
Abstract
Background
Gene editing using CRISPR/Cas9 is a widely used tool for precise gene modification, modulating gene expression and introducing novel proteins, and its use has been reported in various filamentous fungi including the genus Fusarium. The aim of this study was to optimise gene editing efficiency using AMA1 replicator vectors for transient expression of CRISPR constituents in Fusarium venenatum (A3/5), used commercially in the production of mycoprotein (Quorn™).
Results
We present evidence of CRISPR/Cas9 mediated gene editing in Fusarium venenatum, by targeting the endogenous visible marker gene PKS12, which encodes a polyketide synthase responsible for the synthesis of the pigment aurofusarin. Constructs for expression of single guide RNAs (sgRNAs) were cloned into an AMA1 replicator vector incorporating a construct for constitutive expression of cas9 codon-optimised for Aspergillus niger or F. venenatum. Vectors were maintained under selection for transient expression of sgRNAs and cas9 in transformed protoplasts. 100% gene editing efficiency of protoplast-derived isolates was obtained using A. niger cas9 when sgRNA transcription was regulated by the F. venenatum 5SrRNA promoter. In comparison, expression of sgRNAs using a PgdpA-ribozyme construct was much less effective, generating mutant phenotypes in 0–40% of isolates. Viable isolates were not obtained from protoplasts transformed with an AMA1 vector expressing cas9 codon-optimised for F. venenatum.
Conclusions
Using an AMA1 replicator vector for transient expression of A. niger cas9 and sgRNAs transcribed from the native 5SrRNA promoter, we demonstrate efficient gene editing of an endogenous marker gene in F. venenatum, resulting in knockout of gene function and a visible mutant phenotype in 100% of isolates. This establishes a platform for further development of CRISPR/Cas technology in F. venenatum for use as a research tool, for understanding the controls of secondary metabolism and hyphal development and validating prototypes of strains produced using traditional methods for strain improvement.
Similar content being viewed by others

Background
Quorn™ myco-protein is a high fibre, high quality protein with a significantly lower environmental impact compared to that of beef, produced by fermentation of Fusarium venenatum (ATCC 20334, A3/5) utilising wheat-derived glucose in a continuous fermentation process. The organism was selected for its efficient biomass conversion, nutritional value, sparsely branched mycelium which enables processing into a meat-like texture and absence of undesirable secondary metabolites under optimal growth conditions [1,2,3]. F. venenatum has no known sexual cycle and therefore strain improvement is only currently possible thorough clonal mutagenesis. There are multiple opportunities for strain improvement, including use of alternative carbon sources, which may require strain development for optimal growth and mycoprotein quality. These advances could be facilitated by biotechnological innovations including development of gene editing mediated by CRISPR/Cas9, for use currently as a research tool, and possibly for use in future as a method for strain improvement.
The aim of the current study was to optimise CRISPR/Cas9 mediated gene editing in F. venenatum and establish a platform for its use in understanding genetic controls of metabolic pathways and hyphal development. CRISPR/Cas9 is a well-established technology, successfully used in a wide range of organisms for precise gene editing, gene insertion and modulation, facilitating genomic functional analyses and production of novel compounds. The Cas9 enzyme, guided by a sequence-specific RNA complex (sgRNA), generates a double stranded break at the target site in the genome, which can be repaired by either the non-homologous end joining (NHEJ) or microhomology –mediated end joining mechanisms (which are error prone), or by homology-directed repair (HR) in the presence of a repair template [4, 5].
CRISPR/Cas9 mediated gene editing in filamentous fungi was first reported in Aspergillus [6, 7], Neurospora crassa [8] and Trichoderma reesei [9] and subsequent reports include use of CRISPR/Cas9 in Penicillium chrysogenum [10], Ustilago maydis [11] and Magnaporthe oryzae [12]. Its application in Fusarium was reported initially in F. graminearum [13] and F. oxysporum [14] and subsequently in F. proliferatum [15] and F. fujikuroi [16].
Parameters for optimising the methodology in fungi have included use of various delivery systems for CRISPR constituents, evaluation of promoters driving sgRNA expression and codon optimisation of cas9 and its fusion to appropriate protein nuclear localization signals (NLS).
