
Abstract
Background
The bacterial CRISPR/Cas genome editing system has provided a major breakthrough in molecular biology. One use of this technology is within a nuclease-based gene drive. This type of system can install a genetic element within a population at unnatural rates. Combatting of vector-borne diseases carried by metazoans could benefit from a delivery system that bypasses traditional Mendelian laws of segregation. Recently, laboratory studies in fungi, insects, and even mice, have demonstrated successful propagation of CRISPR gene drives and the potential utility of this type of mechanism. However, current gene drives still face challenges including evolved resistance, containment, and the consequences of application in wild populations. Additional research into molecular mechanisms that would allow for control, titration, and inhibition of drive systems is needed.
Results
In this study, we use artificial gene drives in budding yeast to explore mechanisms to modulate nuclease activity of Cas9 through its nucleocytoplasmic localization. We examine non-native nuclear localization sequences (both NLS and NES) on Cas9 fusion proteins in vivo through fluorescence microscopy and genomic editing. Our results demonstrate that mutational substitutions to nuclear signals and combinatorial fusions can both modulate the level of gene drive activity within a population of cells.
Conclusions
These findings have implications for control of traditional nuclease-dependent editing and use of gene drive systems within other organisms. For instance, initiation of a nuclear export mechanism to Cas9 could serve as a molecular safeguard within an active gene drive to reduce or eliminate editing.
Similar content being viewed by others

Background
Control of biological populations is critical to agriculture, ecological conservation, and human health. Numerous methods have been employed to remove invasive species [1, 2], crop-damaging pests [3,4,5,6], or metazoans that harbor diseases [7, 8] including physical barriers, chemical agents, and/or natural predators or competitors. However, the ability to genetically modify an entire species has been hindered by the natural laws of segregation—introduction of a genetic element through natural breeding would require an unattainable number of modified individuals to be released into the wild. Given the introduction of CRISPR/Cas9 as an efficient, convenient, and universal genome editor [9,10,11,12,13,14,15], a mechanism has been developed that is Super-Mendelian in nature: a nuclease “gene drive.”
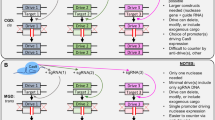
This arrangement of the CRISPR components is simple in design, yet powerful in application. The basic architecture includes a nuclease of choice (usually S. pyogenes Cas9, although many alternatives and engineered variants now exist) and the corresponding single guide RNA (sgRNA) expression cassette integrated within the genome. Placement of Cas9/sgRNA could be at a safe harbor locus or could delete or disrupt an existing endogenous gene. In the case of the former, the gene drive (GD) would likely also contain a “cargo” element—the intended genetic element to be delivered to the entire population. This could include any number of variations including endogenous or exogenous DNA to modify the organism itself (e.g. imposed fitness cost) or to aid in the separation between the host and disease-causing agent. Once expressed, the nuclease is primed by the guide RNA to target the wild-type copy of the gene (or position) on the homologous chromosome within a diploid genome (within the progeny between a gene drive individual and a wild-type individual) to create a double strand break (DSB). The unique arrangement of the GD relative to the DSB allows the expression cassette for Cas9/sgRNA itself to serve as the donor DNA for homology directed repair (HDR). The GD copies itself to the wild-type chromosome to repair the break and replaces the entire endogenous locus; a heterozygous cell (GD/WT) becomes a homozygous (GD/GD) cell. Action of a gene drive within a population would allow the rapid “forced” propagation of any genetic element in a small number of generations and would require only a small number of released GD individuals.
There are numerous applications of gene drive biotechnology to control and alter biological populations including global challenges such as eliminating insect-borne diseases [16,17,18]. Recent experimental [19,20,21,22,23,24] and computational studies [25,26,27] highlight the potential of GD systems. However, there remain many unknowns surrounding implementation and management of this new technology (including accidental or malicious release of such a system without any safeguard or inhibitory mechanism). Release of a GD-organism has the potential to modify a portion of the natural population of the chosen species, even using the current available gene drives (for which GD-resistance is still an ongoing issue) [28]. Therefore, it is critical to identify means to control, titrate, inhibit, or reverse gene drive systems to modulate or slow their progression, and as a failsafe should removal of GD individuals become necessary.
Our previous work focused on examination of conserved components of CRISPR gene drives (e.g. nuclease, guide RNA, DNA repair) in budding yeast to identify modes of control, regulation, and inhibition of drive success in vivo [29, 30]. A variety of molecular mechanisms have been shown to modulate Cas9-based editing including nuclease expression, guide RNA sequence, Cas9–dCas9 fusions, anti-CRISPR mutants, and nucleocytoplasmic shuttling of tagged Cas9. Here, we expanded upon our previous work [29] (which focused on utilizing the SV40 signal) to examine additional NLS and NES combinations appended to S. pyogenes Cas9–eGFP fusion constructs within an artificial GD system. We tested three monopartite NLS sequences, mutated signals, and two NES signals to demonstrate titration of gene drive activity in a diploid yeast model.
