
Abstract
The automated detection of defects in high-angle annular dark-field Z-contrast (HAADF) scanning-transmission-electron microscopy (STEM) images has been a major challenge. Here, we report an approach for the automated detection and categorization of structural defects based on changes in the material’s local atomic geometry. The approach applies geometric graph theory to the already-found positions of atomic-column centers and is capable of detecting and categorizing any defect in thin diperiodic structures (i.e., “2D materials”) and a large subset of defects in thick diperiodic structures (i.e., 3D or bulk-like materials). Despite the somewhat limited applicability of the approach in detecting and categorizing defects in thicker bulk-like materials, it provides potentially informative insights into the presence of defects. The categorization of defects can be used to screen large quantities of data and to provide statistical data about the distribution of defects within a material. This methodology is applicable to atomic column locations extracted from any type of high-resolution image, but here we demonstrate it for HAADF STEM images.
Similar content being viewed by others


Introduction
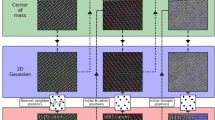
Structural defects can vastly alter the performance of materials so that control of defect distribution and density is an important tool in engineering materials with novel functionalities. Even small concentrations of defects can often change the properties of materials so that it is important to quantify the type and concentration of defects [1,2,3]. Over the last two decades, aberration-corrected scanning-transmission-electron-microscopy (STEM) has become a quantitative structural tool capable of locating atomic columns with picometer-level precision. The ability to achieve sub-pixel precision for the location of the center of an atomic column in STEM images has been demonstrated through image analysis techniques such as finding the center of mass and 2D function fitting with a Gaussian, allowing for accurate, consistent, and repeatable determination of the centers of atomic columns in STEM images [4,5,6,7,8]. Utilizing the crystallographic nomenclature [9], STEM essentially images diperiodic structures where thick structures are often referred to as 3D or bulk-like materials and thinner structures of just a few atomic layers are often termed 2D materials. Within a STEM image, it is possible to visually identify many defects such as impurities, interstitials, stacking faults, and a plethora of other complex defects for both types of diperiodic structures.
Several methods exist to detect and identify defects in STEM images, each having unique benefits and limitations. Defects within atomic columns can be detected by examining deviations in the contrast, looking for deviations in the local atomic-scale structure [10, 11], overlaying an ideal atomic-scale structure on the image [12], and by using vector tracing [13]. These methods include measuring the distance between neighboring atoms in the structure and then using statistics and modeling to detect the presence and depth of a single defect in atomic columns [10, 11]; measuring the relative positions of neighboring atoms and then applying principal component analysis (PCA) followed by K-means clustering to map the ideal atomic-scale structure and statistical deviations from this idealized structure [12]; and using the Fourier transform of the image to determine the crystal structure’s lattice parameter and then overlaying the obtained periodic structure on the atomic coordinates [14]. Alternatively, defects may be detected using cross-correlation between the STEM image and a simulated STEM image based on coordinates obtained by relaxing a model structure via density-functional-theory (DFT) calculations and then detecting defects through areas of low correlation [15]. Determination of the composition for mixed-species atomic columns can also be accomplished through the use of a parametric model based on statistical-parameter-estimation theory and further combined with STEM image simulations to quantitatively improve the model [16, 17]. All of these methods have achieved detection of defects that would not be possible or would be extremely time-intensive with the human eye. Furthermore, these methods provide a framework that is either general across materials or tailored to specific material systems in such a way that transferability is not limited via a known training set as might be the case in machine-learning-based object detection algorithms.