Strategies used for delivery of CRISPR constituents in filamentous fungi including Fusarium include generation of stable transgenic lines for expression of cas9 and sgRNAs [6, 12, 13], introduction of in vitro expressed sgRNAs [9, 16, 17], introduction of Cas9/sgRNA ribonucleic protein (RNP) complexes [10, 12, 14, 15, 17, 18], and transformation with expression vectors for transient production of Cas9 and sgRNAs in vivo [8, 16]. Additionally, sgRNAs produced in vitro have been used in Aspergillus [19] and AMA1 replicator vectors for transient expression of sgRNAs and cas9 in vivo have also been used successfully [7, 10, 11], but their use has not been reported for the genus Fusarium.
PolIII promoters are commonly used to drive sgRNA expression in fungi including Fusarium, such as the yeast SNR52 promoter [6, 8] and endogenous promoters for U6 [9,10,11, 16, 18], tRNA [10], U3 [20] and 5SrRNA [16, 19], and in Aspergillus and Fusarium sgRNAs were embedded in a dual ribozyme driven by a PolII promoter [7, 13].
Successful editing in fungi has been achieved using Cas9 proteins optimised for other organisms, such as human-optimized Cas9 (hSpCas9) [6, 8, 12, 15, 18] and A. niger optimised Cas9 for gene editing in F. oxysporum [14], or Cas9 optimised for the target organism including Fusarium [7, 9, 13, 16].
We report on use of the AMA1 vector system for transient expression of cas9 and sgRNAs via protoplast transformation, which has not previously been reported for Fusarium, and evaluate gene editing efficiency of sgRNAs transcribed using both an endogenous 5SrRNA promoter (PFv5SRNA) and the PolII gdpA promoter (PgdpA) dual ribozyme system. We additionally compare gene editing efficiency of a F. venenatum codon optimised Cas9 (Fv Cas9) with Cas9 optimised for A. niger (A. niger Cas9), which has previously been used for gene editing of Fusarium [12].
Results
CRISPR vectors
sgRNAs targeting the endogenous PKS12 marker gene were assembled with PFv5SRNA or PgdpA (Fig. 1), for transcription from AMA1 vectors expressing either Fvcas9 (pFCFvCas9::5S-PK3/14) or A. niger cas9 (pFC332::5S-PK3/14 and pFC332::PolII-PK3/14).

Schematic maps of single guide RNA (sgRNA) expression constructs and the Fusarium venenatum PKS12 gene indicating the target site used for CRISPR/Cas9 editing. A Fv5SrRNA promoter-sgRNA construct, showing positions of PFv5SrRNA, 20 base target site sequence (yellow and black striped box), scaffold sequence (Sc) and terminator (T). B PgdpA dual ribozyme-sgRNA construct showing positions of PgdpA, inverted repeat (blue arrow) of first six bases in target sequence (yellow and black box), HH and HDV ribozyme sequences and trpC terminator (TtrpC). C F. venenatum PKS12 gene showing exons (yellow arrows) and the location of the CRISPR target sequence (pink arrow) in exon 3, 3-14 (‘14’) antisense strand, sequence position 3’–5’ = 238-257
Transformants obtained using CRISPR vectors, phenotypes and sequence analysis of isogenic isolates
A replicated experiment was performed to obtain transformed protoplast derived colonies, for a comparison of editing efficiency of Fvcas9 with A. niger cas9, and to compare efficacy of sgRNA promoters PFv5SrRNA and PgdpA. For each experiment three replicate transformations were performed with each vector and after 10 days the number of mycelial colonies observed on transformation plates was recorded (Table 1). A total of only 2 colonies developed from protoplasts transformed with empty vector pFCFvCas9 expressing Fvcas9, compared to 35 from protoplasts transformed with empty vector pFC332 expressing A. niger cas9 and only one colony developed from protoplasts transformed with pFCFvCas9::PolII-PK3/14. Using pFC332 vectors, fewer colonies developed when sgRNAs were transcribed from the Fv5SrRNA promoter compared to PgdpA: a total of 12 transformants were obtained using pFC332::5S-PK3/14 compared to a total of 55 following transformation with pFC332::PolII-PK3/14.