Results
Non-native nuclear localization signals can direct Cas9 localization in vivo
Nucleocytoplasmic transport of macromolecules within eukaryotic cells is highly conserved [31,32,33,34,35] and has been recognized as a universal requirement of gene editing in living systems—namely, the intended nuclease must gain access to the interior of the nucleus and genomic content. This intracellular trafficking system involves recognition of nuclear import sequences by karyopherins for transit through the nuclear pore complex [36,37,38]. Use of the CRISPR/Cas system for alteration of the genome typically includes appending one or more NLSs to the nuclease (e.g. Cas9) and the classical NLSSV40 is often used for this purpose [10, 39, 40]. However, several groups have demonstrated that alteration of the nuclear localization of Cas9 can serve as a means to control editing. For instance, the iCas system was constructed to prevent nuclear entry until addition of an external cue [41]. Moreover, design of a split Cas9 included use of a NES sequence to restrict localization of one of the two halves of the nuclease until addition of an exogenous signal [42]. Finally, optimization of CRISPR-based editing in various organisms and cell types has focused on the placement, number, and identity of the included NLS sequence—whether native or non-native to the species of interest [43,44,45,46,47,48,49].
Our previous work in budding yeast demonstrated that the dynamic localization for Cas9 fusions harboring both NLS and NES signals resulted in a variable level of genomic editing (both in haploid and diploid cells) [29]. However, this work focused exclusively on the commonly used NLSSV40 signal. A previous study [50] demonstrated that mutational substitutions to a set of artificially derived NLS signals (from random peptide libraries) allowed for a spectrum of nuclear import efficiencies using a GFP-based reporter system. We sought to test whether alternative nuclear signals could still direct Cas9 to the nucleus and allow for a titration of DSB formation in a CRISPR gene drive diploid strain.
Design of our gene drive system allows for the safe and programmable examination of various CRISPR components. Briefly, two sets of “unique” Cas9 target sites (termed u1 and u2) [51] are positioned flanking both the inducible Cas9-expression cassette (“drive”) and the corresponding locus harboring a selectable marker (“target”). Activation of Cas9 and inclusion of the correct sgRNA fragment (expressed from a plasmid) would cause multiplexing and cleavage of the dual (u1) sites in the target chromosome and repair via homologous recombination using the drive-containing chromosome as the source of donor DNA (Fig. 1). Haploid yeast strains harboring an inducible Cas9–eGFP construct were generated using various C-terminal signals: NLS signals included the classical SV40 and three other monopartite signals with sequences containing either four (NLS-I), three (NLS-II), or two (NLS-III) lysine and/or arginine residues within the motif whereas the export signals (NES-IV and NES-V) were derived from the ΦX3ΦX2ΦXΦ [52] consensus (Fig. 2a). These NLS signals were selected from a previous study [50]: they represented a diverse set of artificially derived peptides found to direct nuclear import across different cellular systems—we chose several that (1) varied in the number and placement of basic residues and (2) had a number of residues within the signal that, when mutated, were found to generate a spectrum of activities through an in vivo cellular assay. Following Cas9 induction in media containing galactose, strains were imaged by fluorescence microscopy (Fig. 2b). Localization was determined by labeling the nuclear periphery with a mCherry-marked integrated copy of Nup188, a nuclear pore complex component. For all three non-native NLSs, Cas9 localized to the nucleus and for both NES-tagged Cas9 fusions, eGFP signal was occluded from the nucleus. However, despite the steady-state localization of the Cas9–eGFP–NES constructs, our previous findings suggested that a small amount of editing (and gene drive activity in diploids) does take place, albeit after significantly longer nuclease induction times [29].

Design of an artificial CRISPR gene drive system in Saccharomyces cerevisiae. a General schematic of a gene drive in a diploid genome. b An artificial gene drive system was constructed at the yeast HIS3 locus. The inducible GAL1/10 promoter drives expression of a codon-optimized S. pyogenes Cas9 containing a C-terminal eGFP fusion followed by a chosen nuclear signal sequence (NLS/NES* see Fig. 2a). An inserted terminator (from CDC10) was placed downstream of the Cas9 coding sequence followed by a selectable marker cassette. This included the non-native MX-based promoter and terminator sequences driving constitutive expression of the C. albicans URA3 gene. The entire gene drive system was flanked by two identical artificial sites, termed (u2), that do not exist in the native genome [29, 51]. The design of the drive also included an engineered “target” cassette (bottom) built within a strain of the opposite mating type at the HIS3 locus. This included an artificial “cargo” gene and a yeast-based terminator (from SHS1). A modified selectable marker cassette included the constitutive CCW12 (cell wall) promoter sequence driving the S. pombe HIS5 gene (functional equivalent to yeast HIS3). Finally, two (u1) sequences were inserted flanking the entire target locus. To complete action of the drive, a high-copy plasmid (pGF-V1220) contained the cassette for the guide RNA (marked with LEU2)

Subcellular localization of S. pyogenes Cas9 tagged with various nuclear localization sequences. a Table of non-native NLS and NES sequences tested; basic residues are underlined for NLSs and hydrophobic residues are underlined for NESs. b Fluorescence microscopy of live yeast cells containing NLS sequences (GFY-3435 to 3437) or NES sequences (GFY-3438, 3439) fused to eGFP-tagged Cas9 (also see Table 1). Yeast were cultured in galactose prior to imaging. An integrated copy of Nup188-mCherry marked the nuclear periphery. Representative images are shown; white dotted lines, outline of selected cells. Scale bar, 3 μm. Triangles indicate the yeast vacuole
Mutational alterations to NLS or NES sequences can modulate gene drive activity
We tested each NLS and NES sequence appended to Cas9–eGFP for its effect on gene drive activity in diploid yeast. The general methodology employed to assay gene drive function in vivo included a variable induction time in galactose followed by recovery on dextrose-containing media (Fig. 3a). Following the formation of single colonies, yeast were transferred to medium lacking histidine to test for the presence of the SpHIS5 marker—equivalent to yeast HIS3—within the target strain. Upon successful action of the drive, the entire target locus is removed, and colonies are unable to grow in the absence of histidine (e.g. SD-HIS plates). Importantly, our artificial system does not impose any selection for or against action of the gene drive unlike other possible arrangements that challenge cells by selecting for successful DSB/repair events. Compared to the NLSSV40-tagged strain, all three artificial NLS signals (I–III) caused a dramatic loss of growth on SD-HIS; constructs harboring the NES (IV, V) retained a large number of viable colonies (Fig. 3b). Previous work suggested that mutations to positions along the length of these artificial signals altered their effectiveness at promoting nuclear import by a fluorescence reporter system in live cells [50]. Therefore, we generated twelve substitutions across NLS(I–III) and examined their gene drive activities in vivo (Fig. 3c). The total percentage of colonies sensitive to the SD-HIS condition was quantified across multiple trials. As predicted, a NLSSV40 tag and a tandem NLSSV40–NLSSV40 tag allowed for > 95% activity at 5 h of Cas9 induction (1, 2). To ensure that high-expression levels from a 5 h induction were not masking subtle effects of the alteration to the NLS signal, we also tested each strain at a lower 2.5 h induction. Many of the substitutions to other classes of NLS (I–III) displayed no change compared to the WT and only four of the twelve substitutions (6, 7, 11, and 14) had a modest decrease in activity. Therefore, these results demonstrate that some of the NLS alterations can partially reduce Cas9 editing in vivo to varying degrees.

