
Abstract
Combined Pulmonary Fibrosis and Emphysema (CPFE) refers to the coexistence of upper lobe predominant emphysema with diffuse pulmonary fibrosis, mainly in the lower lobes. Although initially described in patients with Idiopathic Pulmonary Fibrosis (IPF), since then it has been described in other forms of pulmonary fibrosis, most notably collagen tissue disorder associated interstitial lung diseases. High Resolution Computed Tomography (HRCT) has a pivotal role in diagnosis. Recognizing CPFE is not an academic exercise but has significant clinical implications. Thus, it is important for the treating physician to be familiarized with the radiological characteristics that will establish diagnosis. In this review we will discuss the special physiologic and radiological features of CPFE, the challenges in monitoring the course of the disease, the natural history and also the clinical importance of potential complications.
Similar content being viewed by others


Clinical setting
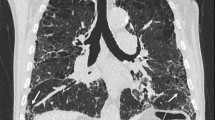
A 74 year old male (current smoker, 50 pack years) presented to our clinic due to progressive dyspnea on exertion and non productive cough. He was diagnosed with COPD about 1 year ago and was treated with tiotropium and indacaterol. Clinical examination revealed the presence of clubbing and velcro like rales with bibasilar symmetric distribution. Pulmonary function tests exhibited an obstructive pattern (FEV1/FVC: 61 %) with a small reduction in TLC (74 % pred) and a disproportionate reduction in DLco (35 % pred). HRCT showed upper lobe paraseptal emphysema with subpleural honeycombing at the lung bases fulfilling the criteria of a definite UIP pattern. A complete clinical and laboratory testing excluded alternative causes and the diagnosis of CPFE in the context of IPF was established (Fig. 1a, b).

1st patient. a HRCT scan at the level of the aortic arch. There is a single layer of subpleural cystic air spaces with no or barely discernible walls that are characteristically bounded by the pleural surface and the interlobular septa. This corresponds to the diagnosis of paraseptal emphysema. b HRCT scan at the level of the dome of the right hemidiaphragm. There is honeycombing (cystic airspaces with well defined walls that are clustered in several layers) with a clear subpleural/peripheral distribution, fulfilling the criteria of definitive UIP pattern
Introduction
CPFE is defined by the co-existence of emphysema and pulmonary fibrosis. Wiggins et al. [1] first described the coexistence of emphysema in the upper lobes and pulmonary fibrosis in the lower lobes on HRCT. The term CPFE was initially introduced by Cottin et al. who described a cohort of 61 patients with both emphysema in the upper zones and diffuse parenchymal lung disease with fibrosis in the lower zones of the lungs on chest HRCT [2]. It is important to note that patients with connective tissue disease, drug-induced interstitial lung disease, pneumoconiosis, hypersensitivity pneumonitis, sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis and eosinophilic pneumonia were excluded [2]. Since then, CPFE has been described not only in the context of IPF but also in the context of other chronic lung fibrotic diseases, such as connective tissue related-interstitial lung diseases [3]. Given the fact that IPF has the worst prognosis in relation to other chronic lung fibrotic diseases, it is important to establish the interstitial lung disease that constitutes the fibrotic component of CPFE. The formation of homogeneous cohorts will allow us to draw comparative conclusions regarding the effect of emphysema on prognosis and therapeutic interventions.
Review
Pathogenesis
The exact pathogenetic mechanisms that lead to the development of CPFE remain elusive. Smoking is believed to play a major role as almost all (98 %) of CPFE patients are current or former smokers [4]. It is intriguing to search for common pathogenetic routes leading to the development of both fibrosis and emphysema. A review on this issue has been published elsewhere [5]. However we must note that several of the studies on pathogenesis, although drawing conclusions on emphysema are based on damage models that result in experimental airspace enlargement that is not equivalent to smoking induced pulmonary emphysema. Also, a lot of the studies explore non-smoking mechanisms. Further research is needed in order to clarify the existence and nature of possible common pathogenetic pathways.