Viability of colonies taken from transformation experiment 2 was subsequently assessed (Table 2): viable cultures were not recovered from transformants with vectors expressing Fvcas9 (pFCFvCas9 or pFCFvCas9::5 SPK-3/14), and viability of transformants expressing A. niger cas9 was less when sgRNAs were transcribed using PFv5SrRNA (7 of 12 pFCFvCas9::5SPK-3/14 transformants were viable) compared to transcription from PgdpA (all of 27 pFC332::PolII-PK3/14 transformants were viable).
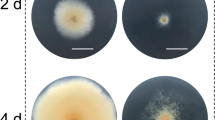
From each of the viable colonies recovered, at least 7 single spore isolates were cultured on non-selective PSA plates for phenotypic screening (Fig. 2). Albino phenotypes, indicative of non-functional PKS12 protein [21] were obtained from all single spore isolates of pFC332::5S-PK3/14 transformed protoplasts, but only 30–40% of isolates, of just 3 of the 11 colonies from pFC332::PolII-PK3/14 transformed protoplasts (Table 3). Sequence data from target site amplicons show a single adenine base insertion at position −1 of the cut site in albino variants (Additional file 1: Table S1), except isolates from colony 5–7 from pFC332::PolII-PK3/14 transformed protoplasts for which no evidence of target site indels was obtained.

Phenotypes on potato sucrose agar of F. venenatum A3/5 (a) and PKS12 gene CRISPR/Cas9 variants (b)
Discussion
This is the first report of CRISPR/Cas mediated gene editing in the industrially important fungus F. venenatum, used in production of mycoprotein. We demonstrate efficient transgene-free target site mutation of the endogenous marker gene PKS12 by transient expression of CRISPR constituents from an AMA1 replicator vector, use of which has been reported for other fungi but not the genus Fusarium.
Use of an autonomously replicating vector for transient expression of CRISPR reagents and selection markers avoids the difficulties associated with in-vitro generated unstable sgRNAs, making this an attractive system for generating transgene-free improved strains of F. venenatum and enables use of the same selection marker for successive gene editing Most isolates obtained were unable to grown on hygromycin after a period of culture on non-selective medium (Additional file 2: Table S2), indicating loss of the AMA1 vector, and the continuing hygromycin resistance observed in a small number of isolates suggests either possible chromosomal integration of vector elements (including the hph gene for hygromycin resistance), or residual extrachromosomal vector in some nuclei (which may be due to higher initial copy number within some isolates). Eventual loss from all nuclei would be expected through continued culture in non-selective conditions, as reported by Leeuwe et al. [22], or after repeated single spore isolation [7].
High efficiency editing is a prerequisite for enabling low input screening of transformants to identify target site variants and may be achieved by optimising levels of sgRNA transcription and nuclear-localised Cas9. Efficiencies of editing in filamentous fungi have been shown to vary depending on the promoter driving sgRNA transcription and between different species and experimental systems, e.g. Nodvig et al.[7, 20] speculate that differences in gene editing efficiency observed in Aspergillus may depend on different requirements for cas9 codon usage and levels of cas9 and sgRNA, which may vary in correlation with AMA1 vector copy number and also promoter, reporting higher efficiencies from sgRNAs expressed from an endogenous PolIII U3 promoter compared to the polymerase II/ribozyme-based system. Likewise, Shi et al.[16] reported enhanced gene editing in F. fujikuroi using promoters for highly expressed PolIII endogenous genes, but they were unsuccessful using the heterologous PolII (PgdpA) dual ribozyme system. We demonstrate similar results in F. venenatum using an AMA1 vector for in vivo production of CRISPR reagents, achieving edits in 100% of recovered isolates when sgRNAs were transcribed from the native Fv5SrRNA promoter and greatly reduced efficacy using the PgdpA ribozyme system, and RT-qPCR results from our preliminary experiments (Additional file 3: Figure S1, Table S3) suggest relatively low levels of transcription from PgdpA in AMA1 vector transformants.