CRISPR gene drives using various NLS and NES fused to Cas9–eGFP. b Schematic of gene drive activation. Following diploid selection, yeast were grown to saturation overnight in media containing raffinose and sucrose lacking leucine. Cultures were back-diluted into rich medium containing galactose for a set number of hours, diluted to approximately 100–500 cells per agar plate (SD-LEU), and incubated for 48 h. Yeast colonies were velvet-transferred to SD-LEU and SD-HIS plates for up to 24 h before imaging. If the GD was successful and removed the target HIS3 locus (harboring SpHIS5), then colonies would be sensitive to the SD-HIS condition. b Haploid yeast strains (GFY-2756, and GFY-3465-3469) were mated to target strains (GFY-3206 and 3207), diploids selected, and gene drives activated for 5 h. Yeast were plated on SD-LEU and transferred to a final SD-LEU plate (control) and SD-HIS plate to assess gene drive activity. c The number of colonies sensitive on SD-HIS provided a measure for “percent gene drive activity”. Diploid gene drives were tested using strains from b and mutational substitutions made to each NLS (GFY-3470, 3443-3447, 3449-3452, and 3454-3456, numbered 1–17) where Cas9 was induced for either 2.5 h or 5 h and quantified for drive activity. Error, SD. NLS(I–III) sequences can be found in Fig. 2a. d Gene drive strains (GFY-3468, 3469, 3471, 3472, 2758, 3716 and 3717) harboring a NES signal in the absence or presence of additional NLSs were tested for 2.5 h, 5 h, 10 h, and 24 h of Cas9 induction and quantified as in (c). Error, SD. Red asterisk, this construct harbors the ADH1(t)-prMX-KanR-MX(t) cassette following Cas9–eGFP. e Top, illustration of the gene drive/target arrangement and the position of oligonucleotides (Additional file 1: Table S1) used. Middle, PCRs were performed on chromosomal DNA from clonal isolates from each drive (5 h). The numbers (1–17) correspond to strains from Fig. 3c. The expected sizes for each PCR a–d are shown along with markers. Images were cropped from independent gels or portions of larger gels and are divided by white lines. Two isolates were obtained with no galactose activation (1’ and 2’; dextrose only treatment) from GFY-2756 (Strain 1). All colonies were tested for ploidy status (diploid) and growth on SD-URA (drive) and SD-HIS (target). Strain GFY-2756 was tested on G418 media. Below, A similar analysis of clonal isolates from the NES-containing strains was performed (24 h). Two isolates each (from strain 18 and 19) were chosen that were either resistant or sensitive to the SD-HIS condition
The two NES signals tested on Cas9–eGFP included a PKI-like sequence [53] (NES-IV) as well as a similar NES with added leucine residues present within the motif (NES-V). A previous study reported that excessive hydrophobic amino acids could interfere with nuclear export signals [52]. Therefore, we tested a NES harboring six total leucine residues as opposed to only three in the PKI-like motif (Fig. 2a)—the expectation was that a reduction in NES effectiveness would manifest in higher nuclear residence time and gene drive efficiency. Our previous work demonstrated that a significantly longer (24 h) induction time was required to observe drive activity for a construct harboring only a single NES signal [29]; to allow for a comparison to the NLS-containing strains, we chose to treat cells in galactose for 2.5, 5, 10, and 24 h (Fig. 3d). For induction times less than 10 h, the gene drive activity (18, 19) remained below 50% (Fig. 3d). We noticed a subtle difference between the effectiveness of the two NES-containing constructs and it appeared that the original PKI-like motif (18) provided less drive activity across multiple time points compared to the sequences with added leucine residues (19). A tandem NES(PKI-like)–NES(PKI-like) signal (20) did not provide any significant change compared to a single NES alone. Previously, we found that a C-terminal fusion of the dual NLS(SV40)–NES(PKI-like) sequence to Cas9–eGFP (22) seemed to phenocopy a fusion of NES(PKI-like) alone [29]. Here, we constructed and tested the reciprocal fusion, NES(PKI-like)–NLS(SV40) (21), to determine its effect in vivo. We observed that both constructs displayed similar gene drive activities to a single NES signal, yet positioning of the NLSSV40 on the extreme C-terminus appeared to provide slightly higher activity (and less contribution from the NES). Finally, we included both NES signals in a construct also harboring two additional NLSSV40 sequences—at the N-terminus and fused between Cas9 and eGFP (23, 24). The competition between two NLS sequences versus one NES sequence provided a high level of gene drive activity, with a slight increase in activity from the construct harboring the leucine-rich NES (24). Together, these data illustrate that changes can also be made to either the primary sequence or placement of nuclear export signals to shift the level of editing in vivo.