In this paper, we report the development of a method that applies cycle analysis from geometric graph theory to the positions of atomic-column centers and is capable of detecting a wide range of defects in STEM images with no prior knowledge of the material. Although graph theoretical techniques have been used previously for the segmentation of spatial regions and identification of voids in imaged materials [18,19,20], these applications do not necessarily provide information on slight structural deviations in the imaged material at an atomic-column by atomic-column level of detail. In graph theory, a cycle is a path between points that connects a point back to itself. Multiple types of cycles exist such as the simple-walk cycle that does not allow any point or connection to be repeated. For this paper, a particular type of cycle is created with the following conditions: no vertices may be repeated, no connecting line may intersect another connecting line, the cycle must enclose a reference atomic column, the cycle must not enclose any additional atomic columns, and, finally, the cycle must be the shortest path connecting the vertices. For every atomic column in an image, a single cycle is found to represent it. Based on the number of vertices and the area of the cycles, it is possible to detect and categorize defects in the STEM image. The concept of pre-filtering wherein crystallographic information can inform the search is also discussed; however, the use of such databases may be limited for real-time analysis during image acquisition since the crystallographic nature of the material may not be known a priori. The approach is applied to STEM images of both thick diperiodic structures (i.e., 3D or bulk-like materials) as well as thin diperiodic structures (i.e., “2D materials”). In bulk-like silicon doped with bismuth, we demonstrate the ability of cycles to detect the Bi dopants in the atomic columns and compare with Z-contrast. In monolayer MoS2 doped with rhenium, sulfur vacancies are detected using two different cycle metrics.
Methods and materials
Image filtering, finding centers of atomic columns
All of the raw STEM data are first processed to identify the centers of atomic columns as follows. At each pixel, a subimage is defined, centered at the pixel and encompassing an area roughly equal to the area per atomic column. These subimages are filtered using PCA to remove noise and surface contamination [21, 22]. The subimages are then passed through a 2D correlation [23] with an ideal atomic column (a 2D Gaussian) defined by:
where A and B are two 2D matrices of the same size while \(\overline{A}\) and \(\overline{B}\) are their determinants, respectively. This process returns a single normalized intensity. From the filtered image, the centers of atomic columns are then found using a simple intensity threshold followed by density-based clustering [24]. Any clusters that do not meet a minimum size requirement are rejected. The center of mass of each cluster is treated as the center of an atomic column. Further refinement of the positions of the atomic-column centers is performed using nonlinear least-squares curve fitting between the raw data and a 2D Gaussian. The center of the fitted Gaussian is then treated as the refined center of the atomic column.
Finding cycles for each atomic column
A cycle is a path that connects atomic-column centers or “vertices” in such a way that it forms a closed loop to the original vertex. First, all possible cycles are found, after which they are filtered based on the previously described restrictions. To find the cycles associated with an atomic column, one considers only its n nearest points to speed up calculation time. The connections between each vertex and its m nearest neighbors are mapped. Typically, n = 40 columns and m = 10 columns. Then, using the nearest neighbor (or one of the equidistant nearest neighbors) as a starting point, all valid connections are followed to neighboring vertices. This process is repeated until the starting vertex is encountered or no connection can be followed without causing a repeating vertex.
Filtering cycles
Once the cycles are found as described above, they must be filtered to find a single cycle to represent each atomic column. This filtering is done by checking every cycle to see if it meets a series of rules. These rules are that no point may be repeated in the cycle, no connecting line may intersect another connecting line, the cycle must enclose the reference atomic column, the cycle must not enclose any other atomic column, the cycle must have no smaller angle then x degrees (x was set at 45o in the current work), and finally the cycle must be the shortest path connecting the points within a tolerance factor (within 1% of shortest path in the current work) (Fig. 1). Once all of the cycles that do not meet these criteria are removed, the cycles with the largest number of points are selected. From these cycles the one with the largest area is chosen as the cycle to represent an atomic column. The reason the largest cycle is used is for reproducibility. To ensure that we choose the same cycle each time, it must have a unique feature. Since the smallest cycle would always be a triangle and provide little information, the largest cycle is used instead. In cases of crystalline symmetry (i.e., rotationally equivalent cycles), a preferred cycle orientation can be chosen.

