
Abstract
The Diels–Alder (DA) reaction refers to a [4 + 2] cycloaddition reaction that falls under the category of pericyclic reactions. It is a reaction that allows regio- and stereo-selective construction of two carbon–carbon bonds simultaneously in a concerted manner to generate a six-membered ring structure through a six-electron cyclic transition state. The DA reaction is one of the most widely applied reactions in organic synthesis, yet its role in biological systems has been debated intensely over the last four decades. A survey of secondary metabolites produced by microorganisms suggests strongly that many of the compounds possess features that are likely formed through DA reactions, and most of them are considered to be catalyzed by enzymes that are commonly referred to as Diels–Alderases (DAases). In recent years, especially over the past 10 years or so, we have seen an accumulation of a substantial body of work that substantiates the argument that DAases indeed exist and play a critical role in the biosynthesis of complex metabolites. This review will cover the DAases involved in the biosynthesis of decalin moieties, which are found in many of the medicinally important natural products, especially those produced by fungi. In particular, we will focus on a subset of secondary metabolites referred to as pyrrolidine-2-one-bearing decalin compounds and discuss the decalin ring stereochemistry and the biological activities of those compounds. We will also look into the genes and enzymes that drive the biosynthetic construction of those complex natural products, and highlight the recent progress made on the structural and mechanistic understanding of DAases, especially regarding how those enzymes exert stereochemical control over the [4 + 2] cycloaddition reactions they catalyze.
Similar content being viewed by others
Introduction
Decalin skeletons have been found frequently among secondary metabolites that are produced by microorganisms such as fungi and actinomycetes. The bicyclic skeleton usually serves as the core structure for various substitutions including an acyl or a pyrrolidinone. As such, decalin-derived compounds exhibit rich structural diversity and exert a wide range of biological activities, such as antibacterial, antiviral, antitumor and antihyperlipidemic activities. Because of the complexity of chemical structures and interesting biological activities of the decalin-based natural products, they have become the target of chemical synthesis and biosynthesis and are being studied all over the world [1, 2].
Based on their biosynthetic origin, decalin ring-containing natural products are broadly classified into two groups: terpenoids and polyketides (PKs). Decalin ring-containing terpenoid compounds mainly belong to sesquiterpenes and diterpenes, and their structures, biological activities and biosynthesis are reviewed in detail elsewhere [3,4,5,6]. While the ring formation reactions in terpenoid biosynthesis is considered to proceed predominantly through a carbocation cyclization cascade [5, 6], it has been proposed that the decalin rings found in many PK-derived compounds are constructed by an enzymatic intramolecular Diels–Alder (IMDA) cycloaddition reaction [1, 7]. The existence of enzymes that can catalyze an IMDA reaction, namely Diels–Alderases (DAases), was experimentally indicated as early as the 1990’s [8] and has been debated extensively over the years [9, 10]. Recently, many reports on the discovery of DAases and characterizations of their detailed biosynthetic mechanisms were published in succession [11]. However, no review, including those that are focused on the DAases that catalyze the construction of decalin rings from PK scaffolds, has been reported recently.
In this review, we will start with the discussion of the pioneering works on the biosynthesis of solanapyrone and monacolins as a brief introduction to the history of the discovery and establishment of the existence of DAases. We will then make a comprehensive account of pyrrolidine-2-one-bearing decalin-forming DAases reported with an emphasis on the stereochemistry of the decalin rings and the biological activities of those compounds. A concise review at this point in time is warranted, as the field has recently seen a substantial advancement in terms of the isolation and characterization of this class of natural products as well as identification and biochemical and structural analysis of lipocalin-type DAases that comprise the subset of DAases responsible for the formation of pyrrolidine-2-one-bearing decalin natural products. Lastly, we will outline the enzymes and their coding genes that are involved in the biosynthesis of pyrrolidine-2-one type decalin natural products and discuss the current structural and mechanistic understanding of DAases and how they catalyze the pericyclic reactions in a stereoselective manner. By providing an overview of the recent studies on decalin natural products and DAases associated with their biosyntheses, we aimed to clarify what has been achieved so far and what outstanding questions still need to be addressed to advance and apply this exciting field of study further.
Decalin ring stereochemistry and its construction
The decalin ring biosynthesized from a PK substrate is considered to be constructed through an IMDA reaction involving a triene in the PK backbone. There are four types of cyclization modes a triene system can undergo during an IMDA reaction to form four stereochemically distinct decalin adducts (Fig. 1). While the use of an IMDA reaction of triene on a straight chain is applied frequently as a means of constructing a decalin ring in chemical synthesis, its stereoselectivity is known to depend on various factors, such as the positioning, properties and conformations of the substituents on the substrate, as well as steric, electronic and solvent effects on the transition state. It is also known that the presence of Lewis acid has a great effect on the cis–trans ratio in the adducts being formed [12,13,14]. In addition, many studies have been conducted on the stereoselective synthesis of decalin rings, because the stereochemistry of the decalin ring has a great influence on the biological activities of the corresponding compounds [15, 16]. Below, we will describe representative groups of compounds whose biosyntheses have been postulated to involve DAases for their decalin construction.

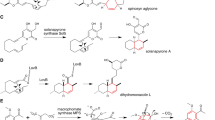
Decalin formation via an intramolecular Diels–Alder reaction, leading to the formation of four stereochemically distinct adducts
Solanapyrones
Natural product solanapyrone is a PK secondary metabolite isolated from the fungus Alternaria solani (Pleosporaceae) which causes a disease called early blight of potato and tomato [17, 18]. Solanapyrone A (Fig. 2) is a known phytotoxin, and its mode of action was determined to be inhibition of DNA polymerases β and λ [19]. The carbon skeleton of the compound was determined to be of PK origin based on feeding experiments with A. solani using labeled precursors [20], and the decalin moiety was thought to be constructed by a biological DA reaction based on the isolation of optically active solanapyrone A reported by Ichihara et al. [21, 22]. To specifically examine the outcome of the cycloaddition step, the linear triene precursors prosolanapyrone I and II labeled with deuterium were synthesized and fed to A. solani (Fig. 2) [22]. The result indicated that prosolanapyrone I was oxidized to prosolanapyrone II followed by a [4 + 2] cycloaddition reaction to selectively form solanapyrone A, the expected exo-axial adduct of the DA reaction, and B upon reduction of the aldehyde to a hydroxymethyl group (Fig. 2).

The proposed biosynthetic pathway of solanapyrones, including the oxidation and the cycloaddition steps that are considered to be catalyzed by Sol5 and the four products resulting from the pathway
In 2010, Fujii et al. isolated the solanapyrone biosynthetic gene cluster (BGC) from the A. solani genome and identified the solanapyrone synthase (SPS, Sol5) as a potential gene encoding the solanapyrone DAase [23]. SPS was initially cloned and functionally expressed in the heterologous host Aspergillus oryzae to confirm its ability to catalyze the formation of the expected cycloaddition product from the linear precursor. Subsequently, SPS was expressed in large scale in Pichia pastoris for a detailed in vitro analysis. The purified enzyme was shown to catalyze an initial oxidation of the achiral prosolanapyrone II to an intermediate prosolanapyrone III before the [4 + 2] cycloaddition reaction takes place to form the endo adduct solanapyrone A and the endo adduct solanapyrone D in an approximate ratio of 7:1. The results from the study suggested that the primary catalytic function of SPS might be an oxidase activity that converts the hydroxymethyl group in prosolanapyrone II into an aldehyde in prosolanapyrone III that promotes the subsequent DA reaction to form the decalin moiety in solanapyrone A and D. The previous finding indicated that the presence of an aldehyde on the pyrone ring lowers the lowest unoccupied molecular orbital (LUMO) energies of the dienophile [24]. Moreover, the DA reaction was shown to be promoted in aqueous media [24], presumably due to the hydrophobic effect on nonpolar substrates in an aqueous environment [25] and water molecules hydrogen-bonding to the carbonyl groups to activate the dienophile [26]. Taken together, SPS is thought to accelerate the DA reaction by activating the substrate by the oxidation of the substituent, providing a hydrophobic environment that confines the straight-chain triene into a catalytically relevant conformation and furnishing hydrogen-bonding partners to the substrate to further activate the dienophile [23].
Monacolins
Monacolins are known as PKs of fungal origin that exhibit a significant inhibition of 3-hydroxy-3-methylglutarylcoenzyme A (HMG-CoA) reductase, which catalyzes important steps in the cholesterol biosynthesis [27]. Monacolin K [28], also known as mevinolin and lovastatin [29], is a decalin-containing nonaketide with an acylated C8 hydroxy group, and the semi-synthetic derivative of monacolin J called simvastatin acts as a potent inhibitor of HMG-CoA reductase that is widely prescribed for the treatment of hypercholesterolemia (Fig. 3) [27]. Those monacolins have a rigid and hydrophobic decalin ring core that is linked to a carboxylic acid side chain whose chemical structure resembles the structure of the HMG moiety of HMG-CoA. The crystal structures of HMG-CoA reductase in complex with HMG and simvastatin and other statin-type inhibitors showed that monacolins occupy the binding site of HMG-CoA, thereby inhibiting the original substrate of HMG-CoA to access the active site [27]. The gene cluster for lovastatin biosynthesis was identified in the genome of the producing fungus A. terreus [30]. Among the biosynthetic enzymes identified, LovB plays an indispensable role of producing lovastatin. LovB, also known as lovastatin-nonaketide synthase (LNKS), is a highly-reducing iterative polyketide synthase (HR-iPKS) that forms the PK carbon framework of the compound [30]. LovB collaborates with the stand-alone enoyl reductase (ER) LovC to produce a nonaketide intermediate, which remains attached to LovB covalently via a thioester bond (Fig. 3) [31, 32]. In the subsequent studies, in vitro reconstitution of LovB, LovC and the stand-alone thioesterase LovG revealed that the straight-chain polyketide intermediate was transformed to bear a trans-decalin ring moiety while attached to LovB, prior to being released by LovG to give the advanced intermediate, dihydromonacolin L [33, 34]. Therefore, unlike the biosynthesis of solanapyrones in which the decalin ring formation is catalyzed by a separate DAase after the PK chain elongation and release by PKS is completed, the lovastatin biosynthesis is considered to accomplish the DA reaction-mediated decalin construction during the process of nonaketide formation catalyzed by the PKS (Fig. 3) [31]. In this respect, LovB also serves as the DAase for the biosynthesis of lovastatin.