Physiology
Patients with CPFE typically present with preserved or slightly reduced lung volumes in relation to the extent of fibrotic changes in the lungs. FVC and TLC are usually within normal limits or slightly reduced. The ratio FEV1/FVC can be normal or reduced (<70 %) and is lower compared to patients with IPF alone. On the other hand, DLco is disproportionately reduced [2, 6–9]. From a physiological point of view the relatively preserved lung volumes are attributed to the counterbalancing effects of fibrosis and emphysema on lung compliance (fibrosis causes a decrease while emphysema causes an increase in lung compliance). However, both processes cause damage to the alveolar-capillary membrane resulting in a significant decrease of DLco. Pulmonary fibrosis can cause increased traction and support of the small airways preventing expiratory airway collapse [10, 11] and resulting in preservation of FEV1 [12] that is sometimes seen in CPFE patients.
Patients with CPFE are frequently hypoxemic with further desaturation after exercise. Hypercarbia is usually not observed [2, 6]. Patients with fibrosis adopt a rapid/shallow pattern of breathing which increases alveolar ventilation and thus reduces the levels of alveolar and blood pCO2.
In clinical practice the above are important for two reasons regarding diagnosis and follow up (Table 1):
First, the finding of preserved or slightly reduced lung volumes does not rule out the presence of fibrosis. Second, in IPF patients the follow up and response to therapy are based on the measurement of FVC and DLco. However, CPFE patients tend to exhibit a delay in the reduction of FVC and DLco which reduces their utility as surrogate markers for disease progression [7, 13]. In addition, a decline in DLco should be viewed cautiously, as it could be the result of development/progression of pulmonary hypertension which is commonly encountered in CPFE. The annual decrease of the ratio FEV1/FVC in CPFE seems to be significantly higher compared to IPF [7, 9].
In a study by Schimdt et al., mortality in CPFE patient was better predicted by the decline in FEV1, while changes in FVC, DLco and Composite Physiological Index (CPI) were not predictive at 12 months follow-up and only FVC was predictive at 6 months [13]. The prognostic validity of FEV1 increased with increasing severity of emphysema in a dose-dependent fashion. On the other hand, FEV1 had no prognostic role in patients with IPF and no emphysema.
Radiology
HRCT plays a critical diagnostic role, as it actually constitutes the examination that establishes the diagnosis. HRCT reveals the coexistence of emphysema and diffuse lung fibrosis (Fig. 2a, b). Emphysema (centrilobular and/or paraseptal) has upper lobe predominance, while fibrosis is observed mainly in the lower lobes. The radiological fibrotic pattern is usually that of UIP (i.e. bilateral reticular pattern with bibasilar, peripheral distribution, with or without the presence of traction bronchiectasis and/or honeycombing) [2].

2nd patient. a HRCT scan right above the upper level of the aortic arch. There is paraseptal emphysema more prominent in the right lung. b There is an irregular reticular pattern with traction bronchiectasis and bronchiolectasis (possible UIP pattern)
Emphysema can be paraseptal, centrilobular or a combination of both. Paraseptal emphysema seems to be more common in the CPFE population than in patients with COPD. In the study by Cottin et al. [2] it was observed in 93 % of patients and was suggested as a hallmark of CPFE. The increased prevalence of paraseptal emphysema in CPFE was also noted, in another study that included a COPD control group [6]. Furthermore, the presence of paraseptal emphysema has been associated with a higher extent of fibrosis in comparison to centrilobular emphysema [14].
The coexistence of emphysema and fibrosis makes the estimation of the extent of fibrosis really difficult. In the transition zone of the emphysematic to the fibrotic areas it is very tricky to make the appropriate distinction. Brillet et al. [15] identified three HRCT patterns in 61 patients with CPFE: i) progressive transition (38 %) with diffuse emphysema (centrilobular and/or bullous) and zone of transition between bullae and honeycombing, ii) paraseptal emphysema (21 %) with predominant subpleural bullae of enlarging size at the bases and iii) separate processes (23 %) with independent areas of fibrosis and emphysema. Eleven patients (18 %) could not be classified. This difficulty is evident even on a histology level. Inomata et al, performed an autopsy study in 3 groups: CPFE, IPF and emphysema. A specific pathological finding of thick walled cystic lesions (TWCLs) was described exclusively in patients with CPFE (16 out if 22, 72,7 %) [16]. TWCLs were not observed in any patients with IPF or emphysema alone. TWCLs are irrelevant to microscopic honeycombing as they are located in the centriacinar/centrilobular region. They involve one or more acini, membranous and respiratory bronchioles with destruction of the alveoli and dense fibrosis of the walls along with occasional fibroblastic foci. The walls of the TWCLs are mainly composed of dense collagen. They are classified as lesions with coexistent fibrosing interstitial pneumonia and emphysema. Radiologically, they correspond to enlarged cysts with thick walls. Whether TWCLs represent just an overlap of the emphysematous and fibrotic processes that evolve in the same area of the lung or a distinct pathological phenotype remains to be clarified by further studies.