a Rejected cycle due to cycle lines crossing each other. b Rejected cycle due to an extra atom enclosed in the cycle. c Rejected cycle due to cycle not being the shortest path connecting the points. d Accepted cycle that meets all parameters. e Accepted cycle that meets all parameters. f Accepted cycle that meets all parameters and is also the cycle with the most points
In order to provide a concrete example of defect detection, we demonstrate in Fig. 2 how the above described algorithm finds a vacancy (i.e., more generally, a missing atomic column) for a 2D material with a hexagonal lattice like graphene (Fig. 2a). In Fig. 2b, three different orientations of a seven-sided cycle are shown. This is the optimal cycle based on the algorithm filter in the absence of defects, while the threefold symmetry is a manifestation of the point-group symmetries of the lattice. In the presence of strain, the length of the three bonds may be differentiated. In Fig. 2c, an atom that serves as a second-nearest neighbor to the vacancy site is shown, having a nine-sided cycle as its optimal cycle. In practical implementations, however, restrictions on the atomic-column search distance may cause the original seven-sided cycle to be found instead. In Fig. 2d, a third-nearest neighbor to the vacancy site retains the original seven-sided cycle (though the threefold symmetry is broken). For the atomic columns adjacent to the vacancy site (Fig. 2e), the optimal cycle is an eight-sided cycle. The resulting cycle mapping of the structure is shown in Fig. 2f, highlighting the location of the vacancy. Due to the atomic-column distance search limits, the purple atomic columns may be grouped with the blue atomic columns; there is no loss of detection of the vacancy due to the uniqueness of the directly neighboring sites. The uniqueness of the neighboring sites implies that the method is, in general, well suited to detection of missing columns with high accuracy in identification. Further classification due to variations in the vertex count of cycles is discussed in Section "Clustering cycles into defects using number of points in cycle".

a Subset of a graphene-like structure in a hexagonal lattice (for the present example, the lattice is assumed to continue outside the drawn boundary). b Three rotationally equivalent seven-sided cycles indicating atoms in defect-free regions. Atomic columns inside such cycles are indicated in blue. c Optimal (nine-sided) and restricted (seven-sided) cycles for atomic columns that are second-nearest neighbor to a vacancy (indicated in purple). d Third-nearest neighbor atomic columns retain the original cycle with loss of rotational symmetry. e Atomic columns adjacent to the vacancy site are described by an eight-sided cycle (indicated in orange). f Final cycle-based color coding of the observed atomic columns based on the algorithm
Constructing cycles using Delaunay triangulation
Finding all possible cycles and checking them is a time-consuming process. A faster way to find a good guess of the best cycle is through the use of Delaunay triangulation [25]. For an atomic column, the positions of the nearest n neighbors (typically n = 40) are put into a Delaunay triangulation algorithm. Using the triangle that encloses the atomic column as the starting cycle, triangles from the Delaunay triangulation are combined with the cycle, testing whether the increased cycle at each step meets the selection criteria, until no further triangles can be added that meet the criteria. This method of finding the cycle for an atomic column is more than an order of magnitude faster than searching all possible cycles. However, it does not always find the true, correct cycle, as determined by a time-consuming exhaustive search, though it is often close. Generally, the fast process does not match the results of an exhaustive search only around defects that change the local structure, which does not affect the ability of this method to correctly detect defects (Fig. 3a, b).

The centers of atomic columns for MoS2 colored based on the number of points in the cycles associated with them for a searching all cycles, b using Delaunay triangulation, and c using Delaunay triangulation plus pre-filtering
Pre-filtering of cycles
To further improve the speed of the algorithm, pre-filtering of cycles was tested. As all defect-free periodic crystalline materials can be described by a Bravais lattice with a given atomic basis and a set of symmetries, it is often possible to know the correct cycle before searching. To take advantage of this, we created a small library of possible cycles. We can overlay a library cycle onto the points by aligning it to the reference point and its nearest neighbor (or one of equidistant nearest neighbors) and scale it to fit the distance between the two points. Starting with the largest cycles in the library, the cycles are overlaid onto the points. If every point in the cycle coincides, within some uncertainty, with a point in the image, it is selected as the correct cycle. This process sometimes yields too small of a cycle, but it has no effect on defect detection (Fig. 3c). The speed improvement of pre-filtering is based on the size of the test library and the percent of defect atomic columns, as this is an additional operation that must be performed on defect atomic columns.
