]. The extracellular form of eNAMPT promotes the production and secretion of IL-6, TNF-α, and CCL2, thereby inducing the M1 polarity of monocytes [102]. In contrast, the ability of eNAMPT to drive M2 polarization in monocytes has also been demonstrated, as treatment with eNAMPT in monocytes upregulated the expression of CD163 and CD206, markers of M2 macrophages [103].
Insulin/insulin-like growth factor (IGF)-1
Insulin resistance is common in obese patients and is potentially regulated by macrophage functions in adipose tissue and carcinogenesis [104]. Congruously, insulin, IGF-1 and IGF-2 have been discovered to be elevated in overweight individuals [105]. Insulin is only generated and released by pancreatic β cells, while IGF-1 is mainly generated in the liver [106]. In the case of insulin resistance, peripheral macrophages are polarized toward an M2-like anti-inflammatory phenotype, showing upregulation of M2 markers but downregulation of proinflammatory factors [107]. Similarly, macrophages undergoing knockout of the insulin receptor gene are equipped with anti-inflammatory behavior, which prevents diet-induced obesity [108, 109]. IGF-1 receptor (IGF-1R) is lower in M1-polarized macrophages; however, genetic deletion of IGF-1R suppresses M1 responses but increases M2 responses [110]. In addition, insulin receptor substrate 2, a substrate of insulin receptor and IGF-1R, inhibits alternative polarization of macrophages in vivo [111]. Overall, the suppression of insulin signals in peripheral macrophages facilitates the M2-like phenotype.
Extracellular vesicles
EVs are a type of vesicle that can be used for cell-to-cell communication to interact with neighboring or distant target cells [112]. Indeed, EVs contain microRNAs, mRNA, proteins, and DNA, among other molecules that can alter the phenotype and function of target cells. EV-specific markers are potential biomarkers. For example, obesity is observed to accelerate the release of EVs, demonstrating that obese patients exhibit higher levels of plasma EVs than normal healthy-weight patients. These EVs are mainly derived from adipocytes [113]. These EVs may potentially be involved in the obesity-induced alteration of macrophages. First, retinol-binding protein 4 is enriched in obesity-associated exosome-like vesicles, which further stimulates the differentiation of peripheral blood monocytes into activated M1 macrophages with increased secretion of IL-6 and TNF-α [114]. In addition, exosomes derived from the tumor-adipocyte microenvironment incorporate some specific microRNAs, including miRNA-126, miRNA-144, and miRNA-155 [61, 115,116,117], which participate in the polarization of macrophages in different manners. More specifically, the miR-126/miR-126* complex suppresses the mRNA expression of stem cell factor 1α (SCF-1α) directly. SCF-1α acts as an upstream regulator of CCL2 in breast cancer cells. Blocking the SCF-1α-CCL2 axis inhibits the recruitment of CCR2+ macrophages into tumor tissues and further suppresses the metastasis of tumor cells [118]. Additionally, overexpression of miR-144 inhibits the viability of macrophages and suppresses the generation of TNF-α, IL-6, and IL-8 by downregulating TLR2 and p-p65 [119]. Finally, exosomal miRNA-155 from tumor-adipocyte crosstalk can upregulate the release of CCL2 and CCL5 from adipocytes, thereby recruiting macrophages around adipocytes and repolarizing macrophages toward the M2-like phenotype [61]. Taken together, these findings demonstrate that adipocytes in the fatty microenvironment may endow their EV-derived contents with the transitive capacity to convert the environment of macrophages into a protumor niche.
Gut microbiota
Recently, some studies have demonstrated that changes in gut microbes have a vital effect on the development of obesity [120]. Compared with normal controls, the intestinal microbiota of obese patients changed considerably, which is particularly related to the associations between bacterial richness, macrophage phenotype, and cancer [121, 122]. Specifically, the gut microbiota may be dominated by potential proinflammatory bacteria, such as Ruminococcus gnavus or Bacteroides, in the case of obesity [123]. Bacteria-derived LPS is a trigger for the onset of obesity-associated inflammation and infiltration of ATMs. For instance, the elevated infiltration of ATMs as well as obesity is caused in normal diet-fed mice treated with LPS for 1 month [124]. In addition, LPS can activate TLR4 in macrophages to upregulate the secretion of proinflammatory cytokines (including TNF-α, IL-6, and CCL2) [125]. The transmission of LPS to tissue cells may be related to the polarization of macrophages from M2 to M1 in the adipose tissue of high-fat diet-fed mice [126]. Additionally, the products derived from gut microbiota induce low-grade inflammation, which activates tissue-resident macrophages and worsens metabolic diseases, such as diabetes, metabolic syndrome, and cancer [127]. For example, trimethylamine-N-oxide has a positive effect on the reverse cholesterol transport pathway in macrophages and promotes M1 polarization via NOD-like receptor (NLR) family protein (NLRP3) inflammasome activation [128]. Furthermore, G-protein-coupled bile acid receptor 1 (GPBAR-1), the receptor of secondary bile acids, is enabled to modulate energy homeostasis as well as macrophage activation. After being activated by secondary bile acids, GPBAR-1 induces a partial switch from the M1 to M2 subtype for macrophages [129]. Short-chain fatty acids (SCFAs) have also been confirmed to mediate macrophage polarization. In the murine alveolar macrophage MH-S cell line, SCFAs (acetate, butyrate, propionate) decrease the protein or mRNA expression of M2-associated genes in a dose-dependent manner. SCFAs inhibit M2 polarization in MH-S cells, likely by activating G-protein-coupled receptor 43 and inhibiting histone deacetylase [130]. Therefore, the potential links between the microbiome, obesity, and cancer show an emerging and promising direction, and macrophages may be a pivotal mediator.
Role of ATMs in cancer progression
As the dominant subpopulation of tissue-resident myeloid cells, macrophages not only have a crucial effect on the maintenance of tissue homeostasis but also have a response to adipose tissue and malignant growth [131, 132]. Macrophages are a heterogeneous cluster of immune cells regarding their origin and biology in different tumor types. Generally, the high levels of macrophages surrounding the tumor are presumed to be derived from monocyte precursors and are generally associated with a poor prognosis [133]. However, tissue-resident macrophages are also involved in malignant progression.