The biosynthetic gene cluster of monacolin and the proposed biosynthetic pathway leading to the formation of the native pathway product lovastatin and the semisynthetic product simvastatin generated upon feeding of α-dimethylbutyryl-N-acetylcysteamine thioester (α-dimethylbutyryl-SNAC) [118]
Pyrrolidine-2-one-bearing decalins
The pyrrolidine-2-one-type moieties are known to exhibit important biological activities and commonly found in fungal metabolites biosynthesized through PKS-nonribosomal peptide synthetase (NRPS) hybrid pathways [2, 35,36,37]. In fact, the lovastatin biosynthetic HR-iPKS LovB carries an inactive domain at its C terminal end that is highly homologous to NRPS-derived condensation domains [30]. This finding, together with the fact that both enzymes are involved in the biosynthesis of decalin-containing products, suggests that LovB might have been derived from a pyrrolidine-2-one-forming PKS–NRPS. In this section, we focus on the pyrrolidine-2-one class of compounds having a decalin ring. Pyrrolidine-2-one derivatives with a decalin ring are composed of two important biosynthetic units. One unit is comprised of a linear carbon fragment of length C14, C16, C18 or longer with several methyl substituents. This linear PK unit can be converted into a decalin ring by an IMDA cycloaddition reaction. The other building block, which is attached to the PK-derived decalin ring, is the amino acid-derived heterocyclic pyrrolidine-2-one moiety that assumes the structure of either a pyrrolin-2-one or a pyrrolidine-2,4-dione. The PK-amino acid product is constructed by a single megaenzyme, a PKS–NRPS hybrid enzyme. Figure 4 illustrates the outline of the biosynthesis of pyrrolidine-2-one class of PK–NRP hybrid compounds by a generic PKS–NRPS enzyme having a typical domain organization. The PK–NRP precursor is released from the PKS–NRPS by the catalytic activity of its terminal reductase (R) domain that yields either pyrrolin-2-one or pyrrolidine-2,4-dione group. The pyrrolin-2-one group is formed via a Knoevenagel condensation after the precursor is released as an aldehyde, whereas the pyrrolidine-2,4-dione group is formed directly by a Dieckmann condensation as the precursor is cleaved from the enzyme (Fig. 4) [38, 39]. Pyrrolidine-2-one-bearing decalin compounds are known to be rich in structural diversity and exhibit various biological activities. In the next section, we will describe the structural diversity and bioactivities of these compounds with an emphasis on the varieties in the pyrrolidine-2-one and decalin moieties and the possible link to the biological activities.

Generalized biosynthetic pathways of pyrrolidine-2-one type natural products catalyzed by a polyketide synthase-nonribosomal synthetase hybrid enzyme. The product can have either a pyrrolin-2one or pyrrolidine-2,4-dione depending on whether the reductase domain-catalyzed cyclorelease reaction proceeds via A aldehyde release followed by Knoevenagel condensation or B direct Dieckmann condensation. KS ketosynthase, AT acyltransferase, DH dehydratase, MT methyltransferase, KR ketoreductase, ACP acyl carrier protein, C condensation, A adenylation, T thiolation, R reduction
Diverse structures and biological activities of tetramic acid-bearing decalin natural products
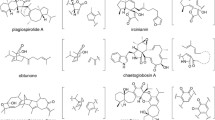
Equisetin (1) is a representative example of tetramic acid-bearing decalin natural products and known to exhibit a wide range of biological activities against various targets, including broad-spectrum antibacterial activities, cytotoxicity, phytotoxicity and most importantly HIV-1 integrase inhibitory activity (Fig. 5) [36, 40]. This compound was first isolated from Fusarium equiseti by Burmeister et al. in 1974 [41, 42]. Compound 1 is characterized by a trisubstituted decalin ring, which also carries a pyrrolidine-2,4-dione moiety derived from a N-methylserine residue. The natural product (+)-fusarisetin A (2) isolated from Fusarium sp. FN080326 was thought to be produced by a similar mechanism with which 1 is biosynthesized based on the similarity in the overall chemical structures of the two compounds [43]. Compound 2 sports a highly unique pentacyclic core structure, and is a potent inhibitor of acinar morphogenesis, cell migration, and invasion in human breast cancer cells [44]. In addition, phomasetin (3), an enantiomer of 1 that was isolated from the fungus Phoma sp. MF6070 (Didymellaceae), exhibited a similar range of IC50 values to 1 in a series of assays for the evaluation of their HIV-1 integrase inhibitory activity [45, 46]. To explore the effect of stereochemistry of the compounds on their bioactivity, a pair of epimers of 1 and 3 that differ in the stereochemistry at the alpha carbon of the N-methylserine residue were prepared and subjected to the same activity tests. The results indicated that all four compounds exhibited similar HIV-1 integrase inhibitory activities [45].

Structural diversity of pyrrolidine-2-one type of compounds categorized according to the stereochemistry at positions 2, 3, 6 and 11 of the decalin moiety that indicates the stereochemical outcome of the decalin-forming Diels–Alder reaction. The top (yellow) and bottom (blue) halves are comprised of trans- and cis-decalin products, respectively. Compounds within parentheses do not have their absolute configurations determined
Cryptocin (4) was isolated from Cryptosporiopsis cf. quercina (Dermateaceae), an endophytic fungus found in the stems of Tripterygium wilfordii, a well-known medicinal herb that has been used in traditional Chinese medicine for a long time [47]. Compound 4 resembles 3 in its chemical structure, but interestingly 4 shows a strong activity against certain phytopathogenic fungi, such as Phytophthora (water molds that frequently cause root rot in various plants), Pyricularia (causative agents of the rice blast disease) and Fusarium (causative agents of wilt, blight and basal rot), showing minimal inhibitory concentration (MIC) values in the range of 0.78 to 1.56 µg/mL. On the other hand, it was not active against human pathogenic fungi such as Candida, Aspergillus and Histoplasma [48].
Altersetin (5) was isolated as an antibiotic from an endophytic fungus Alternaria sp. P 0506 (Pleosporaceae) derived from Vinca minor (common periwinkle) [49]. The antimicrobial activity of 5 significantly suppressed pathogenic Gram-positive bacteria such as Streptococcus and Enterococcus species. In particular, 5 exhibited MIC values of 0.5–1.0 µg/mL against various Staphylococcus aureus strains in a serial agar dilution assay. However, it did not show any activity against Gram-negative bacteria and pathogenic yeast strains such as Klebsiella and Candida (Table 1). Interestingly, a modified 5 with its double bonds in the decalin ring and the aliphatic side chain fully reduced showed almost the same biological activity as 5 (Table 1). The biological activity of 1 was also investigated at the same time because of the structural similarity between 5 and 1. The activity of 1 was similar to that of 5, and the antimicrobial activity of the reduced form of 1 was also hardly affected (Table 1). These results indicate that the contribution of the decalin and aliphatic side chain toward the biological activity of 1 and 5 is minimal, suggesting that the key motif that furnishes the antimicrobial activity of those compounds might be the tetramic acid moiety.
Coniosetin (6) isolated from the fungus Coniochaeta ellipsoidea Udagawa (Coniochaetaceae) had a strong growth-inhibiting activity against Gram-positive bacteria, especially multidrug-resistant S. aureus at a MIC value of 0.3 µg/mL [50]. The compound closely resembles 5, where the only difference is that 6 carries an extra methyl substitution on the decalin ring (Fig. 5). Another equisetin-type compound is trichosetin (7), which was isolated from the co-culture of the fungicidal fungus Trichoderma harzianum with the callus of Catharanthus roseus (Madagascar periwinkle) [51]. It could not be isolated from individual cultures of T. harzianum or the Catharanthus roseus callus. The compound was obtained only when they were co-cultured. Compound 7 is essentially a desmethylequisetin, where the N-methylation at the amide group of the tetramic acid moiety of 1 is missing (Fig. 5). Regarding the biological activity of 7, it showed a strong antimicrobial activity at a MIC value of 1.56 µg/mL against Gram-positive bacteria such as Staphylococcus and Bacillus strains. However, it did not exhibit any activity against Gram-negative bacteria, yeasts or fungi. Because the pattern of biological activities exhibited by 7 was very close to those exhibited by 1, the N-methylation of the tetramic acid moiety was considered to be non-essential in conferring those compounds with their antimicrobial activities [51].
Among the pyrrolidine-2-one-type compounds identified, cissetin (8) isolated from an unidentified fungus OSI 50185 was found to have an unusual cis-decalin ring core (Fig. 5). Biological assays were conducted with cis-decalin-containing 8 and trans-decalin-bearing 1 and 7 to compare their antibiotic activities. However, no significant difference in their antimicrobial activities against Gram-positive bacteria was found. Therefore, the stereochemical conformation of the decalin structure does not appear to play a crucial role in those compounds exhibiting their antibiotic activities. These results suggested that, as the studies on the antibiotic activities of the reduced forms of 1 and 5 also indicated [49], the tetramic acid portion is the key element in conferring to those compounds with their antibiotic activities and the decalin ring is less important in terms of conferring antimicrobial activities to those compounds [52]. Lastly, similar to many of the equisetin-type compounds discussed above, 8 did not show any activity against Gram-negative bacteria as well as yeasts and fungi.
JBIR-22 (9) is a natural product produced discovered after successful isolation of a slow-growing rare fungus Verticillium sp. F21794 (Plectosphaerellaceae) from a soil sample using a screening technique involving the use of daunomycin to suppress growth of ordinary fungi that would grow at a faster rate [53]. Compound 9 was identified as a potent inhibitor of protein–protein interaction involved in the proteasome assembly factor (PAC)1–PAC2 heterodimerization and PAC3 homodimerization that are essential for proper functioning of the 20 S proteasome that is known to sustain high growth rates of cancer cells [54].
Methiosetin (10) isolated from a tropical sooty mold Capnodium sp. F-190679 (Capnodiaceae) found in palm leaf litter from Guatemala was found to possess weak antimicrobial activities at MIC values of 256 and 32 µg/mL against Gram-positive S. aureus and Gram-negative Haemophilus influenzae, respectively, through a screening method involving the use of antisense to elicit target-based hypersensitivity for identifying antimicrobial compounds and their mechanisms of action [55, 56].
Ascosalipyrrolidinones A (11) and B (12) represent another example cis-decalin-bearing secondary metabolites (Fig. 5). Those compounds were isolated from the endophytic obligate marine fungus Ascochyta salicorniae (Didymellaceae) that was isolated from the green alga Ulva sp. and carry an unusual deoxytetramic acid moiety. In addition to antimicrobial activities, 11 and 12 exhibited a strong antiparasitic activity against Trypanosoma cruzi at a MIC value of 1.1 µg/mL [57].
Sch 210972 (13) was isolated from the phytopathogen Chaetomium globosum (Chaetomiaceae) as a natural product with a strong inhibitory activity against the chemokine receptor CCR-5 at a half-maximal inhibitory concentration (IC50) value of 79 nM, exhibiting a potential to be developed into an anti-human immunodeficiency virus type 1 (HIV-1) agent [58]. A series of studies have been conducted on the biosynthesis of this compound to shed light on the mechanism of the DA reaction-mediated decalin formation and the construction of the tetramic acid moiety as discussed in detail below [39, 59].