The importance of the NLS in determining efficiency of Cas9 protein nuclear import and subsequent gene editing success has also been highlighted by several studies. SV40NLS has been used widely for Cas9 nuclear import in eukaryotes including other filamentous fungi [7, 8, 12, 20]; however in Fusarium, Wang et al.[14] and Shi et al.[16] were unsuccessful in obtaining gene edits using Cas9-SV40NLS delivered as a constituent of a RNP or transcribed from an expression vector (but were successful using a Cas9-HBTNLS protein). In our experiments, we were successful in obtaining gene edits using cas9-SV40NLS transcripts, possibly as relatively high levels of transcription can be obtained from genes driven by the tef1 promoter, when expressed from an autonomously replicating vector (Additional file 3: Figure S1, Table S3).
Cas9 optimization is another key factor reported to influence gene editing success in filamentous fungi, and differences in reported effectiveness and toxicity may be ascribed to codon usage as well as potential differences in levels of nuclear Cas9 resulting from the different methodologies employed. Foster et al.[12] reported toxicity of Cas9 optimized for both N. crassa and the target organism in M. oryzae when cas9 was expressed constitutively from integrated transgenes but they were able to recover gene edited mutants using hSpCas9 delivered transiently as RNPs. Fuller et al.[6] found no evidence of toxicity from hSpCas9 in Aspergillus transgenic lines, likewise Shi et al. [16] observed no effect on growth of F. fujikuroi of optimised FuCas9 using expression vectors, and in F. graminearum Gardiner and Kazan [13] were successful with F. graminearum optimised Cas9 produced in transgenic lines. However, in our experiments using an AMA1 vector for transcription of cas9 using the tef1 promoter we were unable to recover viable colonies from protoplasts expressing Cas9 optimised for F. venenatum and recovery rates were low after transformation with vectors expressing A. niger Cas9, which is indicative of potential Cas9 toxicity (see also Additional file 4: Table S4). Additionally, our results show reduced viability of protoplasts and mycelium expressing PFv5SrRNA-sgRNAs compared to those expressing PgdpA-sgRNAs, suggesting a possible interaction between sgRNA transcription levels and Cas9 toxicity.
Conclusions
In conclusion, we have demonstrated 100% gene editing frequency in isolates of F. venenatum by transforming protoplasts with AMA1 replicator vectors expressing A. niger codon-optimised cas9-SV40NLS transcribed from the tef1 promoter and sgRNAs transcribed from the endogenous Fv5SrRNA promoter. Evidence of AMA1 vector loss in the absence of selection makes this an attractive system for generating transgene-free gene edited strains of F. venenatum and for performing successive gene edits using the same selection marker. However, reduced protoplast viability due to potential Cas9 toxicity was observed, which may impede development of further CRISPR/Cas technologies such as gene insertion, base editing and prime editing in F. venenatum. Assessment of the efficacy of alternative promoters and/or use of an inducible system for regulating cas9 expression would enable optimization of Cas9 production and allow re-evaluation of Cas9 codon-optimized for F. venenatum, which was apparently toxic when expressed from the tef1 promoter using an AMA1 vector. Although each of the elements for gene editing reported here have been described previously in fungi, such as use of AMA1 vectors for delivery of CRISPR constituents and use of PolII-ribozyme and 5SrRNA promotors for driving sgRNA expression, this is the first report of using an AMA1 vector for gene editing in Fusarium and the findings reported here should also be of interest more widely for transgene-free gene editing in fungi. Implementation of the gene editing methodology described here, and development of additional CRISPR/Cas technologies will further an understanding of the controls governing growth and metabolite synthesis in F. venenatum, will aid identification of targets for strain improvement using traditional methods and will facilitate innovation in fermentation development.