Tissue-resident macrophages also exhibit substantial heterogeneity in tissue-specific ways [134]. Recent studies have shown that liver-resident macrophages, Kupffer cells, can endocytose malignant cells to suppress tumor growth [135, 136]. Conversely, macrophages in the brain (microglia) and pancreas accelerate tumor proliferation [26, 137]. Mechanistically, tissue-resident macrophages surround cancer cells early during tumorigenesis to activate epithelial-mesenchymal transition, and they also facilitate the function of T regulatory cells to promote tumor immune escape. However, during tumor growth, macrophages are redistributed at the periphery of the TME and are mainly substituted by monocyte-derived macrophages in mouse and human non-small cell lung carcinoma lesions [27]. Hence, the effect of tissue-resident macrophages and monocyte-derived macrophages on cancer development remains complex. Here, we summarize some studies that investigated the effect of ATMs on malignant tumor progression, angiogenesis, and immune escape (Fig. 1c–e).
Proliferation and metastasis
ATMs are a mixed type of macrophages mainly composed of tissue-resident macrophages located in adipose tissue and monocyte-derived macrophages, and they are also associated with poor prognosis and aggressive growth [138, 139]. As a site containing a large amount of visceral adipose tissue, the omentum is one of the most common metastatic sites for gastrointestinal and ovarian cancer [140]. In particular, the omentum is early and robustly disseminated by ovarian cancer cells, which represents a poorer prognosis and higher aggressiveness [141, 142]. Omentum-resident macrophages are mainly of embryonic origin and overexpress CD163 and Tim4. Tim4 is regarded as a phosphatidylserine receptor involved in the endocytosis of apoptotic cells [143] and is a special biomarker of tissue-resident macrophages [134, 144]. CD163+Tim4+ macrophages play a specific role in the invasive progression of metastatic ovarian cancer [139]. The genetic and pharmacological selective depletion of CD163+Tim4+ macrophages in the omentum prevented the progression and metastasis of ovarian cancer [139]. Furthermore, genetic depletion of the autophagic mediator FAK family-interacting protein reduces the infiltration of Tim4+ ATMs and inhibits tumor growth in a T-cell-dependent manner [138].
The TME has fundamental effects on tumor growth and metastasis. The reciprocity between adipocytes and ATMs is regulated under obesity in the TME. Obesity reprograms the metabolic and polarizable phenotype of ATMs. For instance, macrophages remodeled by obesity are predominant ATMs in obese mammary adipose tissue to support tumorigenesis. These macrophages secrete IL-6 to promote stem-like properties via interaction with glycoprotein 130 on triple-negative breast cancer cells [82]. Consistently, weight reduction reverses the roles of obesity on macrophage reprogramming and oncogenesis [82]. On a high-fat diet, ATMs can communicate with white adipocytes via the production of the platelet-derived growth factor (PDGF) ortholog PDGFcc. Indeed, PDGFcc expression is related to poor prognosis in breast cancer [145].
The crosstalk between adipocytes and ATMs in the TME could partly explain the molecular mechanism. Our previous study indicated that CCL2, CCL5, and their receptors linking adipocytes and ATMs promote tumorigenic progression. The elevated expression of CCL2 and CCL5 is mainly secreted by adipocytes, where cancer-derived exosomal miRNA-155 facilitated the production and secretion of CCL2 and CCL5 from adipocytes by modulating the activation of the SOCS6/STAT3 pathway. Consistently, the eliminated macrophages, suppressing the function of STAT3 or chemokines and their receptors, significantly inhibited the tumor proliferation induced by adipocytes [61].
Angiogenesis
Obesity also plays a role in the angiogenesis induced by macrophages. Obesity results in the activation of the NLR family CARD-containing protein 4 inflammasome in macrophages to increase their infiltration and produce IL-1β [146]. In turn, IL-1β contributes to elevated angiogenesis and tumor progression by upregulating angiopoietin-like 4 from adipocytes in an approach dependent on MAP kinase and NF-κB activation [146]. Adipocytes recruit and excite macrophages through the CCL2/IL-1β/CXCL12 pathway during obesity. In turn, activated macrophages increase angiogenesis to promote cancer progression [147].
Immune escape
During the process of immune escape, macrophages with high levels of Tim4 are associated with lower numbers of CD8+ T cells in hydrothorax and ascites from patients with cancer. It has been demonstrated that phosphatidylserine is mainly upregulated in proliferated and cytotoxic antitumor CD8+ T cells, and Tim4+ macrophages can integrate with T cells with overexpressed phosphatidylserine to sequester them away from tumor targets and suppress their proliferation [148]. Furthermore, blocking Tim4 enhances the antitumor effectiveness of programmed cell death-1/programmed cell death-ligand 1 (PD-1/PD-L1) blockers and adoptive T-cell therapy in mouse models [148]. Thus, targeting Tim4+ ATMs through neutralizing antibodies may potentially provide therapeutic benefits and improve the efficacy of immunotherapies in human cancers.
Regarding the metabolic interplay between adipocytes and ATMs in the TME, adipocytes overexpress fatty acid binding protein 4, which promotes lipid transfer from adipocytes to macrophages and activates IL-6/STAT3 signaling through upregulation of the NF-κB/miR-29b pathway, thereby enhancing tumor proliferation and invasiveness [149]. Furthermore, the long-chain unsaturated fatty acids, specific oleate, amplify the immunosuppressive effects of TAMs. Mechanistically, the enriched lipid droplets and oleate induce polarization of macrophages into CD206+ suppressive cells by hyperphosphorylated mTOR complex 2 (mTORC2) at serine 2481 [150].
Role of ATMs in potential cancer therapy
Given the importance of the dynamic interplay between adipocytes and ATMs in cancer progression, potential therapeutic strategies have attracted considerable interest. The main strategies are divided into those that improve the systemic status and those that disrupt intercellular communications in the TME.
