
Abstract
The pathophysiological mechanisms which lead to sudden infant death syndrome (SIDS) are not completely understood. Cardiac channelopathies are a well-established causative factor with long QT syndrome (LQTS) being the most frequent one, accounting for approximately 12% of SIDS cases. The genetic substrate of the above arrhythmogenic syndrome has been thoroughly described but only specific gene mutations or polymorphisms have been identified as SIDS causative. The review will focus on the prevalence of LQTS-induced SIDS or near-SIDS cases and the mutations held responsible. A literature search was performed in PubMed and Scopus electronic databases. Search terms used were: long QT syndrome, channelopathies, QT prolongation, cardiac ion channels. The above-mentioned search terms were always combined with the term: sudden infant death syndrome. Study types considered eligible were: case–control, family pedigree analysis, case reports. The prevalence of LQTS-induced SIDS according to six broad genetic studies ranges from 3.9 to 20.6%, with an average of 12%. Since LQTS can be effectively managed, LQTS-related SIDS cases could be prevented, provided that a screening method is efficient enough to detect all the affected infants.
Similar content being viewed by others


Background
Cardiac ion channelopathies have been found to be implicated as a causative factor of sudden infant death syndrome (SIDS) by numerous studies. The first channelopathy hypothesized to be linked with SIDS was the long QT syndrome (LQTS) in a study conducted in 1976 by Schwarz [1]. Since then, Brugada Syndrome (BrS) and catecholaminergic polymorphic ventricular tachycardia (CPVT) have been listed as possible causes, with short QT syndrome (SQTS) the most recent addition to the list. It is remarkable that long QT syndrome has recently been found to account for almost 12% of SIDS cases [2]. The purpose of this review is to summarize and present major studies (mostly case–control) which investigate the relationship between the two pathological entities up to date. The review will focus on the prevalence of LQTS-induced SIDS or near-SIDS cases and the mutations held responsible. Since LQTS can be adequately managed, LQTS-related SIDS cases could be prevented, provided that a screening method is efficient enough to detect all the affected infants. The systematic classification of the detected gene variations could serve as a basis for discriminating the most common of them and thus facilitate the development of targeted genetic screening in order to prevent the tragic event of sudden death in infancy.
Sudden infant death syndrome (SIDS)
Sudden infant death syndrome was first defined as a pathological entity in 1969 as “the sudden death of any infant or young child, which is unexpected by history, and in which a thorough post-mortem examination fails to demonstrate an adequate cause of death” [3]. However, the definition above was deemed too non-specific since it lacked an age limitation and was based solely on the post-mortem body dissection excluding the autopsy of the surrounding environment. This is the reason why in 1989 the National Institute of Child Health and Human Development (NICHD) set out to redefine SIDS [4]. The revised definition limited the age of the infant to under 1 year old (<1 year) and stated that the diagnosis of SIDS should only be made if a complete autopsy of the death scene and a clinical history (in addition to an unremarkable post-mortem examination) fail to identify any other specific cause of death [5]. In 2003, after years of controversies, Bruce Beckwith, the visionary of a solid definition, pointed out the need of re-evaluating the definition and categorizing SIDS cases so an expert panel was formed in 2004 in San Diego to address the matter [3, 6]. The San Diego definition as named, is therefore the most recent and systematic one and is widely accepted for the classification of SIDS cases [6]. It divides SIDS cases into three categories, according to specific clinical findings and autopsy results and modifies the 1989 definition by adding the condition that the death must occur during sleep.
Several factors have been accused of triggering SIDS such as maternal age, maternal race, alcohol and tobacco use during pregnancy, low birth weight, male gender, bed sharing and prone sleeping position [7–10]. Despite a notable decline of about 50% in SIDS related deaths, SIDS continues to be the leading cause of post-neonatal death in the USA even after the launch of the “Back to Sleep” campaign [11].