Methods
Vector construction
Vector backbones were obtained by restriction digest and component parts for vector inserts were generated by PCR from synthesised DNA or existing vector templates using Q5® High-Fidelity 2X Master Mix (NEB). Vector components were purified using the Monarch® DNA Gel Extraction Kit or the Monarch® PCR & DNA Cleanup Kit (NEB) and assemblies were performed using NEBuilder® HiFi DNA Assembly Master Mix (NEB). For details of primers used in vector construction see Additional file 5: Table S5. Recombinant vectors were recovered by transformation of NEB® 10-beta Competent E. coli (High Efficiency) cells and subsequent plasmid isolation was performed using the NucleoSpin Plasmid kit (Macherey–Nagel). For comparison of Fv Cas9 and A. niger Cas 9 a codon-optimised FvCas9 gene was synthesised and an expression cassette Ptef1-Fvcas9-SV40-Ttef1 was assembled (Additional file 6: Table S6) and ligated with the backbone of AMA1 vector pFC332 [7], replacing the A. niger cas9 cassette to give vector pFCFvCas9. These vectors were used as ‘empty vector’ controls in transformation experiments. The sequence for the target site (‘14’) in exon 3 of the endogenous PKS12 gene was cloned downstream of promoter PgdpA within a dual ribozyme cassette [7] (giving construct PolII-PK3/14) or promoter PFv5SrRNA (giving construct 5S-PK3/14) (Fig. 1, and see Additional file 7: Table S7 for PFv5SrRNA cassette sequence). Promoter-sgRNA constructs were cloned into the USER site of pFC332 to give single sgRNA vectors pFC332::PolII-PK3/14 and pFC332::5S-PK3/14. For comparison of gene editing efficiency with Fvcas9, PolII-PK3/14 was additionally cloned into the USER site of vector pFCFvCas9 to give pFCFvCas9::PolII-PK3/14.
Details of vector components and assembly are as follows: codon usage preference of predicted highly expressed genes for F. venenatum A3/5 was determined (Additional file 8: Table S8) and used to generate a codon-optimised sequence for cas9. Using Optimizer [23] ‘Relative Synonymous Codon Usage’ (RSCU) was performed, selecting options for ‘standard genetic code’ and ‘one amino acid—one codon’. The coding sequence was subsequently domesticated to enable use in Golden Gate cloning if required and DNA for the resulting sequence, (Additional file 6: Table S6), was synthesised (GeneArt, Invitrogen). The AMA1 replication vector backbone (pFCBB) of 10,129 bases was obtained from pFC332 [7] by restriction digest using PacI and BamHI-HF® (NEB), excising part of the Nt.BbvCI–PacI cloning site, the tef1 promoter, cas9 coding sequence and 489 bases (5’-3’) of the tef1 terminator. The residual tef1 terminator (87 bases) was incorporated in recombinant vector assembly by overlap with inserts terminated with Ttef1. The SV40NLS-stop codon, tef1 terminator and tef1 promoter parts were PCR-amplified from template pFC332. FvCas9 was PCR amplified in one part from synthesised DNA and assembled with the tef1 promoter and tef1 terminator parts in Golden Gate vector pICH47751 [24] to give Ptef1-FvCas9-SV40-Ttef1. This was used as a template for generation by PCR of two parts for assembly with pFCBB to give pFCFvCas9: Ptef1 and cas9 bases 1–2141 (5’-3’) were amplified as Part 1 using primers NtPac1_Ptef1 (pFCBB)_F and FvCas9-1_R; and Part 2 was generated by primers FvCas9-2_F and Ttef1_R_no ext, incorporating cas9 bases 2142–4376 (5’-3’) and sequences for SV40-stop codon and Ttef1. The Nt.BbvCI –PacI cloning site was reconstructed by incorporating required bases into the 5’ extension of primer NtPac1_Ptef1 (pFCBB)_F. The PolII/ribozyme sgRNA cassettes were constructed using pFC334 as a template and assembled into the Nt.BbvCI-PacI USER site of pFC332 as described by Nodvig et al.[7]. 5SrRNA sgRNA cassettes were synthesised by IDT (Additional file 7: Table S7) and PCR amplified using primers 5SrRNA_F USER and 5SrRNA_R USER for USER assembly into pFC332 or pFCBB. Details of USER reactions are described in Additional file 9: Table S9.