Previous work has demonstrated that gene drives may induce a DSB but fail to copy the GD cassette to the target chromosome. In such cases, NHEJ causes repair of the broken DNA ends and prevents HR-based propagation of the drive (e.g. GD “resistant” alleles). Therefore, to confirm that yeast sensitive to the SD-HIS condition had lost the target allele at the HIS3 locus, we obtained clonal isolates from each gene drive experiment, tested for each of the included markers, assayed ploidy status, and examined purified genomic DNA by multiple diagnostic PCRs (Fig. 3e). For diploids lacking galactose treatment (0 h), four independent PCR reactions illustrated the presence of both the gene drive and target constructs. However, for isolates subjected to galactose induction (5 h), only the two PCRs corresponding to the gene drive allele (PCR-A, PCR-B) were present; reactions for the target allele were unable to amplify the expected fragment (PCR-C, PCR-D) suggesting the diploid genome was now homozygous for the drive allele (1–17). A similar analysis was performed for eight clonal isolates from the NES strains (18, 19). However, we chose to sample two isolates that were sensitive and two samples resistant to the SD-HIS condition as there were still many colonies displaying each phenotype after 24 h (Additional file 1: Fig. S3). The expectation was that diploid colonies still growing on SD-HIS medium were still heterozygous for the drive and target alleles at the HIS3 locus. Diagnostic PCRs illustrated that the target allele was still present for surviving colonies and absent for colonies sensitive to SD-HIS (Fig. 3e, below). Given that these NES-containing strains had 24 h of galactose induction, the lack of editing (and GD activity) likely results from active nuclear export of Cas9. Collectively, these results demonstrated a continuum of gene drive activity ranging up to 100% in our yeast model across all Cas9–eGFP fusions with both NLS and/or NES signals (Fig. 3).
Discussion
Given that application of the CRISPR/Cas editing biotechnology in eukaryotic systems requires delivery of Cas9/sgRNA to the nucleus, we focused on methodologies that could provide a new suite of molecular tools to control, inhibit, or modulate gene drive systems in vivo, although we recognize these techniques might be used for many types of genomic editing and could apply to alternative uses of the CRISPR system (e.g. dCas9). While there are still few studies on gene drives, the power and potential application for this technology is clear, despite the current challenges and obstacles. The ability to modify an entire population with a genetic element of choice presents numerous advantages including optimizing agricultural crops and animals, prevention of human disease, and ecological control on a large scale. However, ongoing testing of optimal designs including safeguards and controllable drives warrant further research and recent progress has been made in current systems [25, 26, 54,55,56]. Previous efforts (both computational and experimental) have highlighted a variety of components that might serve as a platform for control or inhibition of gene drives [21, 26, 29, 30]. Here, we focused on altering the nucleocytoplasmic localization of Cas9 for titration of editing.
While a popular choice for nuclear import across model systems has been the monopartite NLSSV40 signal, others have found that varying the position, copy number, or identity of the NLS (native or non-native) can alter genomic editing [43, 46, 48, 57, 58]. Given the conservation of nuclear import machinery across eukaryotes and the wide variety of possible nuclear import sequences (native or artificial), this could present a complex platform for tuning or optimizing Cas9 nuclear import in any species of interest. As we have demonstrated here, three artificially generated NLS signals allowed for efficient nuclear entry and subsequent gene drive activity in vivo; also, some mutational substitutions to the NLS primary sequence partially reduced editing. Future iterations might include modifications to signal positioning (N- or C-terminus, or between fusion proteins), the distance from Cas9, and local residue context surrounding the signal.
Previous studies have evaluated the sequence composition of many natural and artificial NLS and NES sequences through mutagenesis [59], in vivo subcellular localization assays [50, 52], in vitro binding experiments [60], and computational analyses [61,62,63]. Consensus sequences have been developed for different classes of NLS (for example, the K–(K/R)–X–(K/R) monopartite signal) that highlight critical residues within the signal. However, there is also a contribution of flanking amino acids (not part of the consensus) that are upstream, downstream, or within the NLS itself [59, 64,65,66]. For NESs, multiple classes of signals have now been defined that are variations of the Φ–X3–Φ–X2–Φ–X–Φ consensus. Previous work has highlighted further preferences and restrictions to NES motifs and these may also be context-dependent within three-dimensional protein structures [52, 61, 62]. For instance, the presence of certain residues (such as proline) may disrupt NESs [52]. Comparison of our NES(IV) versus NES(V) illustrated that the addition of hydrophobic residues (leucine) was less efficient as an export signal, similar to previous findings [52]. Given the large variation in nuclear signals, the contribution of individual residues to NLS or NES function will require experimental validation. Furthermore, the assays used to detect nuclear import and/or export need to be taken into consideration. In the study performed by Kosugi et al. [50], a GFP reporter system in live cells was used to quantify the success of nuclear import. In our gene drive experiments, we assayed the effect of Cas9-dependent DSB formation followed by DNA repair and a resulting growth phenotype. Furthermore, interpretation of nuclear entry or exit may be complicated by additional cryptic signals (enhancement or competition with the signal(s) of interest) or protein size and entry through the nuclear pore complex.