Mitchell et al. found that IPF patients with emphysema exhibited radiologically worse fibrosis and emphysema compared to non-IPF/UIP patients with emphysema. This finding was not explained by key confounding variables and supports the hypothesis that there is synergy among IPF and emphysema [17].
The extent of emphysema and fibrosis on HRCT are independent and significant predictors of DLco [18, 19]. Matsuoka et al. used an objective quantitative method to determine the extent of emphysema and fibrosis on HRCT. In line with previous studies, both extent of emphysema and fibrosis were independent contributors to DLco. However, the extent of fibrosis exhibited a superior predictive power [20]. Finally, the extent of fibrosis on HRCT is an independent predictor of survival in CPFE patients [21].
From a clinical perspective it is important that the presence of emphysema can lead to a false diagnosis of honeycombing. Ground glass opacities surrounding areas of emphysema can give a false appearance of honeycombing [22].
CPFE and pulmonary hypertension
Pulmonary hypertension (PH) appears to be more frequent and severe in CPFE patients than in patients with IPF alone [2, 23–25]. The prevalence of PH in CPFE is estimated to be 28-47 % when assessed by transthoracic echocardiography [2, 23, 25]. Transthoracic echocardiography is an operator dependent imaging examination. Furthermore, the presence of emphysema can add further difficulties in the accurate estimation of RVSP. Right heart catheterization (RHC) remains the gold standard for the diagnosis of pulmonary arterial hypertension. Cottin et al. retrospectively estimated the prevalence of PH in 40 CPFE patients after RHC [24]. In 27 patients (68 %) the mean Ppa was >35 mmHg.
During follow up, CPFE patients can exhibit a worse deterioration of PH compared to patients with sole IPF [25]. Thus, continued vigilance is essential since the development of PH is associated with a worse survival in CPFE [2, 23–25]. In the pivotal study by Cottin et al. [2] the only statistically significant prognostic factor regarding survival was the presence of PH at diagnosis. When PH was confirmed by use of RHC, 1 year survival rate was 60 % [24].
The increased prevalence of PH in CPFE is probably explained by the coexistence of emphysema and fibrosis. Both cause destruction of the pulmonary vasculature bed and of the lung parenchyma. The destruction of the pulmonary vasculature reduces the total cross sectional area. Furthermore, as mentioned CPFE patients are usually hypoxemic due to V/Q mismatching caused by the coexisting emphysema and pulmonary fibrosis. The induced hypoxic pulmonary vasoconstriction is also an important cause of elevated pulmonary arterial pressure. If other pathogenetic pathways are implicated in the development of “out of proportion” PH remains to be clarified. From a clinical point of view the physician should be vigilant for underlying intermittent nocturnal and exercise induced intermittent hypoxia.
Once, pulmonary hypertension is developed it is an equally poor prognosticator in both CPFE and sole IPF patients [26–28].
CPFE and lung cancer
Both emphysema [29–31] and IPF [32] are independent risk factors for the development of lung cancer. Therefore, it is expected that patients with CPFE show increased incidence of lung cancer [32]. Studies suggest that in relation to emphysema, CPFE constitutes a stronger predictor on the occurrence of lung cancer [6, 33–35]. The most common histologic type is that of squamous cell carcinoma [33–35].
CPFE patients are at an increased risk of developing acute exacerbation triggered by surgery, chemotherapy and radiation. In a retrospective study of 101 patients with CPFE the prevalence of acute exacerbation was 19,8 % (postoperative: 27,3 %, during chemotherapy:20 %, during radiation: 16,7 %) [34]. Even after curative resection for NSCLC, CPFE was found to be an independent unfavorable prognostic factor for overall survival (OS) compared to patients with normal lung, emphysema and pulmonary fibrosis. An interesting finding in the above study was that CPFE resulted in an earlier and more frequent recurrence of NSCLC [36]. The above studies highlight the difficulties of managing lung cancer in IPF and CPFE patients.