One strategy to improve the systemic status is caloric restriction mimetics (CRMs). CRMs are defined as compounds that simulate the physiological and biochemical changes of caloric restriction to relieve age-associated diseases, including cancer and obesity [151, 152]. Likewise, CRMs, including isobacachalcone, 3,4-dimethoxychalcone, and picropodophyllin, enhance anticancer immunosurveillance and amplify the efficiency of chemoimmunotherapy [153,154,155]. Caloric restriction benefits metabolism and promotes healthy aging and lifespan in different species [156, 157]. For example, dietary restriction activated p38 signaling and the downstream translation factor ATF-7, which acts as an immune-metabolic pathway that responds to bacterial and nutrient signals, thereby stimulating the conserved innate immunity pathway to extend the lifespan in Caenorhabditis elegans [158]. Caloric restriction downregulates the number of lymphocytes and monocytes in blood without affecting responses to vaccines or infections in healthy humans [159]. Similarly, fasting in humans reduced monocytes and dendritic cells in blood but increased the number of proinflammatory Ly6Chi monocytes in the bone marrow [160]. The very-low-carbohydrate diet promotes the production of ketone bodies in healthy humans, which enhances the activity of CD4+, CD8+, and regulatory T cells, thereby improving human T-cell immunity [161]. Regarding ATMs, the caloric restriction could increase the level of neuropeptide FF in plasma, which promotes the proliferation of ATM and induces M2 activation [162]. Thus, CRMs as a single and/or combinatory approach are potentially applied as an effective strategy against age- or obesity-promoting tumorigenesis.
Other therapeutic targets involved in ATMs and their interaction with other cells have been previously reviewed [22]. Many agents have been explored to regulate ATMs. To target ATM survival, PLX5622, a small-molecule inhibitor of colony-stimulating factor 1 receptor (CSF1R), has been demonstrated to eliminate macrophages regardless of their origin [163]. Combining various CSF1R inhibitors with CD40 agonists effectively depletes macrophages in the TME to inhibit tumor growth [164]. Furthermore, CSF1R inhibition combined with radiation or chemotherapeutic agents markedly improves T-cell responses in animal models [165, 166]. CSF1-CSF1R blockers enhance the therapeutic effectiveness of diverse immunotherapies, such as PD-1/PD-L1 or cytotoxic T lymphocyte antigen 4 antagonists, CD40 agonists, and adoptive T-cell therapy [164, 167, 168]. Given the activation of JAK/STAT signaling and the pathways for angiogenesis in these ATMs [139], inhibition of JAK/STAT signaling or blockade of angiogenesis may be potential therapeutic targets. Finally, epigenetic regulation has an important effect on the proliferation and polarization of ATMs [169]. For example, the transcription factor c-Myc promotes M2 macrophage differentiation by binding to the acetyltransferase p300 [170]. A selective inhibitor of IIa histone deacetylase polarizes macrophages into an antitumor phenotype that sensitizes the tumor to immune checkpoint blockade and chemotherapy in a T-cell-dependent manner [171]. Histone deacetylase 3 deacetylates histone tails in the macrophage genome, suppressing the expression of many IL-4-regulated genes associated with alternative activation of macrophages [172]. Considering the different effects of bone marrow-derived versus tissue-resident macrophages, the deficiency of tissue-resident macrophages may result in some adverse consequences and should be deliberately implemented.
Conclusions
Plasticity and diversity are well-known characteristics of macrophages. ATMs play a vital role in the progression of obesity-related cancers. Progress has been made in defining the surface phenotype, activating signals, and molecular pathways associated with different forms of ATM activation. Although many studies have demonstrated that ATMs profoundly reprogram their functions in obesity-related cancers, ATM-targeted treatments according to different activation mechanisms have not been thoroughly investigated.
Except for scRNA-seq, some new experimental approaches have been applied in the study of microenvironments, including microfluidics, 3D spheroids, and organs-on-chips. Based on droplet microfluidics, spheroids are filled with a novel hydrogel to promote cell adhesion and aggregation, and this system is composed of cancer cells, fibroblasts, and lymphocytes for dynamic analysis of cellular interactions, proliferation, and therapeutic efficacy and has been used in lymphoma research [173]. Compared with the common Transwell coculture system, 3D spheroids model the interactions among multiple cell types in the TME in a better manner [174]. Microfluidic cell culture technology promoted the production of human organs-on-chips, which has been applied to model cancer cell behavior in organ microenvironments in vitro. This approach facilitates the analysis of the effects on tumorigenesis, tumor progression, and responses to therapy [175].
This review discusses four potential mechanisms for ATM polarization in obesity-related cancers: (1) Obesity-induced metabolic alterations regulate the recruitment, differentiation, and polarization of macrophages in the TME. (2) Secreted molecules in adipose tissues drive tumor progression through alternative pathways of ATM infiltration and polarization. (3) Following the transfer of EV cargoes derived from adipose tissues, ATMs undergo polarization shifts in both phenotype and function, thus being more effective in promoting tumor growth. (4) The secretion of inflammatory cytokines, which stimulate the recruitment and polarization of ATMs, is also mediated by the gut microbiota and its metabolites. However, only a few metabolites involved in the process of driving the polarization of ATMs have been studied. Other tumor-secreted or obesity-induced metabolites have not yet been fully investigated.
Furthermore, many questions remain regarding the mechanisms by which cytokines, adipokines, and hormones regulate macrophage polarization. Because of the complex interaction between cytokines and metabolites, there is still much to be explained regarding the physiological response of macrophages in obesity-mediated TME. Further elucidation of the impact of EVs on macrophages in the adipose TME seems to be a promising field for further investigation to address a variety of obesity-associated tumors. The exact effect of gut microbial metabolites on the pathogenesis of obesity-related cancers also remains to be investigated. Hence, a better understanding of ATM diversity and activation will provide new strategies for the treatment and prevention of obesity-related cancers.
Availability of data and materials
Not applicable.