Clustering cycles into defects using number of points in cycle
The first method of detecting defects is by looking at deviations in the number of vertices in the cycles. In “2D materials”, atomic columns near defects that cause changes in the local geometric structure such as vacancies, interstitials, or stacking faults have cycles that contain a different number of points than in the perfect material’s local structure. To detect a defect, we mark as acceptable any atomic column that has a cycle with the same number of vertices in it as a cycle in the perfect crystal. The remaining cycles are clustered together using a density-based clustering algorithm [24]. This algorithm randomly selects a point as the start of a cluster and then adds every point that is within a specified radius into the cluster. This is repeated until no point can be added to the cluster. These clusters are then grouped based on the number of atomic columns in the cluster. This procedure allows for the automatic detection and grouping of defects in STEM data.
Using cycle area to find defects
Another method of using cycles to find defects is to look at changes in the cycle’s area. Using the cycle’s area to look for defects allows for the detection of defects that do not change the local structure’s geometric coordination relative to defect-free regions, such as interstitials and vacancies in bulk materials or substitutional impurities. Any cycle area that is much larger or smaller than the average cycle area within some threshold represents the presence of a defect near that cycle. This works on a similar idea to previous work where single strontium and lanthanum vacancies where detected by measuring changes in the distance to nearest-neighbor atomic columns [10, 11]. The accuracy of using the area to detect such defects is limited by how much deviation in the cycle areas exists in the nominally pristine regions and the threshold for the change in area that is used to classify the defect.
Results and discussion
Here, we demonstrate the method for the detection of defects in 2D and 3D materials and discuss the method’s efficacy at finding such defects.
3D bulk-like materials
The ability to study defects using cycles is first demonstrated in a Mo–V–M–O material system, where M can be one of any number of atomic species [26], with Te being the most likely one in the current sample. The Mo–V–M–O compound is a material that has been studied as a potential catalyst and can display a variety of interesting phases and defects. In certain areas, this material possesses large stacking faults and missing atomic columns making it a good material to demonstrate the steps described in the "Methods" section (Fig. 4). In Mo–V–M–O, we believe that we can see the pooling of vacancies or M atoms under the surface of the material (Fig. 5c). The potential for large-scale vacancy clusters in this material and large stacking faults can be seen in Fig. 4. Ordinarily, if these types of defects occur below the surface layers, they may not be visible in Z-contrast imaging due to the presence of surface contamination which can obscure slight changes in column intensity.

a Original HAADF image of Mo–V–M–O. b Atomic column location colored based on the number of atoms in associated cycles. c Atomic columns whose cycles are not part of the perfect crystal. d Non-perfect crystal atomic columns (i.e., those with differing cycles) grouped into defects and colored based on the number of atoms in a defect (single missing column—yellow; two adjoining missing atomic columns—purple; large staking fault—black)

a Raw HAADF STEM image of Mo–V–M oxide. b Interpolated map of local intensity around every atomic column in panel a with the location of atomic columns shown. c Interpolated map of the cycle size associated with every atomic column in panel a with the location of atomic columns shown. d Raw HAADF STEM image of Bi-doped Si. e Location of every atomic column in panel d colored based on the local intensity around every atomic column. f Location of every atomic column in panel d colored based on the cycle size associated every atomic column
The ability of using cycle area to detect defects within an atomic column was tested using bismuth (Bi) doped silicon (Si) (Fig. 5d). This material was used because the Z-squared difference between Bi and Si makes identifying the Bi locations straightforward as an independent comparison. The ability of cycle size to identify Bi within an atomic column was found to be worse than the reference of Z-squared intensity. Using cycle size, it was only possible to identify the approximate location of roughly 80% of the Bi dopants (Fig. 5e, f). In areas with more than one Bi dopant in close proximity, it is difficult to identify the number and exact location of the dopant. However, for isolated Bi, it is much easier. The 80% identification rate is due to the depth of the defects in the material, as intensity can help identify defects at a greater depth than using the distortion in the local structure on which cycle area identification relies. This result is in line with previous works that have used distortions in the local lattice to identify defects [10, 11].