An accurate pathophysiological model able to explain SIDS is not yet established. Various “triple risk models” have been proposed, correlating environmental and intrinsic risk factors which subsequently lead to a vulnerable infant [12]. Injuries and neurochemical abnormalities of the brainstem have been linked to SIDS. Brainstem gliosis possibly causing hypoxemic events in infant victims was found to be related with maternal smoking [13]. Serotonin transporter gene polymorphisms undermining the infantile brainstem’s response to hypoxia or hypercapnia are also characterized as SIDS risk factors [14, 15]. Bacterial and viral infections as well as immunologic polymorphisms (interleukin-10 over-secretion, interleukin-1β and Vascular Endothelial Growth Factor overexpression) are considered triggering factors [16]. Infants with alterations of the intestinal flora, more specifically infant hosts of enterotoxin producing Staphylococcus aureus, are found to be more susceptible to SIDS than non-toxigenic S. aureus hosts [17]. Finally, arrhythmogenic cardiac ion channelopathies (LQTS, SQTS, BrS and CPVT) are established SIDS causative factors with LQTS being the most frequent one.
Methods
A literature search was performed in PubMed (https://www.ncbi.nlm.nih.gov/pubmed) and Scopus (https://www.scopus.com/home.uri) electronic databases. The latest data retrieval date was December 2015. Search terms used were: long QT syndrome, channelopathies, QT prolongation, cardiac ion channels. The above-mentioned search terms were always combined with the term: sudden infant death syndrome. Abbreviations of the above syndromes were also used in the search. Studies had to satisfy the new definition of SIDS as well as the classification criteria, all of which were introduced in San Diego in 2004. Articles studying a sudden unexplained death population which contained a sub-population of SIDS cases were also included in the review. Study types considered eligible were: case–control, family pedigree analysis, case reports.
Results
We identified 13 case–control studies investigating the prevalence of LQTS-induced SIDS [2, 18–29]. Six of them were broad genetic studies of an unselected SIDS population and screened cases for a majority of the LQTS-causing genes known at the time of the study [18–23], whereas the rest concentrated on one or two specific genes [24–29]. The prevalence of LQTS-induced SIDS according to the six broad studies ranged from 3.9 to 20.6%, with an average of 12% [18–23]. A separate and more detailed discussion of the results, regarding the implication of LQTS in SIDS, is provided right below.
The role of long QT syndrome in sudden infant death syndrome
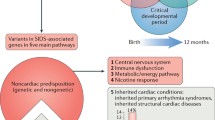
From a historical perspective, in February 1976, Schwartz published the completely novel hypothesis suggesting that LQTS could contribute to a number of SIDS cases [1]. In September of the same year, Maron et al. [30] proposed the connection between LQTS and SIDS. They studied 42 sets of parents who had at least one child with SIDS. In 1998, Schwartz et al. [31] confirmed their initial hypothesis through the results of their study which included 34,442 newborns and lasted for 18 years. They concluded that an evident prolongation of the QT interval on days 3 and 4 of life was associated with a higher risk of SIDS occurrence. Two years later, the first molecular evidence of the appearance of long QT syndrome in SIDS was published [32]. Since then, mutations in 7 genes, KCNQ1, KCNH2/HERG, SCN5A, KCNE2, CAV3, SCN4B, SNTA1 causing the LQTS, have been associated with SIDS cases [2, 18–29]. A brief schematic of the mechanism through which cardiac ion channel mutations lead to the prolongation of the QT interval is provided in Fig. 1.

Functional effect of cardiac ion channel mutations on the repolarization/depolarization capacity of the myocardium
In our review, we divided eligible studies in two separate subsets. The first one consists of broad genetic studies of an unselected SIDS population. Their results are summarized in Table 1. These studies performed mutational analysis of all the LQTS-related genes known at the time they were conducted. The second subset consists of studies investigating the prevalence of SIDS-causative mutations of one specific gene or two, and is presented in Table 2.