Abbreviations
- ATMs:
-
Adipose tissue macrophages
- BCAAs:
-
Branched-chain amino acids
- BCAT:
-
Branched-chain aminotransferase
- BCKA:
-
Branched-chain alpha-keto acid
- CCL2:
-
Chemokine (C-C motif) ligand 2
- CCL5:
-
Chemokine (C-C motif) ligand 5
- CCR2:
-
C-C chemokine receptor 2
- CD36:
-
Cluster of differentiation 36
- CLSs:
-
Crown-like structures
- CRMs:
-
Caloric restriction mimetics
- CSF1R:
-
Colony-stimulating factor 1 receptor
- EVs:
-
Extracellular vesicles
- GPBAR-1:
-
G-protein-coupled bile acid receptor 1
- Gpr132:
-
G-protein-coupled receptor 132
- HIF-1α:
-
Hypoxia-inducible factor 1α
- HSC:
-
Hematopoietic stem cell
- IFN-γ:
-
Interferon gamma
- IGF:
-
Insulin-like growth factors
- IGF-1R:
-
IGF-1 receptor
- IRG1:
-
Immune responsive gene 1
- LAMs:
-
Lipid-associated macrophages
- LPS:
-
Lipopolysaccharide
- Ly6C:
-
Lymphocyte antigen 6C
- MCs:
-
Mast cells
- mTOR:
-
Mammalian target of rapamycin
- mTORC2:
-
mTOR complex 2
- NAMPT:
-
Nicotinamide phosphoribosyltransferase
- NF-κB:
-
Nuclear factor kappa B
- NLRP3:
-
NOD-like receptor (NLR) family protein
- PD1:
-
Programmed cell death-1
- PDGF:
-
Platelet-derived growth factor
- PD-L1:
-
Programmed cell death-Ligand 1
- PPARγ:
-
Peroxisome proliferator-activated receptor-gamma
- SCF-1α:
-
Stem cell factor 1α
- SCFA:
-
Short-chain fatty acids
- scRNA-seq:
-
Single-cell RNA sequencing
- TAM:
-
Tumor-associated macrophage
- TAME:
-
Tumor-adipose microenvironment
- TCA cycle:
-
Tricarboxylic acid cycle
- TLR4:
-
Toll-like receptor 4
- TME:
-
Tumor microenvironment
- TNF-α:
-
Tumor necrosis factor-α
- TNFR:
-
TNF-α receptor
References
Gallagher EJ, Leroith D. Obesity and diabetes: the increased risk of cancer and cancer-related mortality. Physiol Rev. 2015;95(3):727–48.
Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM. Obesity and adverse breast cancer risk and outcome: mechanistic insights and strategies for intervention. CA Cancer J Clin. 2017;67(5):378–97.
Park J, Morley TS, Kim M, Clegg DJ, Scherer PE. Obesity and cancer—mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol. 2014;10(8):455–65.
Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and cancer mechanisms: tumor microenvironment and inflammation. J Clin Oncol. 2016;34(35):4270–6.
Huang M, Narita S, Koizumi A, Nara T, Numakura K, Satoh S, et al. Macrophage inhibitory cytokine-1 induced by a high-fat diet promotes prostate cancer progression by stimulating tumor-promoting cytokine production from tumor stromal cells. Cancer Commun (Lond). 2021;41(5):389–403.
Von Bank H, Kirsh C, Simcox J. Aging adipose: depot location dictates age-associated expansion and dysfunction. Ageing Res Rev. 2021;67:101259.
Cox N, Geissmann F. Macrophage ontogeny in the control of adipose tissue biology. Curr Opin Immunol. 2020;62:1–8.
Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol. 2019;15(3):139–54.
Howe LR, Subbaramaiah K, Hudis CA, Dannenberg AJ. Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin Cancer Res. 2013;19(22):6074–83.
Geng J, Zhang X, Prabhu S, Shahoei SH, Nelson ER, Swanson KS, et al. 3D microscopy and deep learning reveal the heterogeneity of crown-like structure microenvironments in intact adipose tissue. Sci Adv. 2021;7(8):eabe2480.
Cha YJ, Kim ES, Koo JS. Tumor-associated macrophages and crown-like structures in adipose tissue in breast cancer. Breast Cancer Res Treat. 2018;170(1):15–25.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96.
Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. 2016;17(1):2–8.
Shaul ME, Bennett G, Strissel KJ, Greenberg AS, Obin MS. Dynamic. M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet–induced obesity in mice. Diabetes. 2010;59(5):1171–81.
Li B, Yang Q, Li Z, Xu Z, Sun S, Wu Q, et al. Expression of monocarboxylate transporter 1 in immunosuppressive macrophages is associated with the poor prognosis in breast cancer. Front Oncol. 2020;10:574787.
Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20(1):131.
Hernandez GE, Iruela-Arispe ML. The many flavors of monocyte/macrophage–endothelial cell interactions. Curr Opin Hematol. 2020;27(3):181–9.
Pajarinen J, Lin T, Gibon E, Kohno Y, Maruyama M, Nathan K, et al. Mesenchymal stem cell-macrophage crosstalk and bone healing. Biomaterials. 2019;196:80–9.
Zhu D, Johnson TK, Wang Y, Thomas M, Huynh K, Yang Q, et al. Macrophage M2 polarization induced by exosomes from adipose-derived stem cells contributes to the exosomal proangiogenic effect on mouse ischemic hindlimb. Stem Cell Res Ther. 2020;11(1):162.
Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. 2019;10:1462.
Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–62.
Denardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369–82.
Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–55.
Weinstock A, Moura Silva H, Moore KJ, Schmidt AM, Fisher EA. Leukocyte heterogeneity in adipose tissue, including in obesity. Circ Res. 2020;126(11):1590–612.
Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology. 2013;2(12):e26968.
Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47(2):323–38.e6.
Casanova-Acebes M, Dalla E, Leader AM, Leberichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;595(7868):578–84.
Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell. 2019;178(3):686–98.e14.
Liu SQ, Gao ZJ, Wu J, Zheng HM, Li B, Sun S, et al. Single-cell and spatially resolved analysis uncovers cell heterogeneity of breast cancer. J Hematol Oncol. 2022;15(1):19.