Five variants of myceliothermophins, another pyrrolidine-2-one-type natural products, were isolated from a thermophilic fungus Myceliophthora thermophila (Chaetomiaceae). The differences among the group of compounds were concentrated on the leucine residue that makes up the tetramic acid moiety, where myceliothermophins A (14) and B (15) (Fig. 5) have a hydroxyl group at the α carbon, and myceliothermophins C and D have a methoxy group at the same position. Compound 14 and myceliothermophin C are stereoisomers of 15 and D, respectively. In myceliothermophin E, the leucine α–β bond is unsaturated. Of the five compounds, 14 as well as myceliothermophins C and E exhibited cytotoxic effects on four human cancer cell lines A549, Hep3B, MCF-7 and HepG2. However, 15 and myceliothermophin D did not possess such activities. Thus, once again the configuration of the tetramic acid moiety was implicated to play an important role in conferring interesting biological activities to the decalin-bearing natural products [60].
CJ21,058 (16) was isolated from a taxonomically unidentified fungus CL47745 with an antimicrobial activity against Gram-positive bacteria S. aureus and Enterococcus faecalis with a MIC value of 5 µg/mL. Like many other equisetin-type compounds, 16 was also ineffective against Gram-negative strains such as Streptococcus pyogenes and Escherichia coli. It was only moderately cytotoxic against HeLa cells with an IC90 value of 32 µg/mL. The antimicrobial activity is considered likely due to the inhibitory activity of 16 against SecA, an ATPase that is a member of the multisubunit preprotein translocase complex that controls ATP-dependent transport of secreted proteins across the cell membrane [61].
Ascosetin (17) was isolated from an unidentified soil-dwelling ascomycete fungus E-000504855. This pyrrolidine-2-one-type compound carried an unusual carboxylic acid at the beginning of the PK chain and used a leucine residue as the building block for the tetramic acid moiety (Fig. 5). Compound 17 showed antibacterial activities with MIC values of 2–16 µg/mL against various Gram-positive bacteria including S. aureus and Bacillus subtilis. However, like other equisetin-type compounds, 17 was not active against Gram-negative bacteria except for H. influenzae. It was weakly cytotoxic to mammalian cells but not effective against the fungus Candida albicans [62].
Paecilosetin (18) isolated from the endophytic insect-pathogenic fungus Paecilomyces farinosus (Trichocomaceae) is another example of a desmethylequisetin with a substitution of the serine residue with a threonine residue that constructs the tetramic acid moiety (Fig. 5). Compound 18 showed an IC50 value of 3.1 µg/mL against P388 murine leukemia cells. Unlike vast majority of the pyrrolidine-2-one-type compounds, it also exhibited a growth inhibitory activity against the Gram-positive bacteria B. subtilis and the fungi Cladosporium resinae and Trichophyton mentagrophytes [63].
Oteromycin (19) [64], ZG-1494α (20) [65] and talaroconvolutins [66] are unique pyrrolidine-2-one-type compounds possessing an extended PK backbone and a deoxytetramic acid moiety derived from either a phenylalanine residue or possibly a tyrosine residue in 20 and talaroconvolutins (Fig. 5). While the molecular formulae of 20 and talaroconvolutin B are identical, the possibility of them being stereoisomers cannot be ruled out due to lack of stereochemical assignments of the key stereocenters of those compounds. Compound 19 was obtained from unidentified fungal species MF 5810 and MR 5811, while 20 and talaroconvolutins A–D were isolated from Penicillium rubrum and the ascomycete Talaromyces convolutus Udagawa strain NE76-1 (Trichocomaceae), respectively. More recently, another group of compounds called cladosporitins A and B were isolated, together with talaroconvolutin A, from the marine fungus Cladosporium sp. HNWSW-1 (Cladosporiaceae) derived from the mangrove tree Ceriops tagal. Cladosporitins also closely resemble 20 except for the occurrence of a succinimide moiety instead of a tetramic acid moiety [67]. Compound 19 was shown to be a novel antagonist of endothelin receptors that may be useful in treating cardiovascular, respiratory, gastric, renal and urological conditions [64], whereas 20 can inhibit the platelet-activating factor acetyltransferase, the enzyme responsible for the last step of the biosynthesis of platelet-activating factor involved in signaling for physiological reactions such as inflammation, allergic responses and anaphylaxis [65]. Interestingly, unlike most other pyrrolidine-2-one-type compounds, 20 and talaroconvolutins B–D were also shown to exhibit antifungal activities [66], and talaroconvolutin A and cladosporitins A and B were cytotoxic against several human cancer cell lines [67].
An unidentified Fusarium sp. produces TA-289 (21), a very simple type of equisetin-like compounds having only a single methyl group on its decalin ring with the des-N-methylated tetramic acid moiety being derived from an alanine residue (Fig. 5). Compound 21 was shown to be toxic to the yeast Saccharomyces cerevisiae in a pH- and carbon source-dependent manner and caused irreversible blockage of cell cycle without targeting the microtubules. The study also showed that 21 also caused other biological effects that are less commonly seen among the equisetin-like compounds, such as generation of reactive oxygen species (ROS), increased cell wall permeability and ROS-independent abnormal morphology of mitochondrial [68].
An endophytic fungus CR127A collected from Vochysia guatemalensis (white yammer tree), whose internal transcribed spacer (ITS) regions of rDNA was 98% identical in sequence to that of Codinaeopsis gonytrichoides (Chaetosphaeriaceae), produced codinaeopsin (22), an equisetin-like product bearing a previously unreported tetramic acid moiety derived from a tryptophan residue (Fig. 5). Compound 22 showed a growth inhibitory effect against the protozoan human parasite Plasmodium falciparum that causes malaria at an IC50 value of 4.7 µM [69].
Spylidone (23) is unusual in that it not only carries an uncommon cis-decalin ring, but also has an unusual tetracyclic core structure with a spiro ring. Compound 23 is produced by a fungus Phoma sp. FKI-1840 (Didymellaceae) isolated from the soil of Miyakojima Island in Okinawa, Japan [70], along with other previously reported cis-decalin compounds vermisporin (24) [71] and PF1052 (25) [72]. Compound 24 was also isolated from the fungus Ophiobolus vermisporis (Phaeosphaeriaceae) [73]. Instead of the spiro ring of 23, 24 and 25 both contain an epoxide on the butyl side chain of the decalin ring (Fig. 5). More recently, simplicilones A and B, a pair of compounds that closely resemble 23, were isolated from the endophytic fungus Simplicillium subtropicum SPC3 (Cordycipitaceae) found in the fresh bark of the African medicinal plant Duguetia staudtii (Engl. and Diels) Chatrou [74]. While 24 and 25 were found to be antibiotic, 23 did not show any antibiotic activity. Instead, 23 had a biological activity that inhibits mouse macrophages from accumulating lipid droplets [70], which is associated with chronic inflammation that can cause various diseases [75]. On the other hand, vermitrasporin, a possible isomer of 24 isolated from the fungus MSX 105528, was claimed to have an anti-tuberculosis activity [76], and 25 was found to be a neutrophil migration inhibitor [77].
UCS 1025 A (26) is a pyrrolizidinone (azabicyclo [3.3.0] octanone) type of natural product (Fig. 5) that is produced together with the hydroxylated analog UCS 1025B by the fungus Acremonium (formerly known as Cephalosporium) sp. KY4917 (Hypocreaceae). Similar to many other pyrrolidine-2-one-bearing decalins described in this review, 26 was active mainly against Gram-positive bacteria, although it was active against a Gram-negative strain Proteus vulgaris. Similarly, 26 was only moderately cytotoxic against human cancer cell lines [78]. However, it was shown to have a strong telomerase inhibitory activity [79, 80]. In fact, decalin natural products with the pyrrolizidinone skeleton have been shown to exert a variety of biological activities. CJ16264 (27) isolated from CL39457, an unidentified fungus likely an agonomycete strain, was shown to be a broad-spectrum Gram-positive antibiotic with limited activities against Gram-negative bacteria but have a relatively strong cytotoxicity with an IC90 value of 8.0 µg/mL [81]. Similarly, pyrrolizilactone (28), which was also isolated from an uncharacterized fungus, was determined to be cytotoxic to various cancer cell lines with a proteasome inhibitor activity [82, 83]. It is interesting to note from biosynthetic and bioactivity perspectives that 26 carries a trans-decalin moiety while 27 and 28 have a cis-configured decalin moiety (Fig. 5).
New decalin containing compounds isolated by new approaches
In recent years, bioinformatics technology developed rapidly in part due to the advancement of the next-generation sequencing methods that provided vast amounts of genomic information have become available to the scientific community, facilitating the developing new methods and techniques to analyze and exploit the genomic information [84]. Using the so-called Big Data and the continuously advancing array of bioinformatics tools, the new approach of searching for novel secondary metabolites in the genomic information called genome mining has also been practiced actively within the field of natural product biosynthesis in recent years [85]. Such methods have successfully identified several pyrrolidine-2-one-bearing decalin compounds, including oxaleimide A (29) and varicidin A (30) that were found through bioinformatic mining of the genome sequences of Penicillium oxalicum [86] and P. variabile [87], respectively (Fig. 5). Compound 29 along with nine other analogs were discovered by identifying a gene cluster in the P. oxalicum genome that contained a number of genes, including a DAase-coding gene, known to be involved in the biosynthesis of decalin-bearing secondary metabolites [86]. Compound 30 was obtained by heterologously expressing five key genes from the cryptic BGC, including a predicted DAase gene, found in the genome of P. variabile, which was known not to produce any decalin-containing secondary metabolites [87]. Another method that relies on a metabolomic approach to search for new and interesting natural products also led to the identification of novel decalin compounds. Wakodecalines A and B (31, 32) containing a rare cyclopentanone-fused decalin skeleton were isolated from a fungus Pyrenochaetopsis sp. RK10-F058 (Cucurbitariaceae) along with 3 (Fig. 5) using the Natural Products Plot (NPPlot) method [88]. The NPPlot is a library of semipurified microbial metabolites annoted with UV and LC–MS spectral information that has been used successfully to screen for novel secondary metabolites [89, 90]. Wakodecalines are among many examples of compounds that were identified through such screening activities, and were found to have a moderate antimalarial activity against the P. falciparum 3D7 [88]. Wakodecalines might be biosynthesized from 3, which has the same absolute stereochemistry as wakodecalines, through a spiro-intermediate similar to altercrasin A and its derivatives [91, 92]. As discussed further in “Decalin ring stereochemistry and its effect on the biological activity” section below, it is not difficult to imagine that more decalin-bearing secondary metabolites will be discovered by bioinformatic mining of various fungal genome sequences and metabolomic screening of fungi.