It is obvious that the most accurate way to estimate the prevalence of LQTS-induced SIDS is to summarize the results of major genetic studies. Those, include a sufficient enough population to reach safe conclusions, have an extended data collection period and most importantly, as already mentioned, performed mutational analysis on the majority of LQTS-related genes. The functional effect of the detected mutations was confirmed by electrophysiological methods after they were expressed in cell cultures. The study of Arnestad et al. [2] is considered as a significant milestone in the search for a genetic connection between SIDS and channelopathies. It is one of the largest studies conducted; it included 201 Norwegian SIDS cases which were strictly identified as such, according to the Nordic criteria of SIDS [33]. Gene panels were used to detect mutations in cardiac sodium and potassium channel genes. It is remarkable that this study was the first to report that approximately 9.5% of SIDS cases concealed a phenotype of LQTS. More specifically, the majority of the mutations were detected in the SCN5A gene. These mutations caused either an increased persistent sodium current or accelerated inactivation of the sodium channels. Furthermore, it has been noticed that SCN5A mutations serve as a trigger for arrhythmias during sleep [34]. This trigger, in combination with several predisposing factors (prone sleeping position, elevated sympathetic activity during REM sleep, respiratory infections), induced an enhanced cardiac electrical instability state and might subsequently lead to SIDS [35]. Mutations in the KCNQ1 gene (p.G460S) and KCNH2 gene (p.R273Q, p.R954C/p.K897T) led to a loss of function phenotype. Interestingly, certain mutations were capable of causing a fatal QT prolongation without the influence of any other triggering factor, as proven when they were expressed in laboratory animals.
Focusing on the most recent studies, in 2014, Wang et al. [18] separated another large and ethnically diverse cohort of 274 cases of sudden unexplained death in two groups, infants (141 cases) and non-infants (133 cases). Mutations responsible for LQTS were detected in 13.4% of infant cases, most frequently in the SCN5A gene. As in other broad genetic studies SIDS-related mutations were detected in genes which encode the potassium channel complex (KCNQ1, KCNH2, KCNE2), too.
Several studies focused on one single gene, rather than using gene panels. Cronk et al. [26] considered the possibility of CAV3 gene mutations resulting in SIDS. Their hypothesis was based on the observation that CAV3 mutations strongly affect the SCN5A sodium channel by inducing a persistent late sodium current thus serving as a substrate for LQTS [36]. They concluded that 2.2% of cases were due to CAV3 mutations. Tan et al. [24] analyzed another large cohort of 292 SIDS cases, submitted to the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory. They noticed that 1% of cases were due to variants of the SNTA1 gene, which increased peak and late sodium current and led to LQTS type 12. Cheng et al. [25] performed mutational analysis of the SCN4B gene, using the above-mentioned cohort. The variant p.S206L increased late sodium current and possibly led to LQTS type 10.
Conclusions and discussion
The sudden death of an infant is a devastating event, which raises two major concerns for the family. First, the grief for the unexpected loss of an apparently healthy infant and second, the uncertainty for future childbirths. Subsequently, SIDS cases should be managed by a multidisciplinary team of scientists consisting of a variety of specialties such as pediatricians, pediatric cardiologists as well as adult cardiologists, pathologists, coroners, geneticists and psychiatrists. As we can infer from the tables provided, several cardiac ion channel mutations are possibly SIDS causative. However, no solid criteria based on genetic testing have been established in order to point out high risk infants. This is due to the fact that each affected infant has its unique mutational profile regarding the implicated genes. The majority of the studies conducted are based solely on post-mortem genetic analysis and due to ethical regulations the identity of the victims cannot be revealed. As a result, no family pedigree analysis can be performed nor genetic counseling provided. Moreover, no ideal screening method has been proposed, yet. In 2011, the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA), published recommendations concerning the genetic testing of individuals for channelopathies and cardiomyopathies. Family genetic screening is recommended for all first degree relatives of a member affected by LQTS, BrS and CPVT but no universal neonatal screening is discussed or mentioned [37].