Liu Z, Gao Z, Li B, Li J, Ou Y, Yu X, et al. Lipid-associated macrophages in the tumor-adipose microenvironment facilitate breast cancer progression. OncoImmunology. 2022;11(1):2085432.
Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U S A. 2018;115(22):E5096-E105.
Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 2016;17(9):2445–59.
Huggins DN, Larue RS, Wang Y, Knutson TP, Xu Y, Williams JW, et al. Characterizing macrophage diversity in metastasis-bearing lungs reveals a lipid-associated macrophage subset. Cancer Res. 2021;81(20):5284–95.
Hildreth AD, Ma F, Wong YY, Sun R, Pellegrini M, O'sullivan TE. Single-cell sequencing of human white adipose tissue identifies new cell states in health and obesity. Nat Immunol. 2021;22(5):639–53.
Sárvári AK, Van Hauwaert EL, Markussen LK, Gammelmark E, Marcher AB, Ebbesen MF, et al. Plasticity of epididymal adipose tissue in response to diet-induced obesity at single-nucleus resolution. Cell Metab. 2021;33(2):437–53.e5.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–86.
Chylikova J, Dvorackova J, Tauber Z, Kamarad V. M1/M2 macrophage polarization in human obese adipose tissue. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2018;162(2):79–82.
Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol. 2019;106(2):345–58.
Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11(10):936–44.
Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604.
Jackaman C, Yeoh TL, Acuil ML, Gardner JK, Nelson DJ. Murine mesothelioma induces locally-proliferating IL-10(+)TNF-alpha(+)CD206(-)CX3CR1(+) M3 macrophages that can be selectively depleted by chemotherapy or immunotherapy. Oncoimmunology. 2016;5(6):e1173299.
Malyshev I, Malyshev Y. Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage "switch" phenotype. Biomed Res Int. 2015;2015:341308.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69.
Wunderlich CM, Ackermann PJ, Ostermann AL, Adams-Quack P, Vogt MC, Tran ML, et al. Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat Commun. 2018;9(1):1646.
Devericks EN, Carson MS, Mccullough LE, Coleman MF, Hursting SD. The obesity-breast cancer link: a multidisciplinary perspective. Cancer Metastasis Rev. 2022;41(3):607–25.
Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. 2017;8:289.
Mehla K, Singh PK. Metabolic regulation of macrophage polarization in cancer. Trends Cancer. 2019;5(12):822–34.
Goodwin J, Neugent ML, Lee SY, Choe JH, Choi H, Jenkins DMR, et al. The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nat Commun. 2017;8:15503.
Zhang J, Muri J, Fitzgerald G, Gorski T, Gianni-Barrera R, Masschelein E, et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab. 2020;31(6):1136–53.e7.
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–63.
Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–80.
Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J, et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell. 2019;178(1):176 – 89.e15.
Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A. 2017;114(3):580–5.
Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, Mcgettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–42.
Matafome P, Santos-Silva D, Sena CM, Seiça R. Common mechanisms of dysfunctional adipose tissue and obesity-related cancers. Diabetes Metab Res Rev. 2013;29(4):285–95.
Van Kruijsdijk RC, Van Der Wall E, Visseren FL. Obesity and cancer: the role of dysfunctional adipose tissue. Cancer Epidemiol Biomark Prev. 2009;18(10):2569–78.
Xu X, Grijalva A, Skowronski A, Van Eijk M, Serlie MJ, Ferrante AW Jr. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18(6):816–30.
Flaherty SE 3rd, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science. 2019;363(6430):989–93.
Magalhaes MS, Smith P, Portman JR, Jackson-Jones LH, Bain CC, Ramachandran P, et al. Role of Tim4 in the regulation of ABCA1(+) adipose tissue macrophages and post-prandial cholesterol levels. Nat Commun. 2021;12(1):4434.
Huang SC, Everts B, Ivanova Y, O'sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846–55.
Li B, Liu S, Yang Q, Li Z, Li J, Wu J, et al. Macrophages in tumor-associated adipose microenvironment accelerate tumor progression. Adv Biol (Weinh). 2022. doi:https://doi.org/10.1002/adbi.202200161:e2200161.
Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–20.
Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol. 2007;27(1):84–91.
Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24.
Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem. 2010;285(15):11348–56.
Maltais-Payette I, Allam-Ndoul B, Perusse L, Vohl MC, Tchernof A. Circulating glutamate level as a potential biomarker for abdominal obesity and metabolic risk. Nutr Metab Cardiovasc Dis. 2019;29(12):1353–60.
Shanware NP, Bray K, Eng CH, Wang F, Follettie M, Myers J, et al. Glutamine deprivation stimulates mTOR-JNK-dependent chemokine secretion. Nat Commun. 2014;5:4900.
Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients. 2018;10(11):1564.
Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419–30.
Chaves Filho AJM, Lima CNC, Vasconcelos SMM, De Lucena DF, Maes M, Macedo D. IDO chronic immune activation and tryptophan metabolic pathway: a potential pathophysiological link between depression and obesity. Prog Neuropsychopharmacol Biol Psychiatry. 2018;80(Pt C):234–49.
Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–43.
Mccormack SE, Shaham O, Mccarthy MA, Deik AA, Wang TJ, Gerszten RE, et al. Circulating branched-chain amino acid concentrations are associated with obesity and future insulin resistance in children and adolescents. Pediatr Obes. 2013;8(1):52–61.
Lee JH, Park E, Jin HJ, Lee Y, Choi SJ, Lee GW, et al. Anti-inflammatory and anti-genotoxic activity of branched chain amino acids (BCAA) in lipopolysaccharide (LPS) stimulated RAW 264.7 macrophages. Food Sci Biotechnol. 2017;26(5):1371–7.
Papathanassiu AE, Ko JH, Imprialou M, Bagnati M, Srivastava PK, Vu HA, et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat Commun. 2017;8:16040.