Natural history
It is not yet clear whether patients with CPFE have a worse survival than patients with isolated pulmonary fibrosis (Table 2). The studies so far have yielded conflicting results. Mejia et al. found that CPFE patients exhibited a worse survival compared to patients with isolated IPF. This finding was associated with the presence of severe pulmonary arterial hypertension (estimated by Doppler ultrasound systolic pulmonary arterial pressure, eSPAP > 75 mm Hg) [23]. The same findings were reported by Sugino et al. The threshold of eSPAP that was correlated with a worse survival was 30.4 mm Hg [25]. The presence of paraseptal emphysema aggravated prognosis. In contrast, Jankowich and Rounds found no difference in mortality [8]. To make things even more complex, Kurashima et al. and Todd et al., found that patients with CPFE have an improved survival compared to patients with isolated pulmonary fibrosis [37, 38]. The reasons for these conflicting results may be the inclusion in the pulmonary fibrosis group of non IPF patients, the retrospective nature of the studies and genetic unidentified factors, since many of the above studies involved Japanese population. More recently, Ryerson et al. [12] studied the prevalence, clinical features, and prognosis of CPFE in IPF. The advantages of this multicenter study were the strict inclusion of patients with IPF according to current guidelines and the usage of standardized definitions for CPFE and emphysema. Patients with CPFE and IPF and those with sole IPF exhibited similar mortality [12]. Finally, patients with CPFE and positive autoimmune markers exhibited improved survival compared to CPFE patients with a negative autoimmune profile [39]. Prospective studies are needed in order to reach robust conclusions regarding the natural history and outcome of patients with CPFE in comparison to IPF.
Treatment
Smoking is strongly implicated in the pathogenesis of both emphysema and IPF, therefore smoking cessation is a “sine qua non” for the management of CPFE. Following the guidelines that apply for the general population, patients with CPFE should be vaccinated against influenza viruses and Streptococcus pneumonia. Long term oxygen therapy is appropriate in case of hypoxemia by extrapolating current knowledge based on COPD patients [40].
The presence of emphysema per se does not necessarily mean the presence of an obstructive syndrome. Things get even more complicated when emphysema co-exists with fibrosis. In such cases, given the altered physiology it is not known if the 70 % threshold regarding the FEV1/FVC ratio, accurately defines patients with underlying functional obstruction that could benefit from bronchodilator therapy. Treatment with bronchodilators (long acting β2 agonists and/or long acting muscarinic) is a rational option although we lack evidence based on randomized control trials. Patients with fibrosis are usually excluded from COPD clinical trials. Regarding the use of inhaled steroids we express some concerns. Inhaled steroids are beneficial in COPD patients with frequent exacerbations when added to long/ultra acting β2 agonists [41]. In CPFE patients, an event of respiratory deterioration challenges the clinician as the differential diagnosis includes pneumonia, pneumothorax, pulmonary embolism, left heart failure and the probability of acute exacerbation of IPF. The presence of underlying infection is crucial in the majority of these conditions in terms of both pathogenesis and outcome. Given the fact that inhaled steroids increase the incidence of pneumonia in COPD patients [42, 43], we remain very cautious with their use in CPFE.
Currently, there are two approved drugs for the treatment of IPF, pirfenidone [44, 45] and nintedanib [46]. Τhe exclusion criteria in the approval studies included the reduction of the FEV1/FVC ratio <70 % (post bronchodilation) regarding pirfenidone [45] and <80 % (pre bronchodilation) regarding nintedanib [46]. Emphysema on HRCT was not an exclusion criterion. A post-hoc subgroup analysis of pooled data from the INPULSIS trials demonstrated that nintedanib slowed disease progression by reducing the annual rate of FVC decline independent of the presence of emphysema at baseline [47]. The main concern is not whether pirfenidone and nintedanib are efficacious in CPFE, but whether the rate of FVC decline underestimates their efficacy in this specific subpopulation.
Pulmonary arterial hypertension (PAH) appears to be more frequent and severe in CPFE patients than in patients with IPF alone [2, 23, 24] and is associated with decreased survival [23, 25].
According to current guidelines the use of targeted PAH therapy in patients with COPD or ILDs and mean PAP <40 mmHg is currently discouraged as there are no systematic data regarding its safety or efficacy. Vasodilators can aggravate the ventilation/perfusion mismatch and worsen hypoxemia. Patients with “out of proportion” PH due to lung diseases (characterized by dyspnea insufficiently explained by lung mechanical disturbances and mean PAP 40– 45 mmHg at rest) should be referred to expert centers and enrolled in clinical trials targeting PAH-specific drug therapy [48]. Thus, the same recommendation applies for patients with CPFE.