Of note, our data demonstrates that inclusion of a NES (even when not paired with any NLS signal) still allowed for some level of editing as Cas9 may gain entry to the nucleus through diffusion followed by export; editing without any appended NLS has also been observed in previous studies [29, 43]. Therefore, we recommend use of various nuclear signal combinations to modulate and reduce, rather than eliminate, editing. However, nuclear restriction of Cas9 may still be useful when paired with other inhibitory mechanisms such as the AcrIIA2 and AcrIIA4 anti-CRISPR proteins [30], reduced or programmed expression of nuclease transcript [29], or other mechanisms to inhibit editing such as degradation of Cas9 [67].
Restriction of Cas9 nuclear localization has also been demonstrated in other cell types through (1) occlusion of a NLS signal or (2) tethering to the plasma membrane—subsequent release was achieved by activation of a protease to cleave the tethered dCas9–NLS construct and allow transport into the nucleus [41, 68, 69]. However, our study provides experimental evidence for use of this general methodology for titration of gene drive activity. We envision that nuclear occlusion of Cas9 could also be achieved by alternative approaches. Expression of an inducible anti-GFP “nanobody”-containing peptide fused with an export signal might provide temporal control to regulate Cas9–GFP nucleocytoplasmic localization. In this way, activity might be reduced at a later point in time (or to a specific portion of a population) by causing an increase in Cas9 nuclear export. A secondary system that could modulate Cas9 activity—inducible by external stimuli—would provide a suite of new options for controlling gene drive propagation within a population. This could be utilized as a molecular safeguard to slow or inhibit drives, allow for the timely use of anti-drives or other countermeasures, or as a means to cycle gene drive-containing organisms with seasonal or environmental changes. Alternatively, modification of the nuclear pore complex might allow for selective entry of a pool of Cas9 fusion constructs while restricting other variants or orthologs.
Conclusion
In this study, we expanded our analysis of nuclear trafficking to control Cas9 editing within the context of a gene drive system. We predict that a combinatorial approach of altering nuclease (transcript and/or protein) levels, NLS/NES signals, cellular traps, and other inducible/tunable systems might be employed in the design of future gene drive systems that are safe, controllable, and/or reversible.
Materials and methods
Yeast strains and plasmids
Saccharomyces cerevisiae strains used in this study can be found in Table 1. Molecular biology techniques were used to generate all engineered constructs [70]. The general strategy included first constructing a CEN-based plasmid using in vivo assembly in yeast [71] using a modified lithium acetate transformation protocol [72]. Next, PCR amplified DNA of the entire assembled cassette, followed by treatment with DpnI enzyme (to remove template DNA), was integrated into the appropriate haploid yeast genome (for Cas9, the HIS3 locus). For generation of NLS substitutions, a modified PCR mutagenesis protocol was used [73]. To integrate various eGFP–NLS or eGFP–NES combinations, a second integration construct was generated; universal eGFP and MX(t) sequences were used for homologous recombination (Table 1). Finally, selection markers were converted using a third round of integration (e.g. from SpHIS5 to CaURA3) using common CDC10(t) and the MX(t) sequences. The only plasmid used in this study was the high-copy pRS425-based sgRNA(u1) vector (pGF-V1220) [29]. Diagnostic PCRs and Sanger DNA sequencing of all chromosomal modifications was performed to confirm successful integration events (see Additional file 1: Fig. S1).
Culture conditions
Budding yeast were grown on solid agar medium or in liquid cultures; rich media, YPD (2% peptone, 1% yeast extract, 2% dextrose), or synthetic drop-out media (nitrogen base, amino acids, and ammonium sulfate) were used. Prior to galactose metabolism (2%), cultures were grown to saturation in medium containing 2% raffinose and 0.2% sucrose. All sugars were filter sterilized.
Fluorescence microscopy
Haploid yeast cultures were grown to saturation in synthetic complete medium containing raffinose and sucrose overnight. Next, strains were back-diluted into rich medium containing galactose for 4.5 h at 30 °C and prepared on a glass slide [29]. Live cells were examined using a Leica DMI6000 fluorescence microscope (Leica Microsystems, Buffalo Grove, IL). A Leica DFC340 FX camera, 100 × lens, and fluorescence filters (Semrock, GFP- 4050B-LDKM-ZERO, mCherry-C-LDMK-ZERO) were used. Software for image capture and analysis included Leica Microsystems Application Suite and ImageJ (National Institute of Health). Images were taken using identical exposure times; representative cells were chosen for each image set and scaled together. Scale bars, 3 μm. “Merged” images do not contain any additional processing from the two combined channels.
CRISPR gene drives in diploid yeast
Haploid yeast strains harboring the SpCas9 gene drive construct were first transformed with the high-copy LEU2-based sgRNA(u1) plasmid (pGF-V1220) and propagated on SD-LEU plates [29]. Next, Cas9/sgRNA-containing haploids were mated to target yeast strains of the opposite mating type on rich media for 24 h and transferred to diploid selection plates (SD-LEU-HIS) for three consecutive stages of selection. Diploids were cultured overnight to saturation (raffinose/sucrose), grown in rich medium containing galactose for (0 to 24 h) to express Cas9, diluted, spread onto SD-LEU medium at a density of 200–500 cells per plate, and incubated at 30 °C for 48 h. Finally, colonies were replica-plated by velvet transfer to a second SD-LEU and SD-HIS plate for 18–24 h, imaged, and the total surviving colony number was quantified in a single-blind fashion (between 100 and 250 colonies counted for each condition). Gene drive and target genome status were interrogated by diagnostic PCRs (also see Additional file 1: Fig. S2, Table S1) on isolated chromosomal DNA (also confirmed as diploids [29]). Genetic safeguards to contain yeast gene drives included the use of artificial sequences [51] programmed at the HIS3 locus (u1 sites for targeting), a self-excision module (u2 sites) flanking Cas9 constructs [29], an inducible promoter driving Cas9 expression (prGAL1/10), and sgRNA plasmids on an unstable high-copy vector [21, 29].