Protoplast isolation and transformation
A method modified from Moradi et al. [25] was used for protoplast transformation. Media are detailed in Additional file 10: Table S10. To obtain approximately 40 × 100 µl protoplast aliquots, 250 ml CMC medium in a 500 ml Erlenmeyer flask was inoculated with 5 × 4 mm plugs from an agar plate culture and incubated for 6 days on a rotary shaker at 25 °C, shaking at 175 rpm (or to give good aeration). The culture was filtered through 2 layers of sterile Miracloth (Millipore) and the filtrate was centrifuged at 4000×g for 10 min. After decanting the supernatant, the conidia were resuspended in 100 ml of YEPD broth in a 250 ml Erlenmeyer flask and incubated overnight (14 h) in a rotary shaker at 22 °C, shaking at 175 rpm. The mycelium was harvested by filtration using 2 layers of sterile Miracloth and was rinsed with sterile pure water. Excess water was gently removed by blotting, without compacting the mycelium, and the mycelial mat was transferred to 20 ml Protoplasting Buffer in a sterile beaker and gently dispersed to ensure efficient digestion. After 2 h incubation at 28 °C, 80 rpm, the digestion mixture was filtered through sterile Miracloth (3 layers) into 50 ml sterile conical bottomed plastic tubes. The filtrate was centrifuged at RT at 3000×g for 5 min, the supernatant was immediately decanted, and the protoplasts were washed: using a 25 ml sterile filtered serological pipette the pellet was gently dislodged and resuspended (using both manual and air agitation) in 30 ml of STC, before centrifuging at 3,000 × g for 5 min. The wash was repeated (removing an aliquot of the second suspension for protoplast quantification) and the protoplasts were resuspended in STC Buffer to give 1 × 108 protoplasts/ml before dispensing 100 µl aliquots into sterile 2 ml microcentrifuge tubes. For protoplast transformation 1–5 µg DNA was added and the tube was flicked gently 3 times to mix before incubating at RT for 20 min. 1 ml 40% PTC was then added, and the tube was inverted gently three times to mix and incubated at RT for 20 min before decanting into a 50 ml sterile tube containing 5 ml TB3 supplemented with 100 µg ml−1 Hygromycin B and incubated at 21 °C, shaking at 90 rpm for 14–16 h (overnight). Each sample was decanted into a 9 cm petri dish before adding 10 ml Top Agar (cooled to ‘hand hot’ and kept molten in a 50 °C water bath), supplemented with Hygromycin B to give 100 µg ml−1 (one plate was included with no selection for a non-transformed protoplast control), swirling gently to mix. Plates were incubated for 8–10 h at RT before adding 10 ml Top Agar prepared as before and supplemented with 100 µg ml−1 Hygromycin B (except for the control), sealed and incubated at 20 °C. Colonies from transformation plates were transferred after 10 days to PDA supplemented with Hygromycin B (75 µg ml−1) for maintenance of the AMA1 vector, then cultured for 2 weeks at 28 °C. For isolation of isogenic lines, agar plugs from colonies were inoculated in 20 ml CMC medium in a 100 ml Erlenmeyer flask and incubated shaking at 175 rpm for up to 7 days at 25 °C. Spores were collected by filtration through 2 layers of Miracloth, pelleted at 4000xg for 10 min and plated to give a low density on PDA plates. After incubation for 14–15 h at 20 °C single germinating spores were isolated and incubated in the dark at 28 °C on PSA (Additional file 11: Table S11) for identifying albino PKS12 variants.
CRISPR variant analysis
Genomic DNA for use in PCR was extracted from mycelium cultured in 50 ml plastic conical based tubes containing 10 ml of a proprietary medium and incubated shaking at 175 rpm for 5–7 days at 28 °C. The culture was collected on Miracloth, rinsed with autoclaved purified water, blotted to remove excess liquid and added with 2 ball bearings (4 mm chrome steel) to 2 ml microcentrifuge tubes before flash freezing in liquid nitrogen and storage at −80 °C. Mycelium was ground while frozen using a ball mill (Retsch) and DNA was extracted using the DNeasy Plant Mini Kit (Qiagen). PCR amplicons were generated using Q5® High-Fidelity 2X Master Mix (NEB) using primers PKS12_F2 and PKS12_R2, which span the target region (Fig. 1). Amplicons were Sanger sequenced (Eurofins Genomics) using sequencing primers PKS12-Seq_R1 and PKS12-Seq_F2.