Decalin ring stereochemistry and its effect on the biological activity
Among the currently known fungal secondary metabolites that harbor a decalin ring, the majority falls under the trans-decalin type (Fig. 5, top half) and the cis-decalin type (Fig. 5, bottom half) is the minority. One of the reasons for the predominance of trans-decalin adducts among the natural products is that, while the number of substitutions and the type of functional groups on the linear precursor can greatly affect the outcome of the DA cycloaddition reaction, in general an endo cyclization, which leads to the formation of a trans-decalin adduct, is kinetically favored to proceed [93]. However, in terms of biological activities of those compounds, cis-decalin type of natural products have the tendency to exhibit a wider range of interesting biological activities. For example, the cis-decalin-containing diterpene formamide kalihinene X isolated from the marine sponge Acanthella cavernosa is known to inhibit attachment and metamorphosis of the cyprid or final mobile form of the larvae of the barnacle Balanus amphitrite (Fig. 6) [94]. Similarly, vinigrol isolated from the fungus Virgaria nigra F-5408 (Xylariaceae) has a cis-decaline core with a unique 1,5-butylene bridge [95] (Fig. 6). This compound is a potent antihypertensive agent as well as an inhibitor of epinephrine- or platelet activating factor-induced platelet aggregation with an IC50 in the nanomolar range and at the same time an inducer of platelet aggregation with a minimum tolerated concentration in the micromolar range [96, 97]. Interestingly, the cis-clerodane diterpene alkaloid agelasine G (Fig. 6) isolated from the marine sponge Agelus nakamurai collected in Okinawa, Japan, was found to exhibit antileukemic [98] and protein tyrosine phosphatase 1B inhibition [99] activities. However, agelasine B with a trans-decalin core (Fig. 6) but none of the cis-decalin-bearing members acts as a Na+/K+-ATPase inhibitor [100]. Likewise as described in the previous section, cis-decalin-bearing 11 and 12 exert a strong anti-trypanosomal activity [57], and 23 acts as an inhibitor of lipid droplet accumulation in macrophages [70]. However, in some cases the stereochemistry of the decalin moiety does not appear to have a significant effect on the biological activities of the compounds as was the case for the antibiotic 8 and its similar cis counterparts 1 and 7 [49]. Also, the cis-decalin-containing pyrrolizidinone 28 is cytotoxic to various cancer cell lines with a proteasome inhibitor activity [82, 83], but the trans relative 26, while only moderately cytotoxic against human cancer cell lines [78], is a strong telomerase inhibitor [79, 80]. Because there are only a handful of reports of cis-decalin type of pyrrolidine-2-one natural products, it is difficult to draw a clear conclusion on the structure–activity relationship on the stereochemistry of the decalin moieties. Nevertheless, due to the structural complexity of these natural products, it is difficult to synthesize and manipulate those compounds synthetically. Thus, there is a growing interest in devising biosynthetic approaches to producing and engineering decalin-containing natural products and their analogs.

Representative non-pyrrolidine-2-one type natural products containing a cis-decalin core structure
Biosynthetic genes of pyrrolidine-2-one type of decalin ring-containing compounds
As briefly discussed earlier, the backbone core of fungal-derived pyrrolidine-2-one-containing natural products is constructed by a PKS–NRPS hybrid enzyme. Song et al. reported the discovery of a biosynthetic enzyme FusA for the production of fusarin C by Fusarium fungi, the first hybrid enzyme identified that is responsible for the formation of a pyrrolidine-2-one-containing natural product [101]. To date, biosynthetic pathways of various pyrrolidine-2-one-containing natural products have been examined to uncover the enzymes and the mechanisms involved in their biosyntheses [102]. The biosynthetic process starts from the extension of the PK chain by the PKS portion of the PKS–NPRS hybrid enzyme. In the gene clusters for the biosynthesis of fungal natural products containing a PKS–NRPS gene, there is typically a gene encoding a stand-alone ER that functions in trans with the PKS–NRPS [103]. The PKS module and the ER work together to catalyze an iterative elongation of the PK chain. In the meantime, the adenylation (A) domain within the NRPS portion of the hybrid enzyme can activate a preferred free l-amino acid and transfer it to the thiol group at the tip of the phosphopantetheine cofactor covalently linked to the thiolation (T) domain. Once the growing PK chain reaches a pre-set length, the condensation (C) domain of the NRPS module catalyzes the condensation of the PK chain and the thiolation domain-bound amino acid residue to complete the production of the straight-chain precursor PK–NP molecule (Fig. 7). The release of the precursor and the concomitant pyrrolidine-2-one formation catalyzed by the NRPS R domain is as shown in Fig. 4. After the tetramic acid is formed, a [4 + 2] cycloaddition reaction on the triene of the straight polyketide precursor catalyzed by a DAase proceeds to form the decalin moiety. The details of the DAase-catalyzed decalin formation are discussed in the next section.

Generalized biosynthetic pathway of pyrrolidine-2-one decalin-containing metabolites produced by the coordinated action of a polyketide synthase (blue)-nonribosomal synthetase (orange) hybrid enzyme, a stand-alone enoyl reductase (ER, blue) and a Diels–Alderase (green)
Biosynthesis of a complex natural product often involves auxiliary enzymes, such as glycosylases, methyltransferases and redox enzymes, that are required to modify the intermediate compounds to generate the final product. Such enzymes are usually encoded together with the genes for the core enzymes, such as PKSs and NRPSs, and regulatory and resistance factors in a single BGC to code for the complete metabolic pathway for the biosynthesis of the target compound. For biosynthesis of pyrrolidine-2-one type of decalin-containing compounds in fungi, generally a PKS–NRPS, an ER and a DAase are encoded in a BGC as a minimally required set of genes. In other words, if a BGC is found to contain genes for a PKS–NRPS, an ER and a DAase, such a BGC can potentially construct a biosynthetic pathway for the production of a decalin ring-containing compound.
Previous studies have shown that such BGCs are widely found in the genomes of various fungal species [104, 105], and variations among the genes encoded in those BCGs contribute to the structural diversity among the pyrrolidine-2-one type of decalin-containing compounds biosynthesized. As such, it is thought that there exist a vast number of decalin ring-containing compounds that are yet to be discovered in nature. In fact, Tang et al. identified the silenced pvh gene cluster from the genome of Penicillium variabile by using the above set of three genes as query sequence. Upon expressing five genes pdvA (PKS–NRPS), pdvB (DAase), pdvC (ER), pdvD (N-methyltransferase) and pdvE (cytochrome P450) heterologously in A. nidulans, the cis-decalin-containing compound 30 was successfully obtained [87]. Thus, searching for decalin ring-containing compounds by genome mining is an attractive and effective method for discovering novel metabolites. However, cytochalasan-type of compounds are also considered to be biosynthesized by a PKS–NRPS, an ER and a DAase in a manner that is similar to how decalin-containing compounds are biosynthesized [102]. Therefore, it is still difficult to discern with certainty what secondary metabolite a BGC is programed to produce just from the gene composition of the BGC alone. Furthermore, as was the case for the pvh gene cluster discussed above, there are many genetic and epigenetic silencing of secondary metabolite BGCs, especially in fungi. Activation of a silenced BGC through various approaches such as manipulation of transcriptional regulator genes or application of trichostatin A and other small-molecule epigenetic modifiers should facilitate exploration of the untapped potential of fungal biosynthetic capability and identification of various unique biological functions that those secondary metabolites possess [106, 107].
DAase catalyzing decalin ring formation of pyrrolidine-2-one compounds
Regarding the decalin ring-forming DAases involved in the biosynthesis of pyrrolidine-2-one-bearing decalins, Osada et al. reported that Fsa2 present in the BGC of 1 was responsible for the predicted IMDA reaction to form the decalin moiety in 1 [108]. This finding marked the starting point for the research field of DAase-catalyzed decalin biosynthesis. Following the report, Watanabe et al. also showed that CghA, which is also coded in the BGC of 13, is likely a decalin-forming DAase [59]. Initial amino acid sequence analysis indicated that both Fsa2 and CghA belonged to the lipocalin family of proteins with no clear assignment for their functions. One of the typical functions of lipocalin-type proteins is to bind hydrophobic compounds for transport [109]. Thus, it was speculated that Fsa2 and CghA might also function by binding to the hydrophobic polyketide chains. However, when the genes encoding both of these proteins were deleted from their respective biosynthetic pathways, the formation of unnatural products corresponding to the exo adducts of the presumed IMDA reactions was observed in addition to the formation of natural endo adducts (1 and 13, Fig. 8) [59, 108]. Based on those results, it was strongly implicated that Fsa2 and CghA were the enzymes catalyzing stereoselective DA reactions on linear triene polyketide substrates.

Stereoselective enzymatic Diels–Alder reactions catalyzed by CghA (blue) and Fsa2 (orange), showing the preferential generation of the endo adducts by the Diels–Alderases
While observations of lipocalin-type proteins such as Eqx3 for 1 [110], MycB for 14 [111]. UcsH for 26 [112] and PvhB for 30 [87] that carry out DA reactions for the formation of decalin moieties continued to accumulate, how those enzymes catalyze the cycloaddition reactions remained unclear. However, in a recent study the molecular basis of substrate recognition and catalysis of stereoselective pericyclic cycloaddition reaction by the lipocalinic DAase CghA involved in the biosynthesis of 13 was elucidated [39]. Watanabe et al. co-crystallized the recombinant CghA protein purified from E. coli and 13 and determined the enzyme–product complex structure by X-ray crystallography. Based on the structural information, the active site of CghA and the amino acid residues that interact with 13 were clearly delineated (Fig. 9). The large planar sidechain groups from two tryptophan residues Trp183 and Trp235 are coordinated to sandwich the core of the decalin ring, presumably to contribute toward immobilization of the triene polyketide straight chain of the bound substrate within the active site of the enzyme (Fig. 9a). It was also determined that Ser65 and Asn82 formed a water-mediated network of hydrogen bonds with the carbonyl groups of the tetramic acid moiety and the terminal carboxylic acid of the bound ligand. Similarly, Asn364 formed another hydrogen bond with the other carbonyl group of the tetramic acid moiety (Fig. 9a). Since hydrogen-bonding interactions can withdraw electron density and lower the LUMO energy of the dienophile to promote the DA cyclization reaction [26], it was inferred that those residues might also play a catalytic role in accelerating the DA reaction.