2D materials
For 2D materials, we have selected rhenium-doped MoS2 (Fig. 6a). This material was chosen due to the nature of defects available, namely rhenium (Re) dopants at molybdenum (Mo) sites along with single and double sulfur (S) vacancies. We filtered this image and found all the atomic columns using the procedure described in the Methods sections. Using the centers of the atomic columns, the cycles are found for each atomic column more than 2.5 times the average nearest neighbor distance from the edge. The number of points in each cycle are analyzed and the defects are subsequently categorized (Fig. 6b). Using the number of points in the cycle, all of the missing S columns were detected and categorized into single missing columns, two adjacent missing columns, and three adjacent missing columns. In MoS2, a fully missing S column is a divacancy. To find the single S vacancies, the areas of the cycles were used (Fig. 6c). Sulfur vacancies cause a noticeable decrease in the area of cycles. No method was found to identify the Re dopants using cycles because the frequent occurence of S vacancies near the Re makes the use of cycle area unreliable. The presence of S vacancies near Re dopants may be explained by the differing electronic properties of these defects in MoS2. Rhenium dopant atoms are shallow donor defects [27, 28] whereas sulfur vacancies are deep acceptor defects [29, 30]. Therefore, the S vacancies act as electron traps which can compensate for the excess electrons introduced by the Re dopant atoms. The electron transfer leads to an energy gain, making pairing the two defects energetically favorable.

a STEM image of Re-doped MoS2. b Atomic columns identified to be near a defect using number of points in the cycles. c Area of the cycles where smaller areas are due to sulfur vacancies
Conclusions
We described a method to detect structural defects in materials based on the concept of cycles from graph theory. We tested the method on STEM data for both thin and thick diperiodic materials (i.e., 2D and 3D or bulk-like materials, respectively). The method is best suited to finding defects in 2D materials, but it can supply useful information about the presence of defects in thicker materials as well. In 2D materials, we demonstrated the ability of the method to distinguish a number of common defects, including interstitials and vacancies. In practice, the present method provides a relatively fast, non-preconditioned approach to identify and classify defects in atomic-scale images, which can augment existing methods in an ensemble approach.
Availability of data and materials
Data used in this study can be provided by the authors upon request.
Abbreviations
- HAADF:
-
High-angle annular dark-field
- STEM:
-
Scanning transmission electron microscopy
- PCA:
-
Principal component analysis
- DFT:
-
Density functional theory
References
Miranda, E., Dobrosavljević, V.: Disorder-driven non-fermi liquid behaviour of correlated electrons. Rep Prog Phys 68, 2337–2408 (2005). https://doi.org/10.1088/0034-4885/68/10/R02
Abrahams, E., Kotliar, G.: The metal-insulator transition in correlated disordered systems. Science 274(5294), 1853–1854 (1996)
Lee, P.A., Nagaosa, N., Wen, X.G.: Doping a mott insulator: physics of high-temperature superconductivity. Rev Mod Phys (2006). https://doi.org/10.1103/RevModPhys.78.17
Borisevich, A.Y., Chang, H.J., Huijben, M., Oxley, M.P., Okamoto, S., Niranjan, M.K., Burton, J.D., Tsymbal, E.Y., Chu, Y.H., Yu, P., Ramesh, R., Kalinin, S.V., Pennycook, S.J.: Suppression of octahedral tilts and associated changes in electronic properties at epitaxial oxide heterostructure interfaces. Phys Rev Lett 105, 087204 (2010). https://doi.org/10.1103/PhysRevLett.105.087204