Sequencing analysis and software
Nucleotide and protein alignments were performed using Geneious version 10.0.2 (http://www.geneious.com) [26]. Schematic maps were prepared using IBS software [27].
Abbreviations
- Cas9:
-
CRISPR associated DNA-cutting enzyme
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- HDV ribozyme:
-
Hepatitis Delta Virus ribozyme
- HH ribozyme:
-
Hammerhead ribozyme
- indel:
-
Insertion-deletion mutation of genomic sequence
- Kb:
-
Kilobase pairs
- mEGFP:
-
Monomeric enhanced green fluorescent protein
- mRFP:
-
Monomeric red fluorescent protein
- PDA:
-
Potato Dextrose Agar
- PolII:
-
Polymerase II
- PolIII:
-
Polymerase III
- USER:
-
Uracil-Specific Excision Reagent
- PCR:
-
Polymerase chain reaction
- RT-qPCR:
-
Reverse transcription quantitative PCR
References
Trinci APJ. Myco-protein: a twenty-year overnight success story. Mycol Res. 1992;96(1):1–13.
Wiebe MG. Myco-protein from Fusarium venenatum: a well-established product for human consumption. Appl Microbiol Biotechnol. 2002;58(4):421–7.
Whittaker JA, Johnson RI, Finnigan TJA, Avery SV, Dyer PS. The biotechnology of quorn mycoprotein: past, present and future challenges. In: Nevalainen H, editor. Grand challenges in fungal biotechnology. Cham: Springer International Publishing; 2020. p. 59–79.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21.
Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9(1):1911.
Fuller KK, Chen S, Loros JJ, Dunlap JC. Development of the CRISPR/Cas9 System for Targeted Gene Disruption in Aspergillus fumigatus. Eukaryot Cell. 2015;14(11):1073–80.
Nødvig CS, Nielsen JB, Kogle ME, Mortensen UH. A CRISPR-Cas9 System for Genetic Engineering of Filamentous Fungi. PLoS ONE. 2015;10(7):e0133085.
Matsu-Ura T, Baek M, Kwon J, Hong C. Efficient gene editing in Neurospora crassa with CRISPR technology. Fungal Biol Biotechnol. 2015;15(2):4.
Liu R, Chen L, Jiang Y, Zhou Z, Zou G. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov. 2015;12(1):15007.
Pohl C, Kiel JAKW, Driessen AJM, Bovenberg RAL, Nygård Y. CRISPR/Cas9 Based Genome Editing of Penicillium chrysogenum. ACS Synth Biol. 2016;5(7):754–64.
Schuster M, Kahmann R. CRISPR-Cas9 genome editing approaches in filamentous fungi and oomycetes. Fungal Genet Biol. 2019;29(130):43–53.
Foster AJ, Martin-Urdiroz M, Yan X, Wright HS, Soanes DM, Talbot NJ. CRISPR-Cas9 ribonucleoprotein-mediated co-editing and counterselection in the rice blast fungus. Sci Rep. 2018;8(1):14355.
Gardiner DM, Kazan K. Selection is required for efficient Cas9-mediated genome editing in Fusarium graminearum. Fungal Biol. 2018;122(2–3):131–7.
Wang Q, Cobine PA, Coleman JJ. Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes. Fungal Genet Biol. 2018;12(117):21–9.
Ferrara M, Haidukowski M, Logrieco AF, Leslie JF, Mulè G. A CRISPR-Cas9 system for genome editing of Fusarium proliferatum. Sci Rep. 2019;9(1):19836.
Shi T-Q, Gao J, Wang W-J, Wang K-F, Xu G-Q, Huang H, et al. CRISPR/Cas9-based genome editing in the filamentous fungus Fusarium fujikuroi and its application in strain engineering for gibberellic acid production. ACS Synth Biol. 2019;8(2):445–54.
Kuivanen J, Wang YMJ, Richard P. Engineering Aspergillus niger for galactaric acid production: elimination of galactaric acid catabolism by using RNA sequencing and CRISPR/Cas9. Microb Cell Fact. 2016;15(1):210.