The crystal structure of the CghA–Sch 210,972 complex and engineering of CghA to generate a mutant capable of generating the unnatural exo adduct [39]. a The structural arrangement of the amino acid residues in the active site of the wild-type (WT) CghA (carbon atoms colored in green) that interact directly with the bound product Sch 210972 (carbon atoms colored in yellow) as observed in the crystal structure of the CghA–Sch 210972 complex. The nitrogen and oxygen atoms are colored in blue and red, respectively. b Apparent turnover rates of the conversion of a simplified substrate analog by the WT CghA and its mutants for the formation of the endo and exo adducts. c The amino acid residues (carbon atoms colored in blue) in the active site of the WT CghA in the vicinity of the bound Sch 210972. The sulfur atom is colored in yellow. d The amino acid residues (carbon atoms colored in blue) mutagenized within the active site to shift the diastereoselectivity of the Diels–Alder reaction from endo to exo. e Schematic illustrations of the interactions between the side chain groups at residues 242 and 257 and the bound transition state (TS) molecule, indicating the potential steric crashes that can determine the stereoselective outcome of the cycloaddition reaction
To examine these hypotheses on the role of individual active-site residues in catalysis, we prepared CghA with alanine mutation at those residues and analyzed the kinetics of the mutants. Characterization of the mutants confirmed that Trp183 and Trp235 play an important role in catalysis. While the single Trp183Ala mutation did not affect the DAase activity of CghA, the Trp235Ala single mutation reduced the activity by nearly 80%, and the Trp183Ala–Trp235Ala double mutant lost over 94% of the wild-type activity (Fig. 9b) [39]. Similarly, the activities of the Ser65Ala, Asn364Ala and Asn82 mutants were also significantly reduced, suggesting the critical role of hydrogen-bonding interactions at the tetramic acid portion of the molecule in promoting the decalin-forming DA reaction. The loss of activity by the Asn82Ala mutant was particularly interesting, as it indicated that a residue so far removed from the center of reaction could have such a dramatic effect on the catalytic activity of the enzyme. It appeared that Asn82 was important not only for binding the γ-hydroxymethylglutamate side chain to presumably assist in orienting and fixing the substrate within the binding pocket, but also in enhancing the electron-withdrawing ability of Ser65 to accelerate the cycloaddition reaction via the formation of a water-mediate long-range hydrogen-bonding network. The study shed light on the crucial role hydrogen-bonding interactions can play in enhancing the electron attractiveness of the dienophile to accelerate the DA reaction.
The above mutagenesis studies provided some insight into how CghA achieves rate enhancement of the DA reaction. However, they did not address the question of how the enzyme was controlling the stereoselectivity of the cycloaddition reaction. Based on some of the earlier studies of DAase catalytic antibodies [113], it was speculated that the enantioselectivity of CghA was brought about by the conformational constraints placed by the decalin-interfacing amino acid residues on the substrate to facilitate the alkyl chain to preferentially assume the endo transition-state structure. Specifically, it was surmised that residues Ala242 and Met257 around the diene moiety of the substrate were particularly important in establishing the shape complementarity of the active site to favor the endo conformation over the exo conformation (Fig. 9c). Therefore, a Ala242Ser mutation (Fig. 9d) was introduced to increase the steric hindrance against the endo transition state (Fig. 9e) while Met257 was shortened to a valine residue (Fig. 9d) to provide the space to accommodate the exo transition state within the binding pocket (Fig. 9e). By introducing those two mutations along with an additional Val391Leu mutation, CghA was successfully converted into a DAase that favored the formation of the disfavored exo adduct approximately three folds higher than the endo adduct [39]. This experiment indicated clearly that active site shape complementarity is key to the stereoselectivity exerted by DAases, and demonstrated the feasibility to engineer the enzyme to favor the formation of naturally disfavored adduct.
On the other hand, Kato et al. focused on the biosynthesis of 1 and 3 to uncover the mechanism of stereoselectivity imparted by the lipocalinic DAases on the DA reactions they catalyzed, because the decalin rings of the two compounds are enantiomeric to each other [114]. First, the genome sequence of the fungus Pyrenochaetopsis sp. RK10-F058 that produces 3 was searched for a homolog of fsa2 encoding the DAase that forms 1, and phm7 involved in the construction of the decalin ring of 3 was found. Subsequently, by substituting phm7 with fsa2 in Pyrenochaetopsis sp. RK10-F058, they succeeded in producing an unnatural product that was based on 3 with its decalin stereochemistry following that of 1 (i.e., 2 S,3R,6 S,11R), that is, completely opposite of that of 3 (i.e., 2R,3 S,6R,11 S) (Fig. 10). Thus, the study clearly demonstrated that the enzymes Phm7 and Fsa2 control the stereochemical outcome of the decalin-forming [4 + 2] cycloaddition reaction [114]. The result also suggested high substrate tolerance of the lipocalin-type DAase and opened a new way for producing analogs of pyrrolidine-2-one-bearing decalins having different stereochemistry using molecular genetic techniques. Most recently, the apo crystal structures of Fsa2 and Phm7 were reported [11, 115]. Through extensive computational analyses and docking experiments, a rational explanation as to how the two enzymes catalyze [4 + 2] cycloaddition reactions on the same substrate to generate enantiomerically opposite adducts. Furthermore, the study proposes a comprehensive model for the production of four different diastereomeric decalin scaffolds from the same tetramic acid-bearing linear triene substrate by four types of lipocalinic DAases. The key distinction is thought to be made by how the tetramic acid moiety of the substrate is oriented and the flexible triene polyketide chain is preorganized within the binding pocket of lipocalinic DAases [115]. Those studies will pave the way toward answering the long-standing question about naturally occurring DAases and engineering those fascinating catalysts for biotechnological applications.

The phomasetin DAase Phm7 catalyzes the [4 + 2] cycloaddition reaction on the phomasetin-type linear triene substrate to generate desmethylphomasetin with a 2R,3 S,6R,11 S decalin moiety, while the equisetin DAase Fsa2 converts the identical substrate into a equisetin-type adduct, whose 2 S,3R,6 S,11R decalin ring is an enantiomer of that found in desmethylphomasetin, demonstrating the opposite stereoselectivity exerted by those two DAases [114]
Conclusions
In this review, we described the biological activities and biosynthesis of natural products bearing a pyrrolidine-2-one side chain and a PK-derived decalin ring, especially the DAase enzymes that catalyze [4 + 2] cycloaddition reactions for the construction of the decalin ring. This review has focused strictly on lipocalin-type DAases, but there are also other types of DAases that have been discovered to catalyze similar decalin ring constructions. From the recent study on the biosynthesis of ilicicolins, 4-hydroxy-2-pyridone alkaloids, IccD was identified as the enzyme that catalyzes an inverse electron demand Diels–Alder (IEDDA) reaction. Although IccD was annotated initially as a C-methyltransferase based on the sequence information, heterologous expression of iccD in A. nidulans in the presence of a bis-diene linear precursor revealed that IccD was in fact a DAase that preferentially catalyzes an IEDDA reaction rather than a normal electron demand Diels–Alder (NEDDA) [116]. On the other hand, as opposed to the IEDDA reaction involved in the biosynthesis of ilicicolins, zopfiellamide type of compounds are generally considered to be biosynthesized through an NEDDA reaction, have also been reported (Fig. 11) [117]. The enzyme responsible for the formation of the decalin moiety of zopfiellamides or the mechanism of how the NEDDA reaction would be catalyzed by a presumed novel DAase have not been uncovered yet.

Top row: the DAase IccD catalyzing a decalin core-forming reaction on the bis-diene intermediate that proceeds via the inverse-electron demand Diels–Alder (IEDDA) pathway to generate the 4-hydroxy-2-pyridone type compound ilicicolin H [116]. Bottom row: the formation of the decalin core of the natural product zopfiellamide A [117] from its corresponding bis-diene intermediate is thought to proceed via the normal-electron demand Diels–Alder (NEDDA) pathway catalyzed by a yet-to-be-identified DAase
To date, lipocalin-type DAases that generate three out of the four possible structural isomers of decalin adducts that can be produced from a linear polyene substrate via a DA reaction have been identified (Fig. 12) [115]. The only remaining type of lipocalin-type DAase is the one that would produce the 2 S,3 S,6R,11R type of decalin-containing natural products represented by compounds such as 24, 23, 25 (Figs. 5 and 12). However, it will not be long before the last unknown DAase will be discovered to further deepen our knowledge of the lipocalinic DAases. With the ever-increasing rate of genome sequencing of fungi and other microorganisms, the rapid development of bioinformatics technologies and the advancement of molecular genetics in fungi, we expect speedy discovery of a wider variety of decalin ring-containing compounds and their biosynthetic DAases in the future by making full use of the power of genome mining, genetic manipulation, heterologous biosynthesis and enzyme engineering.

Lipocalin-type DAases can catalyze the generation of four diastereomeric decalin cores of pyrrolidine-2-one-bearing decalin natural products from an achiral linear polyene polyketide substrate [115]. Equisetin-forming Fsa2 represents the 2 S,3R,6 S,11R trans-decalin core-forming DAases. Sch 210,972-forming CghA and phomasetin-forming Phm7 represent the 2R,3 S,6R,11 S trans-decalin core-forming DAases, while varicidin A-forming PvhB represents the 2R,3R,6 S,11 S cis-decalin core-forming DAases. Only the 2 S,3 S,6R,11R cis-decalin core-forming DAase remains to be discovered
Availability of data and materials
Not applicable.
Abbreviations
- A:
-
Adenylation
- ACP:
-
Acyl carrier protein
- AT:
-
Acyltransferase
- BGC:
-
Biosynthetic gene cluster
- C:
-
Condensation
- DA:
-
Diels–Alder
- DAase:
-
Diels–Alderase
- DH:
-
Dehydratase
- ER:
-
Enoyl reductase
- HIV-1:
-
Human immunodeficiency virus type 1
- HMG-CoA:
-
3-Hydroxy-3-methylglutarylcoenzyme A
- HR-iPKS:
-
Highly-reducing iterative polyketide synthase
- IC50 :
-
Half-maximal inhibitory concentration
- IC90 :
-
90% maximal inhibitory concentration
- IEDDA:
-
Inverse electron demand Diels–Alder
- IMDA:
-
Intramolecular Diels–Alder
- ITS:
-
Internal transcribed spacer
- KR:
-
Ketoreductase
- KS:
-
Ketosynthase
- LUMO:
-
Lowest unoccupied molecular orbital
- LNKS:
-
Lovastatin-nonaketide synthase
- MIC:
-
Minimal inhibitory concentration
- MT:
-
Methyltransferase
- NEDDA:
-
Normal electron demand Diels–Alder
- NR:
-
Nonribosomal peptide
- NRPS:
-
Nonribosomal peptide synthetase
- PK:
-
Polyketide
- PKS:
-
Polyketide synthase
- PAC:
-
Proteasome assembly factor
- R:
-
Reduction
- ROS:
-
Reactive oxygen species
- T:
-
Thiolation
References
Stocking EM, Williams RM. Chemistry and biology of biosynthetic Diels–Alder reactions. Angew Chem Int Ed Engl. 2003;42(27):3078–115. https://doi.org/10.1002/anie.200200534.
Li G, Kusari S, Spiteller M. Natural products containing ‘decalin’ motif in microorganisms. Nat Prod Rep. 2014;31(9):1175–201. https://doi.org/10.1039/c4np00031e.
Hanson JR. Diterpenoids. Nat Prod Rep. 2009;26(9):1156–71. https://doi.org/10.1039/b807311m.