Patients with CPFE seem to be at greater risk for developing lung cancer than patients with emphysema [6, 33, 34]. Thus, increased vigilance is required for early detection of such lesions. Management of lung cancer in CPFE patients should follow current guidelines [49]. Finally, as for IPF, CPFE patients at increased risk of mortality should be considered for lung transplantation [50].
Stem cell therapy is a promising approach for COPD [51] and IPF [52, 53]. In emphysema [54–56] and IPF [57] there is numerical and functional impairment of regulatory T-cells (Tregs). This impairment is correlated with a decrease in FEV1 in emphysema [54, 55] and a decrease in FVC, DLco and TLC in IPF [57]. Interestingly, one of the beneficial effects of stem cells in chronic lung diseases is thought to be exerted through upregulation of Tregs [58, 59]. Conducting clinical trials of stem cell therapy in CPFE is an intriguing project that could shed further light in the areas of pathogenesis and treatment [60].
Conclusions
CPFE is a syndrome with clinical importance. Diagnosis is not always straightforward as pulmonary function tests usually show preserved or slightly reduced lung volumes and an obstructive pattern is not always observed. Thus, recognizing the typical velcro-like crackles is an important clue that will raise suspicion for an underlying fibrotic lung disease and the need for a subsequent HRCT that will establish the diagnosis. CPFE patients tend to exhibit a delay in the reduction of FVC and DLco and monitoring disease progression and therapeutic response to antifibrotic patients can be especially challenging. Unlike what one would expect based on IPF studies, serial changes in FVC do not have prognostic value in CPFE, while the rate of FEV1 decline is the strongest predictor of mortality. The development of pulmonary hypertension is frequent in the context of CPFE and is associated with reduced survival. Increased vigilance is required for early recognition of pulmonary hypertension that will allow timely referral for lung transplantation.
References
Wiggins J, Strickland B, Turner-Warwick M. Combined cryptogenic fibrosing alveolitis and emphysema: the value of high resolution computed tomography in assessment. Respir Med. 1990;84(5):365–9.
Cottin V, Nunes H, Brillet P, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26:586–93.
Cottin V, Cordier JF. Combined pulmonary fibrosis and emphysema in connective tissue disease. Curr Opin Pulm Med. 2012;18(5):418–27.
Jankowich MD, Rounds SI. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest. 2012;141(1):222–31.
Tzilas V, Bouros D. Pathogenesis of combined pulmonary fibrosis and emphysema. Common pathogenetic pathways. Pneumon. 2015;28(2):133–8.
Kitaguchi Y, Fujimoto K, Hanaoka M, Kawakami S, Honda T, Kubo K. Clinical characteristics of combined pulmonary fibrosis and emphysema. Respirology. 2010;15(2):265–71.
Akagi T, Matsumoto T, Harada T, et al. Coexistent emphysema delays the decrease of vital capacity in idiopathic pulmonary fibrosis. Respir Med. 2009;103(8):1209–15.
Jankowich MD, Rounds S. Combined pulmonary fibrosis and emphysema alters physiology but has similar mortality to pulmonary fibrosis without emphysema. Lung. 2010;188:365–73.
Kim YJ, Shin SH, Park JW, Kyung SY, Kang SM, Lee SP, et al. Annual Change in Pulmonary Function and Clinical Characteristics of Combined Pulmonary Fibrosis and Emphysema and Idiopathic Pulmonary Fibrosis: Over a 3-Year Follow-up. Tuberc Respir Dis. 2014;77(1):18–23.
Strickland NH, Hughes JM, Hart DA, et al. Cause of regional ventilation-perfusion mismatching in patients with idiopathic pulmonary fibrosis: a combined CT and scintigraphic study. Am J Roentgenol. 1993;161(4):719–25.
Schwartz DA, Merchant RK, Helmers RA, et al. The influence of cigarette smoking on lung function in patients with idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1991;144:504–6.
Ryerson CJ, Hartman T, Elicker BM, et al. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest. 2013;144(1):234–40.
Schmidt SL, Nambiar AM, Tayob N, et al. Pulmonary function measures predict mortality differently in IPF versus combined pulmonary fibrosis and emphysema. Eur Respir J. 2011;38(1):176–83.