The usefulness of electrocardiographic (ECG) screening as a universal method for identifying the affected infants was pointed out by Schwartz et al. in 2002 [38]. To their view, a neonatal ECG is commonly unknown territory to the eyes of an adult cardiologist. Therefore, the aim of their report was to provide simple guidelines for its interpretation. The efficacy of ECG screening is addressed by two independent studies, carried out in 2000 and 2006 [39, 40]. Both focused on the LQTS, as it is the most common channelopathy among BrS, CPVT and SQTS and mainly analyzed the cost effectiveness of ECG screening. The first study of Zupancic et al. in 2000 [39], appears to have two possible weaknesses: the underestimation of the prevalence of LQTS (1/10,000) and the time point of ECG screening, which was performed early enough (day 3 of life) to confuse the normal prolongation of the QT interval during the neonatal period with the pathological one. Quaglini et al. in 2006 had more studies investigating the prevalence of LQTS at their disposal and they considered its prevalence to be 1/2500, much closer to reality [40]. Moreover, the ECG screening was performed during the 3rd or 4th week of life, a period more sensitive to identify the affected infants. Both studies suggest that universal neonatal ECG screening, despite the false negative or false positive results (which are unavoidable since the QT interval is not stable throughout the neonatal period and some infants carrying causative mutations may not have ECG findings), remains a cost-effective method.
As far as genetic screening is concerned, efficient mapping of the responsible genes could lead to the development of targeted, rapid genetic tests for the most common ones. In 2009, Redpath et al. [41] published a case report of a 22 year old man with a borderline shortening of the QT interval and proposed the use of rapid direct DNA sequencing in order to facilitate the diagnosis of the SQTS. The test yielded positive results in less than 72 h by identifying a mutation in the KCNH2 gene. Rapid genetic tests for Long QT Syndrome would be more challenging to develop since 16 genes have been found to cause LQTS, out of which more than 7 have been commonly linked to SIDS cases [42]. However, it would be naive to consider that the less common ones are not likely to contribute to SIDS. The most glaring example is represented by mutations on calmodulin, which can clearly lead to deaths that would be interpreted as SIDS [43]. Next generation whole genome sequencing (WGS) seems to achieve promising results. Rapid molecular diagnosis of LQTS at 10 days of life by WGS has been described by Priest et al. [44]. The test provided accurate readings in 8 days. In the case of deceased infants, when family genetic counseling and screening is required, molecular autopsy could be efficiently performed by WGS of DNA extracted from stored dried blood spot cards (also known as Guthrie cards, tiny blood samples obtained by pricking the infant’s heel). Breakthrough techniques allow the amplification of DNA received from such samples and are able of yielding reliable and rapid results [45]. However, parents should be aware that the detection of a mutation by molecular autopsy does not guarantee the appearance of an arrhythmogenic phenotype in the family nor justifies the exact cause of death for the infant. Last but not least, future research should aim to identify the pathogenetic mechanisms at the core of SIDS. This will enrich our knowledge and understanding of such a tragic event and help us develop more efficient preventive measures as well as improved counseling.
Abbreviations
- SIDS:
-
sudden infant death syndrome
- LQTS:
-
long QT syndrome
- BrS:
-
Brugada syndrome
- CPVT:
-
catecholaminergic polymorphic ventricular tachycardia
- SQTS:
-
short QT syndrome
- WGS:
-
whole genome sequencing
References
Schwartz PJ. Cardiac sympathetic innervation and the sudden infant death syndrome. A possible pathogenetic link. Am J Med. 1976;60:167–72.
Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–7.
Beckwith JB. Defining the sudden infant death syndrome. Arch Pediatr Adolesc Med. 2003;157:286–90.
Bajanowski T, Brinkmann B, Vennemann M. The San Diego definition of SIDS: practical application and comparison with the GeSID classification. Int J Legal Med. 2006;120:331–6.