Ganta VC, Choi MH, Kutateladze A, Fox TE, Farber CR, Annex BH. A microRNA93-interferon regulatory factor-9-immunoresponsive gene-1-itaconic acid pathway modulates M2-like macrophage polarization to revascularize ischemic muscle. Circulation. 2017;135(24):2403–25.
Liu S, Li L, Lou P, Zhao M, Wang Y, Tang M, et al. Elevated branched-chain α-keto acids exacerbate macrophage oxidative stress and chronic inflammatory damage in type 2 diabetes mellitus. Free Radic Biol Med. 2021;175:141–54.
Silva LS, Poschet G, Nonnenmacher Y, Becker HM, Sapcariu S, Gaupel AC, et al. Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. 2017;18(12):2172–85.
Sun K, Tordjman J, Clement K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18(4):470–7.
Korbecki J, Kojder K, Barczak K, Simińska D, Gutowska I, Chlubek D, et al. Hypoxia alters the expression of CC chemokines and CC chemokine receptors in a tumor: a literature review. Int J Mol Sci. 2020;21(16):5647.
Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012;38(7):904–10.
Braune J, Weyer U, Hobusch C, Mauer J, Bruning JC, Bechmann I, et al. IL-6 regulates M2 polarization and local proliferation of adipose tissue macrophages in obesity. J Immunol. 2017;198(7):2927–34.
Tiwari P, Blank A, Cui C, Schoenfelt KQ, Zhou G, Xu Y, et al. Metabolically activated adipose tissue macrophages link obesity to triple-negative breast cancer. J Exp Med. 2019;216(6):1345–58.
Batra R, Suh MK, Carson JS, Dale MA, Meisinger TM, Fitzgerald M, et al. IL-1beta (interleukin-1beta) and TNF-alpha (tumor necrosis factor-alpha) impact abdominal aortic aneurysm formation by differential effects on macrophage polarization. Arterioscler Thromb Vasc Biol. 2018;38(2):457–63.
Pamir N, Mcmillen TS, Kaiyala KJ, Schwartz MW, Leboeuf RC. Receptors for tumor necrosis factor-alpha play a protective role against obesity and alter adipose tissue macrophage status. Endocrinology. 2009;150(9):4124–34.
Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b + peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem. 2009;284(49):34342–54.
Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014;192(8):3858–67.
Faas M, Ipseiz N, Ackermann J, Culemann S, Grüneboom A, Schröder F, et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity. 2021;54(11):2531–46.e5.
Bidault G, Virtue S, Petkevicius K, Jolin HE, Dugourd A, Guénantin AC, et al. SREBP1-induced fatty acid synthesis depletes macrophages antioxidant defences to promote their alternative activation. Nat Metab. 2021;3(9):1150–62.
Friedman J. The long road to leptin. J Clin Invest. 2016;126(12):4727–34.
Naylor C, Petri WA Jr. Leptin regulation of immune responses. Trends Mol Med. 2016;22(2):88–98.
Souza-Almeida G, Palhinha L, Liechocki S, Da Silva Pereira JA, Reis PA, Dib PRB, et al. Peripheral leptin signaling persists in innate immune cells during diet-induced obesity. J Leukoc Biol. 2021;109(6):1131–8.
Kiguchi N, Maeda T, Kobayashi Y, Fukazawa Y, Kishioka S. Leptin enhances CC-chemokine ligand expression in cultured murine macrophage. Biochem Biophys Res Commun. 2009;384(3):311–5.
Zhou Y, Yu X, Chen H, Sjoberg S, Roux J, Zhang L, et al. Leptin deficiency shifts mast cells toward anti-inflammatory actions and protects mice from obesity and diabetes by polarizing M2 macrophages. Cell Metab. 2015;22(6):1045–58.
Maya-Monteiro CM, Almeida PE, D'avila H, Martins AS, Rezende AP, Castro-Faria-Neto H, et al. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J Biol Chem. 2008;283(4):2203–10.
Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6(10):772–83.
Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem. 2010;285(9):6153–60.
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409(6818):307–12.
Park HK, Kwak MK, Kim HJ, Ahima RS. Linking resistin, inflammation, and cardiometabolic diseases. Korean J Intern Med. 2017;32(2):239–47.
Xu W, Yu L, Zhou W, Luo M. Resistin increases lipid accumulation and CD36 expression in human macrophages. Biochem Biophys Res Commun. 2006;351(2):376–82.
Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ. Human resistin stimulates the pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Biochem Biophys Res Commun. 2005;334(4):1092–101.
Travelli C, Colombo G, Mola S, Genazzani AA, Porta C. NAMPT: a pleiotropic modulator of monocytes and macrophages. Pharmacol Res. 2018;135:25–36.
Bermudez B, Dahl TB, Medina I, Groeneweg M, Holm S, Montserrat-De La Paz S, et al. Leukocyte overexpression of intracellular NAMPT attenuates atherosclerosis by regulating PPARγ-dependent monocyte differentiation and function. Arterioscler Thromb Vasc Biol. 2017;37(6):1157–67.
Audrito V, Serra S, Brusa D, Mazzola F, Arruga F, Vaisitti T, et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood. 2015;125(1):111–23.
Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132(6):2169–80.
Nam SY, Lee EJ, Kim KR, Cha BS, Song YD, Lim SK, et al. Effect of obesity on total and free insulin-like growth factor (IGF)-1, and their relationship to IGF-binding protein (BP)-1, IGFBP-2, IGFBP-3, insulin, and growth hormone. Int J Obes Relat Metab Disord. 1997;21(5):355–9.
Giustina A, Berardelli R, Gazzaruso C, Mazziotti G. Insulin and GH-IGF-I axis: endocrine pacer or endocrine disruptor? Acta Diabetol. 2015;52(3):433–43.
Ieronymaki E, Theodorakis EM, Lyroni K, Vergadi E, Lagoudaki E, Al-Qahtani A, et al. Insulin resistance in macrophages alters their metabolism and promotes an M2-like phenotype. J Immunol. 2019;202(6):1786–97.
Senokuchi T, Liang CP, Seimon TA, Han S, Matsumoto M, Banks AS, et al. Forkhead transcription factors (FoxOs) promote apoptosis of insulin-resistant macrophages during cholesterol-induced endoplasmic reticulum stress. Diabetes. 2008;57(11):2967–76.
Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Bluher M, et al. Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet. 2010;6(5):e1000938.
Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV, et al. IGF1 shapes macrophage activation in response to immunometabolic challenge. Cell Rep. 2017;19(2):225–34.
Dasgupta P, Dorsey NJ, Li J, Qi X, Smith EP, Yamaji-Kegan K, et al. The adaptor protein insulin receptor substrate 2 inhibits alternative macrophage activation and allergic lung inflammation. Sci Signal. 2016;9(433):ra63.
Wu Q, Zhang H, Sun S, Wang L, Sun S. Extracellular vesicles and immunogenic stress in cancer. Cell Death Dis. 2021;12(10):894.
Eguchi A, Lazic M, Armando AM, Phillips SA, Katebian R, Maraka S, et al. Circulating adipocyte-derived extracellular vesicles are novel markers of metabolic stress. J Mol Med (Berl). 2016;94(11):1241–53.
Deng ZB, Poliakov A, Hardy RW, Clements R, Liu C, Liu Y, et al. Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes. 2009;58(11):2498–505.
Wu Q, Sun S, Li Z, Yang Q, Li B, Zhu S, et al. Tumour-originated exosomal miR-155 triggers cancer-associated cachexia to promote tumour progression. Mol Cancer. 2018;17(1):155.
Wu Q, Sun S, Li Z, Yang Q, Li B, Zhu S, et al. Breast cancer-released exosomes trigger cancer-associated cachexia to promote tumor progression. Adipocyte. 2019;8(1):31–45.
Wu Q, Li J, Li Z, Sun S, Zhu S, Wang L, et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J Exp Clin Cancer Res. 2019;38(1):223.
Zhang Y, Yang P, Sun T, Li D, Xu X, Rui Y, et al. miR-126 and miR-126* repress recruitment of mesenchymal stem cells and inflammatory monocytes to inhibit breast cancer metastasis. Nat Cell Biol. 2013;15(3):284–94.
Zhou G, Li Y, Ni J, Jiang P, Bao Z. Role and mechanism of miR-144-5p in LPS-induced macrophages. Exp Ther Med. 2020;19(1):241–7.
Woting A, Blaut M. The intestinal microbiota in metabolic disease. Nutrients. 2016;8(4):202.
Zitvogel L, Daillere R, Roberti MP, Routy B, Kroemer G. Anticancer effects of the microbiome and its products. Nat Rev Microbiol. 2017;15(8):465–78.
Wu Q, Yu X, Li J, Sun S, Tu Y. Metabolic regulation in the immune response to cancer. Cancer Commun (Lond). 2021;41(8):661–94.
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–6.
Saad MJ, Santos A, Prada PO. Linking gut microbiota and inflammation to obesity and insulin resistance. Physiology (Bethesda). 2016;31(4):283–93.
Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. 2013;182(2):375–87.
Hersoug LG, Moller P, Loft S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: implications for inflammation and obesity. Obes Rev. 2016;17(4):297–312.
Belizário JE, Faintuch J, Garay-Malpartida M. Gut microbiome dysbiosis and immunometabolism: new frontiers for treatment of metabolic diseases. Mediat Inflamm. 2018;2018:2037838.
Man AWC, Zhou Y, Xia N, Li H. Involvement of gut microbiota, microbial metabolites and interaction with polyphenol in host immunometabolism. Nutrients. 2020;12(10):3054.
Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. 2018;15(2):111–28.
Huang C, Du W, Ni Y, Lan G, Shi G. The effect of short chain fatty acids on M2 macrophages polarization in vitro and in vivo. Clin Exp Immunol. 2021;207(1):53–64.
Nobs SP, Kopf M. Tissue-resident macrophages: guardians of organ homeostasis. Trends Immunol. 2021;42(6):495–507.
Cox N, Crozet L, Holtman IR, Loyher PL, Lazarov T, White JB, et al. Diet-regulated production of PDGFcc by macrophages controls energy storage. Science. 2021;373(6550):eabe9383.
Cox N, Pokrovskii M, Vicario R, Geissmann F. Origins, biology, and diseases of tissue macrophages. Annu Rev Immunol. 2021;39:313–44.
Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–95.
Kimura Y, Inoue A, Hangai S, Saijo S, Negishi H, Nishio J, et al. The innate immune receptor Dectin-2 mediates the phagocytosis of cancer cells by Kupffer cells for the suppression of liver metastasis. Proc Natl Acad Sci U S A. 2016;113(49):14097–102.
Gül N, Babes L, Siegmund K, Korthouwer R, Bögels M, Braster R, et al. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest. 2014;124(2):812–23.
Dumas AA, Pomella N, Rosser G, Guglielmi L, Vinel C, Millner TO, et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. EMBO J. 2020;39(15):e103790.
Xia H, Li S, Li X, Wang W, Bian Y, Wei S, et al. Autophagic adaptation to oxidative stress alters peritoneal residential macrophage survival and ovarian cancer metastasis. JCI Insight. 2020;5(18):e141115.
Etzerodt A, Moulin M, Doktor TK, Delfini M, Mossadegh-Keller N, Bajenoff M, et al. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J Exp Med. 2020;217(4):e20191869.
Meza-Perez S, Randall TD. Immunological functions of the omentum. Trends Immunol. 2017;38(7):526–36.
Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17(11):1498–503.
Sun C, Li X, Guo E, Li N, Zhou B, Lu H, et al. MCP-1/CCR-2 axis in adipocytes and cancer cell respectively facilitates ovarian cancer peritoneal metastasis. Oncogene. 2020;39(8):1681–95.
Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–9.
Shaw TN, Houston SA, Wemyss K, Bridgeman HM, Barbera TA, Zangerle-Murray T, et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J Exp Med. 2018;215(6):1507–18.
Roswall P, Bocci M, Bartoschek M, Li H, Kristiansen G, Jansson S, et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat Med. 2018;24(4):463–73.