Hanson JR. Diterpenoids of terrestrial origin. Nat Prod Rep. 2013;30(10):1346–56. https://doi.org/10.1039/c3np70046a.
Baunach M, Franke J, Hertweck C. Terpenoid biosynthesis off the beaten track: unconventional cyclases and their impact on biomimetic synthesis. Angew Chem Int Ed Engl. 2015;54(9):2604–26. https://doi.org/10.1002/anie.201407883.
Christianson DW. Structural and chemical biology of terpenoid cyclases. Chem Rev. 2017;117(17):11570–648. https://doi.org/10.1021/acs.chemrev.7b00287.
Kelly WL. Intramolecular cyclizations of polyketide biosynthesis: mining for a “Diels–Alderase”? Org Biomol Chem. 2008;6(24):4483–93. https://doi.org/10.1039/b814552k.
Oikawa H, Katayama K, Suzuki Y, Ichihara A. Enzymatic activity catalysing exo-selective Diels–Alder reaction in solanapyrone biosynthesis. J Chem Soc Chem Commun. 1995. https://doi.org/10.1039/C39950001321.
Klas K, Tsukamoto S, Sherman DH, Williams RM. Natural Diels–Alderases: elusive and irresistable. J Org Chem. 2015;80(23):11672–85. https://doi.org/10.1021/acs.joc.5b01951.
Minami A, Oikawa H. Recent advances of Diels–Alderases involved in natural product biosynthesis. J Antibiot. 2016;69(7):500–6. https://doi.org/10.1038/ja.2016.67.
Chi C, Wang Z, Liu T, Zhang Z, Zhou H, Li A, et al. Crystal structures of Fsa2 and Phm7 catalyzing [4 + 2] cycloaddition reactions with reverse stereoselectivities in equisetin and phomasetin biosynthesis. ACS Omega. 2021;6(19):12913–22. https://doi.org/10.1021/acsomega.1c01593.
Brieger G, Bennett JN. The intramolecular Diels–Alder reaction. Chem Rev. 1980;80(1):63–97. https://doi.org/10.1021/cr60323a004.
Craig D. Stereochemical aspects of the intramolecular Diels–Alder reaction. Chem Soc Rev. 1987;16(0):187–238. https://doi.org/10.1039/cs9871600187.
Roush WR. Intramolecular Diels–Alder reactions. In: Trost BM, Fleming I, Paquette LA, editors. Comprehensive organic synthesis. Oxford: Pergamon Press; 1991. p. 513–50. https://doi.org/10.1016/B978-0-08-052349-1.00131-1.
Varner MA, Grossman RB. Annulation routes to trans-decalins. Tetrahedron. 1999;55(49):13867–86. https://doi.org/10.1016/s0040-4020(99)00851-0.
Singh V, Iyer SR, Pal S. Recent approaches towards synthesis of cis-decalins. Tetrahedron. 2005;61(39):9197–231. https://doi.org/10.1016/j.tet.2005.06.102.
Ichihara A, Tazaki H, Sakamura S. Solanapyrones A, B and C, phytotoxic metabolites from the fungus Alternaria solani. Tetrahedron Lett. 1983;24(48):5373–6. https://doi.org/10.1016/s0040-4039(00)87872-7.
Oikawa H, Yokota T, Sakano C, Suzuki Y, Naya A, Ichihara A. Solanapyrones, phytotoxins produced by Alternaria solani: biosynthesis and isolation of minor components. Biosci Biotechnol Biochem. 1998;62(10):2016–22. https://doi.org/10.1271/bbb.62.2016.
Mizushina Y, Kamisuki S, Kasai N, Shimazaki N, Takemura M, Asahara H, et al. A plant phytotoxin, solanapyrone A, is an inhibitor of DNA polymerase beta and lambda. J Biol Chem. 2002;277(1):630–8. https://doi.org/10.1074/jbc.M105144200.
Oikawa H, Yokota T, Abe T, Ichihara A, Sakamura S, Yoshizawa Y, et al. Biosynthesis of solanapyrone A, a phytotoxin of Alternaria solani. J Chem Soc Chem Commun. 1989. https://doi.org/10.1039/C39890001282.
Oikawa H, Yokota T, Ichihara A, Sakamura S. Structure and absolute configuration of solanapyrone D: a new clue to the occurrence of biological Diels–Alder reactions. J Chem Soc Chem Commun. 1989. https://doi.org/10.1039/C39890001284.
Oikawa H, Suzuki Y, Naya A, Katayama K, Ichihara A. First direct evidence in biological Diels–Alder reaction of incorporation of diene-dienophile precursors in the biosynthesis of solanapyrones. J Am Chem Soc. 1994;116(8):3605–6. https://doi.org/10.1021/ja00087a059.
Kasahara K, Miyamoto T, Fujimoto T, Oguri H, Tokiwano T, Oikawa H, et al. Solanapyrone synthase, a possible Diels–Alderase and iterative type I polyketide synthase encoded in a biosynthetic gene cluster from Alternaria solani. Chembiochem. 2010;11(9):1245–52. https://doi.org/10.1002/cbic.201000173.
Oikawa H, Kobayashi T, Katayama K, Suzuki Y, Ichihara A. Total synthesis of (–)-solanapyrone A via enzymatic Diels–Alder reaction of prosolanapyrone. J Org Chem. 1998;63(24):8748–56. https://doi.org/10.1021/jo980743r.
Breslow R. Hydrophobic effects on simple organic reactions in water. Acc Chem Res. 1991;24(6):159–64. https://doi.org/10.1021/ar00006a001.
Zheng Y, Thiel W. Computational insights into an enzyme-catalyzed [4 + 2] cycloaddition. J Org Chem. 2017;82(24):13563–71. https://doi.org/10.1021/acs.joc.7b02794.
Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292(5519):1160–4. https://doi.org/10.1126/science.1059344.
Endo A, Monacolin K. A new hypocholesterolemic agent produced by a Monascus species. J Antibiot. 1979;32(8):852–4. https://doi.org/10.7164/antibiotics.32.852.
Moore RN, Bigam G, Chan JK, Hogg AM, Nakashima TT, Vederas JC. Biosynthesis of the hypocholesterolemic agent mevinolin by Aspergillus terreus. Determination of the origin of carbon, hydrogen, and oxygen atoms by carbon-13 NMR and mass spectrometry. J Am Chem Soc. 1985;107(12):3694–701. https://doi.org/10.1021/ja00298a046.
Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science. 1999;284(5418):1368–72.
Auclair K, Sutherland A, Kennedy J, Witter DJ, Van den Heever JP, Hutchinson CR, et al. Lovastatin nonaketide synthase catalyzes an intramolecular Diels–Alder reaction of a substrate analogue. J Am Chem Soc. 2000;122(46):11519–20. https://doi.org/10.1021/ja003216+.
Ames BD, Nguyen C, Bruegger J, Smith P, Xu W, Ma S, et al. Crystal structure and biochemical studies of the trans-acting polyketide enoyl reductase LovC from lovastatin biosynthesis. Proc Natl Acad Sci USA. 2012;109(28):11144–9. https://doi.org/10.1073/pnas.1113029109.
Ma SM, Li JW, Choi JW, Zhou H, Lee KK, Moorthie VA, et al. Complete reconstitution of a highly reducing iterative polyketide synthase. Science. 2009;326(5952):589–92. https://doi.org/10.1126/science.1175602.
Xu W, Chooi YH, Choi JW, Li S, Vederas JC, Da Silva NA, et al. LovG: the thioesterase required for dihydromonacolin L release and lovastatin nonaketide synthase turnover in lovastatin biosynthesis. Angew Chem Int Ed Engl. 2013;52(25):6472–5. https://doi.org/10.1002/anie.201302406.
Royles BJL. Naturally occurring tetramic acids: structure, isolation, and synthesis. Chem Rev. 1995;95(6):1981–2001. https://doi.org/10.1021/cr00038a009.
Schobert R, Schlenk A. Tetramic and tetronic acids: an update on new derivatives and biological aspects. Bioorg Med Chem. 2008;16(8):4203–21. https://doi.org/10.1016/j.bmc.2008.02.069.
Nay B, Riache N, Evanno L. Chemistry and biology of non-tetramic gamma-hydroxy-gamma-lactams and gamma-alkylidene-gamma-lactams from natural sources. Nat Prod Rep. 2009;26(8):1044–62. https://doi.org/10.1039/b903905h.
Sims JW, Schmidt EW. Thioesterase-like role for fungal PKS–NRPS hybrid reductive domains. J Am Chem Soc. 2008;130(33):11149–55. https://doi.org/10.1021/ja803078z.
Sato M, Kishimoto S, Yokoyama M, Jamieson CS, Narita K, Maeda N, et al. Catalytic mechanism and endo-to-exo selectivity reversion of an octalin-forming natural Diels–Alderase. Nat Catal. 2021;4(3):223–32. https://doi.org/10.1038/s41929-021-00577-2.
Wheeler MH, Stipanovic RD, Puckhaber LS. Phytotoxicity of equisetin and epi-equisetin isolated from Fusarium equiseti and F. pallidoroseum. Mycol Res. 1999;103(8):967–73. https://doi.org/10.1017/S0953756298008119.
Burmeister HR, Bennett GA, Vesonder RF, Hesseltine CW. Antibiotic produced by Fusarium equiseti NRRL 5537. Antimicrob Agents Chemother. 1974;5(6):634–9. https://doi.org/10.1128/AAC.5.6.634.
Vesonder RF, Tjarks LW, Rohwedder WK, Burmeister HR, Laugal JA. Equisetin, an antibiotic from Fusarium equiseti NRRL 5537, identified as a derivative of N-methyl-2,4-pyrollidone. J Antibiot. 1979;32(7):759–61. https://doi.org/10.7164/antibiotics.32.759.
Yin J, Kong L, Wang C, Shi Y, Cai S, Gao S. Biomimetic synthesis of equisetin and (+)-fusarisetin A. Chem Eur J. 2013;19(39):13040–6. https://doi.org/10.1002/chem.201302163.
Jang JH, Asami Y, Jang JP, Kim SO, Moon DO, Shin KS, et al. Fusarisetin A, an acinar morphogenesis inhibitor from a soil fungus, Fusarium sp. FN080326. J Am Chem Soc. 2011;133(18):6865–7. https://doi.org/10.1021/ja1110688.
Singh SB, Zink DL, Goetz MA, Dombrowski AW, Polishook JD, Hazuda DJ. Equisetin and a novel opposite stereochemical homolog phomasetin, two fungal metabolites as inhibitors of HIV-1 integrase. Tetrahedron Lett. 1998;39(16):2243–6. https://doi.org/10.1016/s0040-4039(98)00269-x.
Hazuda D, Blau CU, Felock P, Hastings J, Pramanik B, Wolfe A, et al. Isolation and characterization of novel human immunodeficiency virus integrase inhibitors from fungal metabolites. Antivir Chem Chemother. 1999;10(2):63–70. https://doi.org/10.1177/095632029901000202.