Zhang C, Meng X, Wei X, Lu L. Highly efficient CRISPR mutagenesis by microhomology-mediated end joining in Aspergillus fumigatus. Fungal Genet Biol. 2016;86:47–57.
Zheng X, Zheng P, Zhang K, Cairns TC, Meyer V, Sun J, et al. 5S rRNA promoter for guide RNA expression enabled highly efficient CRISPR/Cas9 genome editing in Aspergillus niger. ACS Synth Biol. 2019;8(7):1568–74.
Nødvig CS, Hoof JB, Kogle ME, Jarczynska ZD, Lehmbeck J, Klitgaard DK, et al. Efficient oligo nucleotide mediated CRISPR-Cas9 gene editing in Aspergilli. Fungal Genet Biol. 2018;115:78–89.
Kim J-E, Han K-H, Jin J, Kim H, Kim J-C, Yun S-H, et al. Putative polyketide synthase and laccase genes for biosynthesis of aurofusarin in Gibberella zeae. Appl Environ Microbiol. 2005;71(4):1701–8.
van Leeuwe TM, Arentshorst M, Ernst T, Alazi E, Punt PJ, Ram AFJ. Efficient marker free CRISPR/Cas9 genome editing for functional analysis of gene families in filamentous fungi. Fungal Biol Biotechnol. 2019;21(6):13.
Puigbò P, Guzmán E, Romeu A, Garcia-Vallvé S. OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007;35(Web Server issue):W126-31.
Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S. A modular cloning system for standardized assembly of multigene constructs. PLoS ONE. 2011;6(2):e16765.
Moradi S. A modified method for transformation of Fusarium graminearum. J Crop Prot. 2013;2(3):297–304.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9.
Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, et al. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31(20):3359–61.
Acknowledgements
The authors thank the following: Marlow Foods for financial support and participation at project inception and throughout the duration of the project, and for provision of strains and technical advice; the vectors pFC332 (Addgene Plasmid #87845) and pFC334 (Addgene Plasmid #87846) were a gift from the Uffe Mortensen laboratory, Technical University of Denmark; synthesised DNA for Fvcas9 was kindly provided through the services of SCLU; A.D. Armitage annotated the F. venenatum genome and produced the codon usage table for F. venenatum; J. Bennett performed RNA extractions and qRT-PCRs including data analysis.
Funding
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) and (BB/P020364/1) and Marlow Foods.
Author information
Authors and Affiliations
Contributions
FMW and RJH devised the research plan. FMW planned the experiments, designed and constructed the CRISPR cassettes and vectors, performed transformation experiments and conducted phenotypic and molecular analyses of variants. FMW wrote the manuscript with input from RJH and Marlow Foods. Both authors reviewed the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
PKS12 gene target site sequence data for phenotypic variants.
Additional file 2: Table S2.
Persistence of hygromycin tolerance in CRISPR variants.
Additional file 3: Figure S1.
RT-qPCR analysis of F. venenatum AMA1 vector cas9 transcripts expressed in F. venenatum; Table S3. Primers used in RT-qPCR analysis.
Additional file 4: Table S4
Viability of protoplasts transformed with AMA1 vectors expressing mEGFP and cas9.
Additional file 5: Table S5
. Primers used in vector construction and PCR analysis of PKS12 gene variants.
Additional file 6: Table S6.
Expression cassette Ptef1-FvCas9-SV40-Ttef1 used in this study.
Additional file 7: Table S7.
Sequence of PFv5SrRNA-sgRNA cassettes used in this study.
Additional file 8: Table S8.
Codon usage table for highly expressed genes in F. venenatum.
Additional file 9: Table S9.
Method for USER cloning of promoter-sgRNA cassettes into AMA1 vectors.
Additional file 10: Table S10
. Media used for making protoplasts and protoplast transformation.
Additional file 11: Table S11.
Method for making Potato Sucrose Agar (PSA).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wilson, F.M., Harrison, R.J. CRISPR/Cas9 mediated editing of the Quorn fungus Fusarium venenatum A3/5 by transient expression of Cas9 and sgRNAs targeting endogenous marker gene PKS12. Fungal Biol Biotechnol 8, 15 (2021). https://doi.org/10.1186/s40694-021-00121-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40694-021-00121-8