Abbreviations
- CRISPR:
-
clustered regularly interspaced short palindromic repeats
- GD:
-
gene drive
- DSB:
-
double strand break
- HR:
-
homologous recombination
- HDR:
-
homology directed repair
- NHEJ:
-
non-homologous end joining
- sgRNA:
-
single guide RNA fragment
- NLS:
-
nuclear localization sequence
- NES:
-
nuclear export signal
- GPCR:
-
G-protein coupled receptor
- DIC:
-
differential interference contrast
- Pr:
-
promoter
- (t):
-
terminator
- S:
-
synthetic complete media
References
Bajwa AA, et al. Biology and management of two important Conyza weeds: a global review. Environ Sci Pollut Res Int. 2016;23:24694–710. https://doi.org/10.1007/s11356-016-7794-7.
Neilson BJ, Wall CB, Mancini FT, Gewecke CA. Herbivore biocontrol and manual removal successfully reduce invasive macroalgae on coral reefs. PeerJ. 2018;6:e5332. https://doi.org/10.7717/peerj.5332.
Kergunteuil A, et al. Biological control beneath the feet: a review of crop protection against insect root herbivores. Insects. 2016. https://doi.org/10.3390/insects7040070.
Khan Z, Midega CA, Hooper A, Pickett J. Push-pull: chemical ecology-based integrated pest management technology. J Chem Ecol. 2016;42:689–97. https://doi.org/10.1007/s10886-016-0730-y.
Stenberg JA, Heil M, Ahman I, Bjorkman C. Optimizing crops for biocontrol of pests and disease. Trends Plant Sci. 2015;20:698–712. https://doi.org/10.1016/j.tplants.2015.08.007.
Peterson JA, Ode PJ, Oliveira-Hofman C, Harwood JD. Integration of plant defense traits with biological control of arthropod pests: challenges and opportunities. Front Plant Sci. 2016;7:1794. https://doi.org/10.3389/fpls.2016.01794.
Delves MJ, Angrisano F, Blagborough AM. Antimalarial transmission-blocking interventions: past, present, and future. Trends Parasitol. 2018. https://doi.org/10.1016/j.pt.2018.07.001.
Tizifa TA, et al. Prevention efforts for malaria. Curr Trop Med Rep. 2018;5:41–50. https://doi.org/10.1007/s40475-018-0133-y.
Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. https://doi.org/10.1126/science.1225829.
Jinek M, et al. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. https://doi.org/10.7554/elife.00471.
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–7. https://doi.org/10.1038/nature13011.
Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR–Cas9. Science. 2014;346:1258096. https://doi.org/10.1126/science.1258096.
Estrela R, Cate JH. Energy biotechnology in the CRISPR–Cas9 era. Curr Opin Biotechnol. 2016;38:79–84. https://doi.org/10.1016/j.copbio.2016.01.005.
Sternberg SH, Doudna JA. Expanding the biologist’s toolkit with CRISPR–Cas9. Mol Cell. 2015;58:568–74. https://doi.org/10.1016/j.molcel.2015.02.032.
Wright AV, Nunez JK, Doudna JA. Biology and applications of CRISPR systems: harnessing nature’s toolbox for genome engineering. Cell. 2016;164:29–44. https://doi.org/10.1016/j.cell.2015.12.035.
Esvelt KM, Gemmell NJ. Conservation demands safe gene drive. PLoS Biol. 2017;15:e2003850. https://doi.org/10.1371/journal.pbio.2003850.
Prowse TAA, et al. Dodging silver bullets: good CRISPR gene-drive design is critical for eradicating exotic vertebrates. Proc Biol Sci. 2017. https://doi.org/10.1098/rspb.2017.0799.
Courtier-Orgogozo V, Morizot B, Boete C. Using CRISPR-based gene drive for agriculture pest control. EMBO Rep. 2017. https://doi.org/10.15252/embr.201744822.
Roggenkamp E, et al. CRISPR-UnLOCK: multipurpose Cas9-based strategies for conversion of yeast libraries and strains. Front Microbiol. 2017;8:1773. https://doi.org/10.3389/fmicb.2017.01773.
Shapiro RS, et al. A CRISPR–Cas9-based gene drive platform for genetic interaction analysis in Candida albicans. Nat Microbiol. 2018;3:73–82. https://doi.org/10.1038/s41564-017-0043-0.
DiCarlo JE, Chavez A, Dietz SL, Esvelt KM, Church GM. Safeguarding CRISPR–Cas9 gene drives in yeast. Nat Biotechnol. 2015;33:1250–5. https://doi.org/10.1038/nbt.3412.
Gantz VM, et al. Highly efficient Cas9-mediated gene drive for population modification of the malaria vector mosquito Anopheles stephensi. Proc Natl Acad Sci USA. 2015;112:E6736–43. https://doi.org/10.1073/pnas.1521077112.
Hammond A, et al. A CRISPR–Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol. 2016;34:78–83. https://doi.org/10.1038/nbt.3439.
Grunwald HA, et al. Super-Mendelian inheritance mediated by CRISPR/Cas9 in the female mouse germline. bioRxiv. 2018. https://doi.org/10.1101/362558.