Ishikawa, R., Lupini, A.R., Hinuma, Y., Pennycook, S.J.: Large-angle illumination STEM: toward three-dimensional atom-by-atom imaging. Ultramicroscopy 151, 122–129 (2015). https://doi.org/10.1016/j.ultramic.2014.11.009
Krivanek, O.L., Chisholm, M.F., Nicolosi, V., Pennycook, T.J., Corbin, G.J., Dellby, N., Murfitt, M.F., Own, C.S., Szilagyi, Z.S., Oxley, M.P., Pantelides, S.T., Pennycook, S.J.: Atom-by-atom structural and chemical analysis by annular dark-field electron microscopy. Nature 464, 571–574 (2010). https://doi.org/10.1038/nature08879
Shen, X., Hernández-Pagan, E.A., Zhou, W., Puzyrev, Y.S., Idrobo, J.-C., Macdonald, J.E., Pennycook, S.J., Pantelides, S.T.: Interlaced crystals having a perfect Bravais lattice and complex chemical order revealed by real-space crystallography. Nat Commun 5, 5431 (2014). https://doi.org/10.1038/ncomms6431
Zhou, W., Lee, J., Nanda, J., Pantelides, S.T., Pennycook, S.J., Idrobo, J.-C.: Atomically localized plasmon enhancement in monolayer graphene. Nat Nanotechnol 7, 161–165 (2012). https://doi.org/10.1038/nnano.2011.252
Nespolo, M.: Lattice versus structure, dimensionality versus periodicity: a crystallographic Babel? J Appl Crystallogr 52, 451–456 (2019). https://doi.org/10.1107/S1600576719000463
Feng, J., Kvit, A. V., Zhang, C., Hoffman, J., Bhattacharya, A., Morgan, D., Voyles, P.M.: Imaging of single La vacancies in LaMnO3. arXiv:1711.06308 (2017)
Kim, H., Zhang, J.Y., Raghavan, S., Stemmer, S.: Direct observation of Sr vacancies in SrTiO3 by quantitative scanning transmission electron microscopy. Phys Rev X 6, 041063 (2016). https://doi.org/10.1103/PhysRevX.6.041063
Belianinov, A., He, Q., Kravchenko, M., Jesse, S., Borisevich, A., Kalinin, S.V.: Identification of phases, symmetries and defects through local crystallography. Nat Commun (2015). https://doi.org/10.1038/ncomms8801
Liu, Z., Suenaga, K., Wang, Z., Shi, Z., Okunishi, E., Iijima, S.: Identification of active atomic defects in a monolayered tungsten disulphide nanoribbon. Nat Commun (2011). https://doi.org/10.1038/ncomms1224
Borisevich, A.Y., Ovchinnikov, O.S., Chang, H.J., Oxley, M.P., Yu, P., Seidel, Ќ.J., Eliseev, E.A., Morozovska, A.N., Ramesh, R., Pennycook, Ќ.S.J., Kalinin, S.V.: Mapping octahedral tilts and polarization across a domain wall in BiFeO3 from Z-contrast scanning transmission electron microscopy image atomic column shape analysis. ACS Nano 4, 6071–6079 (2010). https://doi.org/10.1021/nn1011539
Ovchinnikov, O.S., Hara, A., Nicholl, R.J., Hachtel, J.A., Bolotin, K., Lupini, A., Jesse, S., Baddorf, A.P., Kalinin, S.V., Borisevich, A.Y., Pantelides, S.T.: Theory-assisted determination of nano-rippling and impurities in atomic resolution images of angle-mismatched bilayer graphene. 2D Materials 5, 041008 (2018). https://doi.org/10.1088/2053-1583/aadb5f
Van Aert, S., Verbeeck, J., Erni, R., Bals, S., Luysberg, M., Dyck, D.Van, Tendeloo, G.Van: Quantitative atomic resolution mapping using high-angle annular dark field scanning transmission electron microscopy. Ultramicroscopy 109, 1236–1244 (2009). https://doi.org/10.1016/j.ultramic.2009.05.010
Martinez, G.T., Rosenauer, A., De Backer, A., Verbeeck, J., Van Aert, S.: Quantitative composition determination at the atomic level using model-based high-angle annular dark field scanning transmission electron microscopy. Ultramicroscopy 137, 12–19 (2014). https://doi.org/10.1016/j.ultramic.2013.11.001