Oikonomou A, Mintzopoulou P, Tzouvelekis A, et al. Pulmonary fibrosis and emphysema: Is the emphysema type associated with the pattern of fibrosis? World J Radiol. 2015;7(9):294–30.
Brillet PY, Cottin V. Letoumelin Pet al., Combined apical emphysema and basal fibrosis syndrome (emphysema/fibrosis syndrome): CT imaging features and pulmonary function tests. J Radiol. 2009;90(1 Pt 1):43–51.
Inomata M, Ikushima S, Awano N, et al. An autopsy study of combined pulmonary fibrosis and emphysema: correlations among clinical, radiological, and pathological features. BMC Pulm Med. 2014;14:104.
Mitchell PD, Das JP, Murphy DJ, et al. Idiopathic pulmonary fibrosis with emphysema: evidence of synergy among emphysema and idiopathic pulmonary fibrosis in smokers. Respir Care. 2015;60(2):259–68.
Mura M, Zompatori M, Pacilli AM, et al. The presence of emphysema further impairs physiologic function in patients with idiopathic pulmonary fibrosis. Respir Care. 2006;51:257–65.
Ando K, Sekiya M, Tobino K, et al. Relationship between quantitative CT metrics and pulmonary function in combined pulmonary fibrosis and emphysema. Lung. 2013;191:585–91.
Matsuoka S, Yamashiro T, Matsushita S, et al. Quantitative CT evaluation in patients with combined pulmonary fibrosis and emphysema: correlation with pulmonary function. Acad Radiol. 2015;22(5):626–31.
Choi SH, Lee HY, Lee KS, et al. The value of CT for disease detection and prognosis determination in combined pulmonary fibrosis and emphysema (CPFE). PLoS One. 2014;9(9):e107476.
Akira M, Inoue Y, Kitaichi M, Yamamoto S, Arai T, Toyokawa K. Usual interstitial pneumonia and nonspecific interstitial pneumonia with and without concurrent emphysema: thin-section CT findings. Radiology. 2009;251:271–9.
Mejıa M, Carrillo G, Rojas-Serrano J, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009;136:10–5.
Cottin V, Le Pavec J, Prévot G, et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010;35:105–11.
Sugino K, Ishida F, Kikuchi N, et al. Comparison of clinical characteristics and prognostic factors of combined pulmonary fibrosis and emphysema versus idiopathic pulmonary fibrosis alone. Respirology. 2014;19(2):239–45.
Corte TJ, Wort SJ, Gatzoulis MA, et al. Pulmonary vascular resistance predicts early mortality in patients with diffuse fibrotic lung disease and suspected pulmonary hypertension. Thorax. 2009;64:883–8.
Hamada K, Nagai S, Tanaka S, et al. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest. 2007;131:650–6.
Lettieri CJ, Nathan SD, Barnett SD, et al. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129:746–52.
de Torres JP, Bastarrika G, Wisnivesky JP, Alcaide AB, Campo A, Seijo LM, et al. Assessing the relationship between lung cancer risk and emphysema detected on low-dose CT of the chest. Chest. 2007;132:1932–8.
Wilson DO, Weissfeld JL, Balkan A, Schragin JG, Fuhrman CR, Fisher SN, et al. Association of radiographic emphysema and airflow obstruction with lung cancer. Am J Respir Crit Care Med. 2008;178(7):738e44.
De-Torres JP, Wilson DO, Sanchez-Salcedo P, Weissfeld JL, Berto J, Campo A, et al. Lung cancer in patients with chronic obstructive pulmonary disease. Development and validation of the COPD lung cancer screening score. Am J Respir Crit Care Med. 2015;191(3):285–91.
Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis a population-based cohort study. Am J Respir Crit Care Med. 2000;161(1):5e8.
Kwak N, Park CM, Lee J, et al. Lung cancer risk among patients with combined pulmonary fibrosis and emphysema. Respir Med. 2014;108:524e30.
Usui K, Tanai C, Tanaka Y, Noda H, Ishihara T. The prevalence of pulmonary fibrosis combined with emphysema in patients with lung cancer. Respirology. 2011;16:326–31.