Liu R, Li X, Huang N, Fan M, Sun R. Toxicity of traditional Chinese medicine herbal and mineral products. Adv Pharmacol. 2020;87:301–46. https://doi.org/10.1016/bs.apha.2019.08.001.
Li JY, Strobel G, Harper J, Lobkovsky E, Clardy J. Cryptocin, a potent tetramic acid antimycotic from the endophytic fungus Cryptosporiopsis cf. quercina. Org Lett. 2000;2(6):767–70. https://doi.org/10.1021/ol000008d.
Hellwig V, Grothe T, Mayer-Bartschmid A, Endermann R, Geschke FU, Henkel T, et al. Altersetin, a new antibiotic from cultures of endophytic Alternaria spp. Taxonomy, fermentation, isolation, structure elucidation and biological activities. J Antibiot. 2002;55(10):881–92. https://doi.org/10.7164/antibiotics.55.881.
Segeth MP, Bonnefoy A, Bronstrup M, Knauf M, Schummer D, Toti L, et al. Coniosetin, a novel tetramic acid antibiotic from Coniochaeta ellipsoidea DSM 13856. J Antibiot. 2003;56(2):114–22. https://doi.org/10.7164/antibiotics.56.114.
Marfori EC, Kajiyama S, Fukusaki E, Kobayashi A. Trichosetin, a novel tetramic acid antibiotic produced in dual culture of Trichoderma harzianum and Catharanthus roseus callus. Z Naturforsch C J Biosci. 2002;57(5–6):465–70. https://doi.org/10.1515/znc-2002-5-611.
Boros C, Dix A, Katz B, Vasina Y, Pearce C. Isolation and identification of cissetin—a setin-like antibiotic with a novel cis-octalin ring fusion. J Antibiot. 2003;56(10):862–5. https://doi.org/10.7164/antibiotics.56.862.
Hashimoto J, Watanabe T, Seki T, Karasawa S, Izumikawa M, Seki T, et al. Novel in vitro protein fragment complementation assay applicable to high-throughput screening in a 1536-well format. J Biomol Screen. 2009;14(8):970–9. https://doi.org/10.1177/1087057109341406.
Izumikawa M, Hashimoto J, Hirokawa T, Sugimoto S, Kato T, Takagi M, et al. JBIR-22, an inhibitor for protein–protein interaction of the homodimer of proteasome assembly factor 3. J Nat Prod. 2010;73(4):628–31. https://doi.org/10.1021/np900788e.
Donald RG, Skwish S, Forsyth RA, Anderson JW, Zhong T, Burns C, et al. A Staphylococcus aureus fitness test platform for mechanism-based profiling of antibacterial compounds. Chem Biol. 2009;16(8):826–36. https://doi.org/10.1016/j.chembiol.2009.07.004.
Herath K, Jayasuriya H, Zink DL, Sigmund J, Vicente F, de la Cruz M, et al. Isolation, structure elucidation, and antibacterial activity of methiosetin, a tetramic acid from a tropical sooty mold (Capnodium sp.). J Nat Prod. 2012;75(3):420–4. https://doi.org/10.1021/np200857y.
Osterhage C, Kaminsky R, König GM, Wright AD. Ascosalipyrrolidinone A, an antimicrobial alkaloid, from the obligate marine fungus Ascochyta salicorniae. J Org Chem. 2000;65(20):6412–7. https://doi.org/10.1021/jo000307g.
Yang S-W, Mierzwa R, Terracciano J, Patel M, Gullo V, Wagner N, et al. Chemokine receptor CCR-5 inhibitors produced by Chaetomium globosum. J Nat Prod. 2006;69(7):1025–8. https://doi.org/10.1021/np060121y.
Sato M, Yagishita F, Mino T, Uchiyama N, Patel A, Chooi YH, et al. Involvement of lipocalin-like CghA in decalin-forming stereoselective intramolecular [4 + 2] cycloaddition. Chembiochem. 2015;16(16):2294–8. https://doi.org/10.1002/cbic.201500386.
Yang Y-L, Lu C-P, Chen M-Y, Chen K-Y, Wu Y-C, Wu S-H. Cytotoxic polyketides containing tetramic acid moieties isolated from the fungus Myceliophthora thermophila: elucidation of the relationship between cytotoxicity and stereoconfiguration. Chem Eur J. 2007;13(24):6985–91. https://doi.org/10.1002/chem.200700038.
Sugie Y, Inagaki SAE, Kato Y, Nishida H, Pang C-H, Saito T, et al. CJ-21,058, a new SecA inhibitor isolated from a fungus. J Antibiot. 2002;55(1):25–9. https://doi.org/10.7164/antibiotics.55.25.
Ondeyka JG, Smith SK, Zink DL, Vicente F, Basilio A, Bills GF, et al. Isolation, structure elucidation and antibacterial activity of a new tetramic acid, ascosetin. J Antibiot. 2014;67(7):527–31. https://doi.org/10.1038/ja.2014.33.
Lang G, Blunt JW, Cummings NJ, Cole AL, Munro MH. Paecilosetin, a new bioactive fungal metabolite from a New Zealand isolate of Paecilomyces farinosus. J Nat Prod. 2005;68(5):810–1. https://doi.org/10.1021/np0500979.
Singh SB, Goetz MA, Jones ET, Bills GF, Giacobbe RA, Herranz L, et al. Oteromycin: a novel antagonist of endothelin receptor. J Org Chem. 1995;60(21):7040–2. https://doi.org/10.1021/jo00126a071.
West RR, Van Ness J, Varming AM, Rassing B, Biggs S, Gasper S, et al. ZG-1494 alpha, a novel platelet-activating factor acetyltransferase inhibitor from Penicillium rubrum, isolation, structure elucidation andbiological activity. J Antibiot. 1996;49(10):967–73. https://doi.org/10.7164/antibiotics.49.967.
Suzuki S, Hosoe T, Nozawa K, Kawai K, Yaguchi T, Udagawa S. Antifungal substances against pathogenic fungi, talaroconvolutins, from Talaromyces convolutus. J Nat Prod. 2000;63(6):768–72. https://doi.org/10.1021/np990371x.
Wang P, Cui Y, Cai C, Chen H, Dai Y, Chen P, et al. Two new succinimide derivatives cladosporitins A and B from the mangrove-derived fungus Cladosporium sp. HNWSW-1. Mar Drugs. 2018;17(1):4. https://doi.org/10.3390/md17010004.
Quek NC, Matthews JH, Bloor SJ, Jones DA, Bircham PW, Heathcott RW, et al. The novel equisetin-like compound, TA-289, causes aberrant mitochondrial morphology which is independent of the production of reactive oxygen species in Saccharomyces cerevisiae. Mol Biosyst. 2013;9(8):2125–33. https://doi.org/10.1039/c3mb70056a.
Kontnik R, Clardy J. Codinaeopsin, an antimalarial fungal polyketide. Org Lett. 2008;10(18):4149–51. https://doi.org/10.1021/ol801726k.
Koyama N, Nagahiro T, Yamaguchi Y, Ohshiro T, Masuma R, Tomoda H, et al. Spylidone, a novel inhibitor of lipid droplet accumulation in mouse macrophages produced by Phoma sp. FKI-1840. J Antibiot. 2005;58(5):338–45. https://doi.org/10.1038/ja.2005.42.
Mikawa T, Chiba N, Ogishi H, Sato Y, Miyaji S, Sezaki M. Antimicrobial formulations containing vermisporin. Jpn Kokai Tokkyo Koho. 1990;JP-02040328.
Sasaki T, Takagi M, Yaguchi M, Nishiyama K, Yaguchi T, Koyama M. Novel antibiotic PF1052 and its manufacture with Phoma species. Jpn Kokai Tokkyo Koho. 1992;JP-04316578.
Chin NX, Neu HC. In vitro antimicrobial activity of the new antibiotic vermisporin. Eur J Clin Microbiol Infect Dis. 1992;11(8):755–7. https://doi.org/10.1007/bf01989986.
Anoumedem EGM, Mountessou BYG, Kouam SF, Narmani A, Surup F. Simplicilones A and B isolated from the endophytic fungus Simplicillium subtropicum SPC3. Antibiotics. 2020;9(11):753. https://doi.org/10.3390/antibiotics9110753.
Onal G, Kutlu O, Gozuacik D, Dokmeci Emre S. Lipid droplets in health and disease. Lipids Health Dis. 2017;16(1):128. https://doi.org/10.1186/s12944-017-0521-7.
Hwang CH, Jaki BU, Napolitano JG, Lankin D, McAlpine JB, Franzblau SG, et al. Biological and chemical assessment of vermitrasporin, an anti-TB active metabolite from the fungus MSX 105528. Planta Med. 2012;78(11):PI90. https://doi.org/10.1055/s-0032-1320777.
Wang X, Robertson AL, Li J, Chai RJ, Haishan W, Sadiku P, et al. Inhibitors of neutrophil recruitment identified using transgenic zebrafish to screen a natural product library. Dis Model Mech. 2014;7(1):163–9. https://doi.org/10.1242/dmm.012047.
Nakai R, Ogawa H, Asai A, Ando K, Agatsuma T, Matsumiya S, et al. UCS1025A, a novel antibiotic produced by Acremonium sp. J Antibiot. 2000;53(3):294–6. https://doi.org/10.7164/antibiotics.53.294.
Agatsuma T, Akama T, Nara S, Matsumiya S, Nakai R, Ogawa H, et al. UCS1025A and B, new antitumor antibiotics from the fungus Acremonium species. Org Lett. 2002;4(25):4387–90. https://doi.org/10.1021/ol026923b.
Nakai R, Ishida H, Asai A, Ogawa H, Yamamoto Y, Kawasaki H, et al. Telomerase inhibitors identified by a forward chemical genetics approach using a yeast strain with shortened telomere length. Chem Biol. 2006;13(2):183–90. https://doi.org/10.1016/j.chembiol.2005.11.010.
Sugie Y, Hirai H, Kachi-Tonai H, Kim YJ, Kojima Y, Shiomi Y, et al. New pyrrolizidinone antibiotics CJ-16,264 and CJ-16,367. J Antibiot. 2001;54(11):917–25. https://doi.org/10.7164/antibiotics.54.917.
Nogawa T, Kawatani M, Uramoto M, Okano A, Aono H, Futamura Y, et al. Pyrrolizilactone, a new pyrrolizidinone metabolite produced by a fungus. J Antibiot. 2013;66(10):621–3. https://doi.org/10.1038/ja.2013.55.
Futamura Y, Kawatani M, Muroi M, Aono H, Nogawa T, Osada H. Identification of a molecular target of a novel fungal metabolite, pyrrolizilactone, by phenotypic profiling systems. ChemBioChem. 2013;14(18):2456–63. https://doi.org/10.1002/cbic.201300499.