Noble C, Olejarz J, Esvelt KM, Church GM, Nowak MA. Evolutionary dynamics of CRISPR gene drives. Sci Adv. 2017;3:e1601964. https://doi.org/10.1126/sciadv.1601964.
Vella MR, Gunning CE, Lloyd AL, Gould F. Evaluating strategies for reversing CRISPR–Cas9 gene drives. Sci Rep. 2017;7:11038. https://doi.org/10.1038/s41598-017-10633-2.
Dhole S, Vella MR, Lloyd AL, Gould F. Invasion and migration of spatially self-limiting gene drives: a comparative analysis. Evol Appl. 2018;11:794–808. https://doi.org/10.1111/eva.12583.
Noble C, Adlam B, Church GM, Esvelt KM, Nowak MA. Current CRISPR gene drive systems are likely to be highly invasive in wild populations. eLife. 2018. https://doi.org/10.7554/elife.33423.
Roggenkamp E, et al. Tuning CRISPR–Cas9 gene drives in Saccharomyces cerevisiae. G3 (Bethesda). 2018;8:999–1018. https://doi.org/10.1534/g3.117.300557.
Basgall EM, et al. Gene drive inhibition by the anti-CRISPR proteins AcrIIA2 and AcrIIA4 in Saccharomyces cerevisiae. Microbiology. 2018;164:464–74. https://doi.org/10.1099/mic.0.000635.
Kim YH, Han ME, Oh SO. The molecular mechanism for nuclear transport and its application. Anat Cell Biol. 2017;50:77–85. https://doi.org/10.5115/acb.2017.50.2.77.
Bauer NC, Doetsch PW, Corbett AH. Mechanisms regulating protein localization. Traffic. 2015;16:1039–61. https://doi.org/10.1111/tra.12310.
Sorokin AV, Kim ER, Ovchinnikov LP. Nucleocytoplasmic transport of proteins. Biochem Biokhimiia. 2007;72:1439–57.
Poon IK, Jans DA. Regulation of nuclear transport: central role in development and transformation? Traffic. 2005;6:173–86. https://doi.org/10.1111/j.1600-0854.2005.00268.x.
Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol. 2007;8:195–208. https://doi.org/10.1038/nrm2114.
DeGrasse JA, et al. Evidence for a shared nuclear pore complex architecture that is conserved from the last common eukaryotic ancestor. Mol Cell Proteom MCP. 2009;8:2119–30. https://doi.org/10.1074/mcp.M900038-MCP200.
Wente SR, Rout MP. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol. 2010;2:a000562. https://doi.org/10.1101/cshperspect.a000562.
O’Reilly AJ, Dacks JB, Field MC. Evolution of the karyopherin-beta family of nucleocytoplasmic transport factors; ancient origins and continued specialization. PLoS ONE. 2011;6:e19308. https://doi.org/10.1371/journal.pone.0019308.
Glass Z, Lee M, Li Y, Xu Q. Engineering the delivery system for CRISPR-based genome editing. Trends Biotechnol. 2018;36:173–85. https://doi.org/10.1016/j.tibtech.2017.11.006.
Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984;39:499–509.
Liu KI, et al. A chemical-inducible CRISPR–Cas9 system for rapid control of genome editing. Nat Chem Biol. 2016;12:980–7. https://doi.org/10.1038/nchembio.2179.
Zetsche B, Volz SE, Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol. 2015;33:139–42. https://doi.org/10.1038/nbt.3149.
Hu P, Zhao X, Zhang Q, Li W, Zu Y. Comparison of various nuclear localization signal-fused Cas9 proteins and Cas9 mRNA for genome editing in Zebrafish. G3 (Bethesda). 2018;8:823–31. https://doi.org/10.1534/g3.117.300359.
Menoret S, et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Sci Rep. 2015;5:14410. https://doi.org/10.1038/srep14410.
Torres-Ruiz R, et al. Efficient recreation of t(11;22) EWSR1-FLI1+ in human stem cells using CRISPR/Cas9. Stem Cell Rep. 2017;8:1408–20. https://doi.org/10.1016/j.stemcr.2017.04.014.
Staahl BT, et al. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat Biotechnol. 2017;35:431–4. https://doi.org/10.1038/nbt.3806.
Weninger A, Glieder A, Vogl T. A toolbox of endogenous and heterologous nuclear localization sequences for the methylotrophic yeast Pichia pastoris. FEMS Yeast Res. 2015. https://doi.org/10.1093/femsyr/fov082.
Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. https://doi.org/10.1126/science.1231143.
Shen B, et al. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013;23:720–3. https://doi.org/10.1038/cr.2013.46.
Kosugi S, et al. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J Biol Chem. 2009;284:478–85. https://doi.org/10.1074/jbc.M807017200.
Finnigan GC, Thorner J. mCAL: a new approach for versatile multiplex action of Cas9 using one sgRNA and loci flanked by a programmed target sequence. G3 (Bethesda). 2016;6:2147–56.
Kosugi S, Hasebe M, Tomita M, Yanagawa H. Nuclear export signal consensus sequences defined using a localization-based yeast selection system. Traffic. 2008;9:2053–62. https://doi.org/10.1111/j.1600-0854.2008.00825.x.
Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–73.
Hammond AM, et al. The creation and selection of mutations resistant to a gene drive over multiple generations in the malaria mosquito. PLoS Genet. 2017;13:e1007039. https://doi.org/10.1371/journal.pgen.1007039.