Willinger M, James LS, Catz C. Defining the sudden infant death syndrome (SIDS): deliberations of an expert panel convened by the National Institute of Child Health and Human Development. Pediatr Pathol. 1991;11:677–84.
Krous HF, Beckwith JB, Byard RW, Rognum TO, Bajanowski T, Corey T, et al. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics. 2004;114:234–8.
Zhang K, Wang X. Maternal smoking and increased risk of sudden infant death syndrome: a meta-analysis. Leg Med (Tokyo). 2013;15:115–21.
Hakeem GF, Oddy L, Holcroft CA, Abenhaim HA. Incidence and determinants of sudden infant death syndrome: a population-based study on 37 million births. World J Pediatr. 2015;11:41–7.
Trachtenberg FL, Haas EA, Kinney HC, Stanley C, Krous HF. Risk factor changes for sudden infant death syndrome after initiation of Back-to-Sleep campaign. Pediatrics. 2012;129:630–8.
Highet AR, Goldwater PN. Maternal and perinatal risk factors for SIDS: a novel analysis utilizing pregnancy outcome data. Eur J Pediatr. 2013;172:369–72.
Moon RY, Horne RSC, Hauck FR. Sudden infant death syndrome. Lancet. 2007;370:1578–87.
Guntheroth WG, Spiers PS. The triple risk hypotheses in sudden infant death syndrome. Pediatrics. 2002;110:e64.
Storm H, Nylander G, Saugstad O. The amount of brainstem gliosis in sudden infant death syndrome (SIDS) victims correlates with maternal cigarette smoking during pregnancy. Acta Paediatr. 1999;88:13–8.
Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pahol. 2009;4:517–50.
Weese-Mayer DE, Berry-Kravis EM, Maher BS, Silvestri JM, Curran ME, Marazita ML. Sudden infant death syndrome: association with a promoter polymorphism of the serotonin transporter gene. Am J Med Genet A. 2003;117:268–74.
Highet AR. An infectious aetiology of sudden infant death syndrome. J Appl Microbiol. 2008;105:625–35.
Highet AR, Goldwater PN. Staphylococcal enterotoxin genes are common in Staphylococcus aureus intestinal flora in Sudden Infant Death Syndrome (SIDS) and live comparison infants. FEMS Immunol Med Microbiol. 2009;57:151–5.
Wang D, Shah KR, Um SY, Eng LS, Zhou B, Lin Y, et al. Cardiac channelopathy testing in 274 ethnically diverse sudden unexplained deaths. Forensic Sci Int. 2014;237:90–9.
Glengarry JM, Crawford J, Morrow PL, Stables SR, Love DR, Skinner JR. Long QT molecular autopsy in sudden infant death syndrome. Arch Dis Child. 2014;99:635–40.
Horigome H, Nagashima M, Sumitomo N, Yoshinaga M, Ushinohama H, Iwamoto M, et al. Clinical characteristics and genetic background of congenital long-QT syndrome diagnosed in fetal, neonatal, and infantile life: a nationwide questionnaire survey in Japan. Circ Arrhythm Electrophysiol. 2010;3:10–7.
Millat G, Kugener B, Chevalier P, Chahine M, Huang H, Malicier D, et al. Contribution of long-QT syndrome genetic variants in sudden infant death syndrome. Pediatr Cardiol. 2009;30:502–9.
Otagiri T, Kijima K, Osawa M, Ishii K, Makita N, Matoba R, et al. Cardiac ion channel gene mutations in sudden infant death syndrome. Pediatr Res. 2008;64:482–7.
Kato K, Makiyama T, Wu J, Ding WG, Kimura H, Naiki N, et al. Cardiac channelopathies associated with infantile fatal ventricular arrhythmias: from the cradle to the bench. J Cardiovasc Electrophysiol. 2014;25:66–73.
Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, et al. Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm. 2010;7:771–8.