Girard N, Marchand-Adam S, Naccache JM, et al. Groupe d’Etudes et de Recherche sur les Maladies "Orphelines" Pulmonaires (GERM"O"P). Lung cancer in combined pulmonary fibrosis and emphysema: a series of 47 Western patients. J Thorac Oncol. 2014;9(8):1162–70.
Kumagai S, Marumo S, Yamanashi K, Tokuno J, Ueda Y, Shoji T, et al. Prognostic significance of combined pulmonary fibrosis and emphysema in patients with resected non-small-cell lung cancer: a retrospective cohort study. Eur J Cardiothorac Surg. 2014;46(6):e113–9.
Kurashima K, Takayanagi N, Tsuchiya N, et al. The effect of emphysema on lung function and survival in patients with idiopathic pulmonar fibrosis. Respirology. 2010;15(5):843–8.
Todd NW, Jeudy J, Lavania S, et al. Centrilobulal emphysema combined with pulmonary fibrosis results in improved survival. Fibrogenesis Tissue Repair. 2011;4(1):6.
Tzouvelekis A, Zacharis G, Oikonomou A, et al. Increased incidence of autoimmune markers in patients with combined pulmonary fibrosis and emphysema. BMC Pulm Med. 2013;13:31.
Crockett AJ, Cranston JM, Moss JR, Alpers JH. A review of long-term oxygen therapy for chronic obstructive pulmonary disease. Respir Med. 2001;95:437–43.
Nannini LJ, Poole P, Milan SJ, Kesterton A. Combined corticosteroid and long-acting beta(2)-agonist in one inhaler versus inhaled corticosteroids alone for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2013;8:CD006826.
Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax. 2013;68(11):1029–36.
Kew KM, Seniukovich A. Inhaled steroids and risk of pneumonia for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2014;3:CD010115.
Noble PW, Albera C, Bradford WZ, CAPACITY Study Group, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet. 2011;377(9779):1760–9.
King Jr TE, Bradford WZ, Castro-Bernardini S, ASCEND Study Group, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
Richeldi L, du Bois RM, Raghu G. et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82.
Pfeifer M et al. Effect of baseline emphysema on reduction in FVC decline with nintedanib in the INPULSIS trials. Pneumologie. 2015;69:P254.
Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46(4):903–75.
Detterbeck FC, Lewis SZ, Diekemper R, et al. Executive Summary: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):7S–37S.
Raghu G et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
Tzouvelekis A, Laurent G, Bouros D. Stem cell therapy in chronic obstructive pulmonary disease. Seeking the Prometheus effect. Curr Drug Targets. 2013;14(2):246–52.
Tzouvelekis A, Antoniadis A, Bouros D. Stem cell therapy in pulmonary fibrosis. Curr Opin Pulm Med. 2011;17(5):368–73.
Tzouvelekis A, Paspaliaris V, Koliakos G, et al. A prospective, non-randomized, no placebo-controlled, phase Ib clinical trial to study the safety of the adipose derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. J Transl Med. 2013;11:171.
Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking–induced emphysema. Nat Med. 2007;13:567–9.
Isajevs S, Taivans I, Strazda G, et al. Decreased FOXP3 expression in small airways of smokers with COPD. Eur Respir J. 2009;33:61–7.
Hou J, Sun Y, Hao Y, et al. Imbalance between subpopulations of regulatory T cells in COPD. Thorax. 2013;68:1131–9.
Kotsianidis I, Nakou E, Bouchliou I, et al. Global impairment of CD4 + CD25 + FOXP3+ regulatory T cells in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179(12):1121–30.
Ghannam S et al. Immunosuppression by mesenchymal stem cells: mechanisms and clinical applications. Stem Cell Res Ther. 2010;1(1):2.
Sueblinvong V, Weiss DJ. Stem cells and cell therapy approaches in lung biology and diseases. Transl Res. 2010;156(3):188–205.
Tzilas V, Bouros D et al. Prospective phase 1 open clinical trial to study the safety of adipose derived mesenchymal stem cells (ADMSCs) in COPD and combined pulmonary fibrosis and emphysema (CPFE). ERJ. 2015, 46 (suppl 59). DOI: 10.1183/13993003.congress-2015. OA 1970.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Both authors contributed equally to the writing of this manuscript. Both authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tzilas, V., Bouros, D. Combined Pulmonary Fibrosis and Emphysema, a clinical review. COPD Res Pract 2, 2 (2016). https://doi.org/10.1186/s40749-016-0018-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40749-016-0018-1