Drury DW, Dapper AL, Siniard DJ, Zentner GE, Wade MJ. CRISPR/Cas9 gene drives in genetically variable and nonrandomly mating wild populations. Sci Adv. 2017;3:e1601910. https://doi.org/10.1126/sciadv.1601910.
Champer J, et al. Reducing resistance allele formation in CRISPR gene drive. Proc Natl Acad Sci USA. 2018. https://doi.org/10.1073/pnas.1720354115.
Wang Y, et al. A ‘suicide’ CRISPR–Cas9 system to promote gene deletion and restoration by electroporation in Cryptococcus neoformans. Sci Rep. 2016;6:31145. https://doi.org/10.1038/srep31145.
Osakabe Y, et al. Optimization of CRISPR/Cas9 genome editing to modify abiotic stress responses in plants. Sci Rep. 2016;6:26685. https://doi.org/10.1038/srep26685.
Makkerh JP, Dingwall C, Laskey RA. Comparative mutagenesis of nuclear localization signals reveals the importance of neutral and acidic amino acids. Curr Biol CB. 1996;6:1025–7.
Tarendeau F, et al. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat Struct Mol Biol. 2007;14:229–33. https://doi.org/10.1038/nsmb1212.
la Cour T, et al. NESbase version 1.0: a database of nuclear export signals. Nucl Acids Res. 2003;31:393–6.
Xu D, Farmer A, Collett G, Grishin NV, Chook YM. Sequence and structural analyses of nuclear export signals in the NESdb database. Mol Biol Cell. 2012;23:3677–93. https://doi.org/10.1091/mbc.E12-01-0046.
Kosugi S, Yanagawa H, Terauchi R, Tabata S. NESmapper: accurate prediction of leucine-rich nuclear export signals using activity-based profiles. PLoS Comput Biol. 2014;10:e1003841. https://doi.org/10.1371/journal.pcbi.1003841.
Xiao CY, Hubner S, Jans DA. SV40 large tumor antigen nuclear import is regulated by the double-stranded DNA-dependent protein kinase site (serine 120) flanking the nuclear localization sequence. J Biol Chem. 1997;272:22191–8.
Fontes MR, et al. Role of flanking sequences and phosphorylation in the recognition of the simian-virus-40 large T-antigen nuclear localization sequences by importin-alpha. Biochem J. 2003;375:339–49. https://doi.org/10.1042/bj20030510.
Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci USA. 2009;106:10171–6. https://doi.org/10.1073/pnas.0900604106.
Maji B, et al. Multidimensional chemical control of CRISPR–Cas9. Nat Chem Biol. 2017;13:9–11. https://doi.org/10.1038/nchembio.2224.
Kipniss NH, et al. Engineering cell sensing and responses using a GPCR-coupled CRISPR–Cas system. Nat Commun. 2017;8:2212. https://doi.org/10.1038/s41467-017-02075-1.
Baeumler TA, Ahmed AA, Fulga TA. Engineering synthetic signaling pathways with programmable dCas9-based chimeric receptors. Cell Rep. 2017;20:2639–53. https://doi.org/10.1016/j.celrep.2017.08.044.
Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001.
Finnigan GC, Thorner J. Complex in vivo ligation using homologous recombination and high-efficiency plasmid rescue from Saccharomyces cerevisiae. Bio-protocol. 2015;5: e1521. http://www.bio-protocol.org/e1521.
Eckert-Boulet N, Pedersen ML, Krogh BO, Lisby M. Optimization of ordered plasmid assembly by gap repair in Saccharomyces cerevisiae. Yeast. 2012;29:323–34. https://doi.org/10.1002/yea.2912.
Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. https://doi.org/10.1093/nar/gnh110.
Brachmann CB, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–32. https://doi.org/10.1002/(sici)1097-0061(19980130)14:2%3c115:aid-yea204%3e3.0.co;2-2.
Authors’ contributions
MEG, EMB, ICL, SCG, YY, MH, and GCF built all reagents (plasmids and yeast) and performed all experiments. MEG, EMB, ICL, SCG, and GCF performed all data analyses and figure preparation. GCF wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Rachael Giersch, Emily Roggenkamp, and Madison Schrock (Kansas State University) for useful comments and laboratory assistance.
Competing interests
GC.F. (Kansas State University) has filed for a provisional patent on September 29, 2017 (U.S. Provisional Patent Application Serial No. 62/565,651, “Programmed modulation of CRISPR/Cas9 activity”) followed by patent filing on January 31, 2018 regarding the data presented in this work. A patent was filed (G.C.F.) on April 20, 2017 by the University of California, Berkeley, “Methods and Compositions for Genomic Editing” International Application No. PCT/US2017/028676 and published as No. WO 2017/189336 A1 on November 2, 2017, for the artificial target sites (u1/u2) used in this study. GCF declares no non-financial competing interests. MEG, EMB, ICL, SCG, YY, and MH declare no competing interests of any kind.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This project was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grant Number P20 GM103418 (G.C.F and E.M.B). This work was also supported by the USDA National Institute of Food and Agriculture, Hatch Project 1013520 (to G.C.F.) and by an Innovative Research Award (to G.C.F.) from the Johnson Cancer Research Center (Kansas State University). Also, Undergraduate Research Awards from the College of Arts and Sciences (Kansas State University) funded this work (to M.E.G and I.C.L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institute of Health.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1.
Supplementary information.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Goeckel, M.E., Basgall, E.M., Lewis, I.C. et al. Modulating CRISPR gene drive activity through nucleocytoplasmic localization of Cas9 in S. cerevisiae. Fungal Biol Biotechnol 6, 2 (2019). https://doi.org/10.1186/s40694-019-0065-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40694-019-0065-x