Baral B, Akhgari A, Metsä-Ketelä M. Activation of microbial secondary metabolic pathways: avenues and challenges. Synth Syst Biotechnol. 2018;3(3):163–78. https://doi.org/10.1016/j.synbio.2018.09.001.
Chevrette MG, Gavrilidou A, Mantri S, Selem-Mojica N, Ziemert N, Barona-Gómez F. The confluence of big data and evolutionary genome mining for the discovery of natural products. Nat Prod Rep. 2021;38(11):2024–40. https://doi.org/10.1039/d1np00013f.
Sato M, Dander JE, Sato C, Hung YS, Gao SS, Tang MC, et al. Collaborative biosynthesis of maleimide- and succinimide-containing natural products by fungal polyketide megasynthases. J Am Chem Soc. 2017;139(15):5317–20. https://doi.org/10.1021/jacs.7b02432.
Tan D, Jamieson CS, Ohashi M, Tang MC, Houk KN, Tang Y. Genome-mined Diels–Alderase catalyzes formation of the cis-octahydrodecalins of varicidin A and B. J Am Chem Soc. 2019;141(2):769–73. https://doi.org/10.1021/jacs.8b12010.
Nogawa T, Kato N, Shimizu T, Okano A, Futamura Y, Takahashi S, et al. Wakodecalines A and B, new decaline metabolites isolated from a fungus Pyrenochaetopsis sp. RK10-F058. J Antibiot. 2018;71(1):123–8. https://doi.org/10.1038/ja.2017.103.
Osada H, Nogawa T. Systematic isolation of microbial metabolites for natural products depository (NPDepo). Pure Appl Chem. 2011;84(6):1407–20. https://doi.org/10.1351/PAC-CON-11-08-11.
Kato N, Takahashi S, Nogawa T, Saito T, Osada H. Construction of a microbial natural product library for chemical biology studies. Curr Opin Chem Biol. 2012;16(1–2):101–8. https://doi.org/10.1016/j.cbpa.2012.02.016.
Yamada T, Kikuchi T, Tanaka R, Altercrasin A. A novel decalin derivative with spirotetramic acid, produced by a sea urchin-derived Alternaria sp. Tetrahedron Lett. 2015;56(10):1229–32. https://doi.org/10.1016/j.tetlet.2015.01.066.
Yamada T, Tanaka A, Nehira T, Nishii T, Kikuchi T. Altercrasins A–E, decalin derivatives, from a sea-urchin-derived Alternaria sp.: isolation and structural analysis including stereochemistry. Mar Drugs. 2019;17(4):218. https://doi.org/10.3390/md17040218.
Sauer J, Sustmann R. Mechanistic aspects of Diels–Alder reactions: a critical survey. Angew Chem Int Ed Engl. 1980;19(10):779–807. https://doi.org/10.1002/anie.198007791.
Okino T, Yoshimura E, Hirota H, Fusetani N. Antifouling kalihinenes from the marine sponge Acanthella cavernosa. Tetrahedron Lett. 1995;36(47):8637–40. https://doi.org/10.1016/0040-4039(95)01861-b.
Uchida I, Ando T, Fukami N, Yoshida K, Hashimoto M, Tada T, et al. The structure of vinigrol, a novel diterpenoid with antihypertensive and platelet aggregation-inhibitory activities. J Org Chem. 1987;52(23):5292–3. https://doi.org/10.1021/jo00232a048.
Ando T, Tsurumi Y, Ohata N, Uchida I, Yoshida K, Okuhara M. Vinigrol, a novel antihypertensive and platelet aggregation inhibitory agent produced by a fungus, Virgaria nigra. I. Taxonomy, fermentation, isolation, physicochemical and biological properties. J Antibiot. 1988;41(1):25–30. https://doi.org/10.7164/antibiotics.41.25.
Ando T, Yoshida K, Okuhara M. Vinigrol, a novel antihypertensive and platelet aggregation inhibitory agent produced by a fungus, Virgaria nigra. II. Pharmacological characteristics. J Antibiot. 1988;41(1):31–5. https://doi.org/10.7164/antibiotics.41.31.
Ishida K, Ishibashi M, Shigemori H, Sasaki T, Kobayashi J, Agelasine G. A new antileukemic alkaloid from the Okinawan marine sponge Agelas sp. Chem Pharm Bull. 1992;40(3):766–7. https://doi.org/10.1248/cpb.40.766.
Yamazaki H, Kanno SI, Abdjul DB, Namikoshi M. A bromopyrrole-containing diterpene alkaloid from the Okinawan marine sponge Agelas nakamurai activates the insulin pathway in Huh-7 human hepatoma cells by inhibiting protein tyrosine phosphatase 1B. Bioorg Med Chem Lett. 2017;27(10):2207–9. https://doi.org/10.1016/j.bmcl.2017.03.033.
Kobayashi M, Nakamura H, Wu HM, Kobayashi J, Ohizumi Y. Mode of inhibition of brain Na+, K+-ATPase by agelasidines and agelasines from a sea sponge. Arch Biochem Biophys. 1987;259(1):179–84. https://doi.org/10.1016/0003-9861(87)90484-x.
Song Z, Cox RJ, Lazarus CM, Simpson TT. Fusarin C biosynthesis in Fusarium moniliforme and Fusarium venenatum. ChemBioChem. 2004;5(9):1196–203. https://doi.org/10.1002/cbic.200400138.
Li H, Booth TJ, Chooi YH. Fungal polyketide–nonribosomal peptide synthetases and their associated natural products. In: Liu H-W, Begley T, editors. Comprehensive natural products III. San Diego: Elsevier Science; 2020. p. 415–44. https://doi.org/10.1016/B978-0-12-409547-2.14677-3.
Boettger D, Hertweck C. Molecular diversity sculpted by fungal PKS–NRPS hybrids. Chembiochem. 2013;14(1):28–42. https://doi.org/10.1002/cbic.201200624.
Bushley KE, Turgeon BG. Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol Biol. 2010;10:26. https://doi.org/10.1186/1471-2148-10-26.
Li YF, Tsai KJS, Harvey CJB, Li JJ, Ary BE, Berlew EE, et al. Comprehensive curation and analysis of fungal biosynthetic gene clusters of published natural products. Fungal Genet Biol. 2016;89:18–28. https://doi.org/10.1016/j.fgb.2016.01.012.
Kjærbølling I, Mortensen UH, Vesth T, Andersen MR. Strategies to establish the link between biosynthetic gene clusters and secondary metabolites. Fungal Genet Biol. 2019;130:107–21. https://doi.org/10.1016/j.fgb.2019.06.001.
Keller NP, Turner G, Bennett JW. Fungal secondary metabolism—from biochemistry to genomics. Nat Rev Microbiol. 2005;3(12):937–47. https://doi.org/10.1038/nrmicro1286.
Kato N, Nogawa T, Hirota H, Jang JH, Takahashi S, Ahn JS, et al. A new enzyme involved in the control of the stereochemistry in the decalin formation during equisetin biosynthesis. Biochem Biophys Res Commun. 2015;460(2):210–5. https://doi.org/10.1016/j.bbrc.2015.03.011.
Flower DR, North ACT, Sansom CE. The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta. 2000;1482(1–2):9–24. https://doi.org/10.1016/s0167-4838(00)00148-5.
Kakule TB, Jadulco RC, Koch M, Janso JE, Barrows LR, Schmidt EW. Native promoter strategy for high-yielding synthesis and engineering of fungal secondary metabolites. ACS Synth Biol. 2015;4(5):625–33. https://doi.org/10.1021/sb500296p.
Li L, Yu P, Tang MC, Zou Y, Gao SS, Hung YS, et al. Biochemical characterization of a eukaryotic decalin-forming Diels–Alderase. J Am Chem Soc. 2016;138(49):15837–40. https://doi.org/10.1021/jacs.6b10452.
Li L, Tang MC, Tang S, Gao S, Soliman S, Hang L, et al. Genome mining and assembly-line biosynthesis of the UCS1025A pyrrolizidinone family of fungal alkaloids. J Am Chem Soc. 2018;140(6):2067–71. https://doi.org/10.1021/jacs.8b00056.
Cannizzaro CE, Ashley JA, Janda KD, Houk KN. Experimental determination of the absolute enantioselectivity of an antibody-catalyzed Diels–Alder reaction and theoretical explorations of the origins of stereoselectivity. J Am Chem Soc. 2003;125(9):2489–506. https://doi.org/10.1021/ja020879d.
Kato N, Nogawa T, Takita R, Kinugasa K, Kanai M, Uchiyama M, et al. Control of the stereochemical course of [4 + 2] cycloaddition during trans-decalin formation by Fsa2-family enzymes. Angew Chem Int Ed Engl. 2018;57(31):9754–8. https://doi.org/10.1002/anie.201805050.
Fujiyama K, Kato N, Re S, Kinugasa K, Watanabe K, Takita R, et al. Molecular basis for two stereoselective Diels–Alderases that produce decalin skeletons. Angew Chem Int Ed Engl. 2021;60(41):22401–10. https://doi.org/10.1002/anie.202106186.
Zhang Z, Jamieson CS, Zhao YL, Li D, Ohashi M, Houk KN, et al. Enzyme-catalyzed inverse-electron demand Diels–Alder reaction in the biosynthesis of antifungal ilicicolin H. J Am Chem Soc. 2019;141(14):5659–63. https://doi.org/10.1021/jacs.9b02204.
Daferner M, Anke T, Sterner O. Zopfiellamides A and B, antimicrobial pyrrolidinone derivatives from the marine fungus Zopfiella latipes. Tetrahedron. 2002;58(39):7781–4. https://doi.org/10.1016/s0040-4020(02)00942-0.
Xie X, Watanabe K, Wojcicki WA, Wang CC, Tang Y. Biosynthesis of lovastatin analogs with a broadly specific acyltransferase. Chem Biol. 2006;13(11):1161–9. https://doi.org/10.1016/j.chembiol.2006.09.008.
Acknowledgements
This review was written in response to an invite of “Beyond the assembly line—showcasing the complexities of fungal natural product biosynthesis” in 2022.
Funding
This work was supported in part by the Japan Society for the Promotion of Science (JSPS) (K.W., G2604, 19K22291, 19H02898, 19KK0150; M.S., 19K07136; H.O., 17H06412, 21H04720) and Innovative Areas from MEXT, Japan (K.W., 16H06449).
Author information
Authors and Affiliations
Contributions
HO, MS and KW conceptualized and co-drafted the manuscript. MS designed and prepared the table and figures. HO, MS and KW discussed the manuscript. HO, MS and KW revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Watanabe, K., Sato, M. & Osada, H. Recent advances in the chemo-biological characterization of decalin natural products and unraveling of the workings of Diels–Alderases. Fungal Biol Biotechnol 9, 9 (2022). https://doi.org/10.1186/s40694-022-00139-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40694-022-00139-6