Kolb R, Kluz P, Tan ZW, Borcherding N, Bormann N, Vishwakarma A, et al. Obesity-associated inflammation promotes angiogenesis and breast cancer via angiopoietin-like 4. Oncogene. 2019;38(13):2351–63.
Arendt LM, Mccready J, Keller PJ, Baker DD, Naber SP, Seewaldt V, et al. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013;73(19):6080–93.
Chow A, Schad S, Green MD, Hellmann MD, Allaj V, Ceglia N, et al. Tim-4(+) cavity-resident macrophages impair anti-tumor CD8(+) T cell immunity. Cancer Cell. 2021;39(7):973–88.e9.
Hao J, Yan F, Zhang Y, Triplett A, Zhang Y, Schultz DA, et al. Expression of adipocyte/macrophage fatty acid-binding protein in tumor-associated macrophages promotes breast cancer progression. Cancer Res. 2018;78(9):2343–55.
Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C, et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med. 2019. doi:https://doi.org/10.15252/emmm.201910698:e10698.
Madeo F, Carmona-Gutierrez D, Hofer SJ, Kroemer G. Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metab. 2019;29(3):592–610.
Madeo F, Pietrocola F, Eisenberg T, Kroemer G. Caloric restriction mimetics: towards a molecular definition. Nat Rev Drug Discov. 2014;13(10):727–40.
Chen G, Xie W, Nah J, Sauvat A, Liu P, Pietrocola F, et al. 3,4-Dimethoxychalcone induces autophagy through activation of the transcription factors TFE3 and TFEB. EMBO Mol Med. 2019;11:e10469.
Wu Q, Tian AL, Li B, Leduc M, Forveille S, Hamley P, et al. IGF1 receptor inhibition amplifies the effects of cancer drugs by autophagy and immune-dependent mechanisms. J Immunother Cancer. 2021;9(6):e002722.
Wu Q, Tian AL, Durand S, Aprahamian F, Nirmalathasan N, Xie W, et al. Isobacachalcone induces autophagy and improves the outcome of immunogenic chemotherapy. Cell Death Dis. 2020;11(11):1015.
Pak HH, Haws SA, Green CL, Koller M, Lavarias MT, Richardson NE, et al. Fasting drives the metabolic, molecular and geroprotective effects of a calorie-restricted diet in mice. Nat Metab. 2021;3(10):1327–41.
Wu Q, Gao Z-J, Yu X, Wang P. Dietary regulation in health and disease. Signal Transduct Targeted Ther. 2022;7(1):252.
Wu Z, Isik M, Moroz N, Steinbaugh MJ, Zhang P, Blackwell TK. Dietary restriction extends lifespan through metabolic regulation of innate immunity. Cell Metab. 2019;29(5):1192–205.e8.
Lee AH, Dixit VD. Dietary regulation of immunity. Immunity. 2020;53(3):510–23.
Jordan S, Tung N, Casanova-Acebes M, Chang C, Cantoni C, Zhang D, et al. Dietary intake regulates the circulating inflammatory monocyte pool. Cell. 2019;178(5):1102–4.e17.
Hirschberger S, Strauß G, Effinger D, Marstaller X, Ferstl A, Müller MB, et al. Very-low-carbohydrate diet enhances human T-cell immunity through immunometabolic reprogramming. EMBO Mol Med. 2021;13(8):e14323.
Waqas SFH, Hoang AC, Lin YT, Ampem G, Azegrouz H, Balogh L, et al. Neuropeptide FF increases M2 activation and self-renewal of adipose tissue macrophages. J Clin Invest. 2017;127(7):2842–54.
Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. 2019;10(1):3758.
Wiehagen KR, Girgis NM, Yamada DH, Smith AA, Chan SR, Grewal IS, et al. Combination of CD40 agonism and CSF-1R blockade reconditions tumor-associated macrophages and drives potent antitumor immunity. Cancer Immunol Res. 2017;5(12):1109–21.
Seifert L, Werba G, Tiwari S, Giao Ly NN, Nguy S, Alothman S, et al. Radiation therapy induces macrophages to suppress T-Cell responses against pancreatic tumors in mice. Gastroenterology. 2016;150(7):1659–72.e5.
Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73(9):2782–94.
Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci Transl Med. 2018;10(436):eaan3311.
Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74(1):153–61.
Daskalaki MG, Tsatsanis C, Kampranis SC. Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. J Cell Physiol. 2018;233(9):6495–507.
Tikhanovich I, Zhao J, Bridges B, Kumer S, Roberts B, Weinman SA. Arginine methylation regulates c-Myc-dependent transcription by altering promoter recruitment of the acetyltransferase p300. J Biol Chem. 2017;292(32):13333–44.
Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543(7645):428–32.
Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011;25(23):2480–8.
Sabhachandani P, Sarkar S, Mckenney S, Ravi D, Evens AM, Konry T. Microfluidic assembly of hydrogel-based immunogenic tumor spheroids for evaluation of anticancer therapies and biomarker release. J Control Release. 2019;295:21–30.
Zhuang P, Chiang YH, Fernanda MS, He M. Using spheroids as building blocks towards 3d bioprinting of tumor microenvironment. Int J Bioprint. 2021;7(4):444.
Sontheimer-Phelps A, Hassell BA, Ingber DE. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer. 2019;19(2):65–81.
Acknowledgements
We thank professional language editing by American Journal Experts.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities (2042021kf0102, 2042021kf0083). This work was also supported by two National Natural Science Foundation of China (NSFC) Grants (81903166, 82203629) and a Natural Science Foundation of Hubei (2018CKB916). This work was also sponsored by Shanghai Pujiang Program (22PJD054).
Author information
Authors and Affiliations
Contributions
QW and SRS are responsible for collecting and collating documents. QW and BL are responsible for writing this review, SS and JPY are responsible for the revision, and JJL is responsible for editing and submission. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, B., Sun, S., Li, JJ. et al. Adipose tissue macrophages: implications for obesity-associated cancer. Military Med Res 10, 1 (2023). https://doi.org/10.1186/s40779-022-00437-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40779-022-00437-5