Cheng J, Van Norstrand DW, Medeiros-Domingo A, Valdivia C, Tan B-H, Ye B, et al. α1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current. Circ Arrhythm Electrophysiol. 2009;2:667–76.
Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, et al. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4:161–6.
Plant LD, Bowers PN, Liu Q, Morgan T, Zhang T, State MW, et al. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J Clin Invest. 2006;116:430–5.
Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Long QT syndrome in neonates: conduction disorders associated with HERG mutations and sinus bradycardia with KCNQ1 mutations. J Am Coll Cardiol. 2004;43:826–30.
Ackerman MJ, Siu BL, Sturner WQ, Tester DJ, Valdivia CR, Makielski JC, et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 2001;286:2264–9.
Maron BJ, Clark CE, Goldstein RE, Epstein SE. Potential role of QT interval prolongation in sudden infant death syndrome. Circulation. 1976;54:423–30.
Schwartz PJ, Stramba-Badiale M, Segantini A, Austoni P, Bosi G, Giorgetti R, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338:1709–14.
Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, et al. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med. 2000;343:262–7.
Vege A, Rognum TO. Use of new Nordic criteria for classification of SIDS to re-evaluate diagnoses of sudden unexpected infant death in the Nordic countries. Acta Paediatr. 1997;86:391–6.
Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95.
Schwartz PJ. The quest for the mechanisms of the sudden infant death syndrome: doubts and progress. Circulation. 1987;75:677–83.
Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–12.
Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011;13:1077–109.
Schwartz PJ, Garson A Jr, Paul T, Stramba-Badiale M, Vetter V, Villain E, et al. Guidelines for the interpretation of the neonatal electrocardiogram. Eur Heart J. 2002;23:1329–44.
Zupancic JA, Triedman JK, Alexander M, Walsh EP, Richardson DK, Berul CI. Cost-effectiveness and implications of newborn screening for prolongation of QT interval for the prevention of sudden infant death syndrome. J Pediatr. 2000;136:481–9.
Quaglini S, Rognoni C, Spazzolini C, Priori SG, Mannarino S, Schwartz PJ. Cost-effectiveness of neonatal ECG screening for the long QT syndrome. Eur Heart J. 2006;27:1824–32.
Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol. 2009;25:e133–5.
Schwartz PJ, Ackerman MJ, George AL Jr, Wilde AAM. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013;62:169–80.
Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009–17.
Priest JR, Ceresnak SR, Dewey FE, Malloy-Walton LE, Dunn K, Grove ME, et al. Molecular diagnosis of long QT syndrome at 10 days of life by rapid whole genome sequencing. Heart Rhythm. 2014;11:1707–13.
Winkel BG, Hollegaard MV, Olesen MS, Svendsen JH, Haunsø S, Hougaard DM, et al. Whole-genome amplified DNA from stored dried blood spots is reliable in high resolution melting curve and sequencing analysis. BMC Med Genet. 2011;12:22.
Authors’ contributions
All the authors have equally contributed to the conception of the review, data acquisition as well as to the drafting and critical revision of the article. The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Acknowledgements
The authors gratefully acknowledge the invaluable assistance of Dr. Ourania Kariki for the collection of part of the data and retrieval of cited journal articles. They would also like to acknowledge the assistance of Mr. Pantelis Mintekidis—mathematician and statistician—on the interpretation and definition of statistical terms as well as on drafting the manuscript in LaTeX.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The current article is a review. Data presented are properly cited and can be obtained from already published original research articles, which are available on electronic databases (e.g. PubMed etc.)
Ethics approval and consent to participate
Not applicable to this article. The current article is a review. It reports on already published human data.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ioakeimidis, N.S., Papamitsou, T., Meditskou, S. et al. Sudden infant death syndrome due to long QT syndrome: a brief review of the genetic substrate and prevalence. J of Biol Res-Thessaloniki 24, 6 (2017). https://doi.org/10.1186/s40709-017-0063-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40709-017-0063-1