Frangakis, A.S., Hegerl, R.: Segmentation of two- and three-dimensional data from electron microscopy using eigenvector analysis. J Struct Biol 138, 105–113 (2002). https://doi.org/10.1016/S1047-8477(02)00032-1
Wodo, O., Tirthapura, S., Chaudhary, S., Ganapathysubramanian, B.: A graph-based formulation for computational characterization of bulk heterojunction morphology. Org Electron 13, 1105–1113 (2012). https://doi.org/10.1016/j.orgel.2012.03.007
Sridharamurthy, R., Masood, T.B., Doraiswamy, H., Patel, S., Varadarajan, R., Natarajan, V.: Extraction of robust voids and pockets in proteins. In: Linsen, L., Hamann, B., Hege, H.C. (eds.) Visualization in Medicine and Life Sciences III, pp. 329–349. Mathematics and Visualization, Springer, Cham (2016)
Somnath, S., Smith, C.R., Kalinin, S.V., Chi, M., Borisevich, A., Cross, N., Duscher, G., Jesse, S.: Feature extraction via similarity search: application to atom finding and denoising in electron and scanning probe microscopy imaging. Adv Struct Chem Imaging 4, 3 (2018). https://doi.org/10.1186/s40679-018-0052-y
Bonacum, J.P., O’Hara, A., Bao, D.-L., Ovchinnikov, O.S., Zhang, Y.-F., Gordeev, G., Arora, S., Reich, S., Idrobo, J.-C., Haglund, R.F., Pantelides, S.T., Bolotin, K.I.: Atomic-resolution visualization and doping effects of complex structures in intercalated bilayer graphene. Phys Rev Mater 3(6), (2019)
McNemar, Q.: Note on the sampling error of the difference between correlated proportions or percentages. Psychometrika 12, 153–157 (1947). https://doi.org/10.1007/BF02295996
Ester, M., Kriegel, H.P., Sander, J., Xu, X.: A density-based algorithm for discovering clusters in large spatial databases with noise. In: Proceedings of the 2nd international conference on knowledge discovery and data mining, pp. 226–231 (1996)
Lee, D.T., Schachter, B.J.: Two algorithms for constructing a Delaunay triangulation. Int J Comput Inf Sci 9, 219–242 (1980). https://doi.org/10.1007/BF00977785
Shiju, N.R., Guliants, V.V.: Recent developments in catalysis using nanostructured materials. Appl Catal A Gen 356, 1–17 (2009). https://doi.org/10.1016/j.apcata.2008.11.034
Yang, S.-Z., Gong, Y., Manchanda, P., Zhang, Y.-Y., Ye, G., Chen, S., Song, L., Pantelides, S.T., Ajayan, P.M., Chisholm, M.F., Zhou, W.: Rhenium-doped and stabilized MoS 2 atomic layers with basal-plane catalytic activity. Adv Mater 30, 1803477 (2018). https://doi.org/10.1002/adma.201803477
Yang, S.-Z., Sun, W., Zhang, Y.-Y., Gong, Y., Oxley, M.P., Lupini, A.R., Ajayan, P.M., Chisholm, M.F., Pantelides, S.T., Zhou, W.: Direct cation exchange in monolayer MoS2 via recombination-enhanced migration. Phys Rev Lett 122, 106101 (2019). https://doi.org/10.1103/PhysRevLett.122.106101
Liu, D., Guo, Y., Fang, L., Robertson, J.: Sulfur vacancies in monolayer MoS 2 and its electrical contacts. Appl Phys Lett 103, 183113 (2013). https://doi.org/10.1063/1.4824893
Houssa, M., Iordanidou, K., Pourtois, G., Afanas’ev, V.V., Stesmans, A.: Point defects in MoS 2: comparison between first-principles simulations and electron spin resonance experiments. Appl Surf Sci 416, 853–857 (2017). https://doi.org/10.1016/j.apsusc.2017
Acknowledgements
Data analysis was supported by the US Department of Energy, Office of Science, Basic Energy Sciences, Materials Science and Engineering Division (OSO, SJ, SVK, AYB). Part of the data analysis was supported by US Department of Energy Grant DE-FG-02-09ER46554 and by the Vanderbilt McMinn Endowment (AO, STP). Electron microscopy was supported by the US Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division (BMH, SZY, ARL, MFC, WZ). Experiments were in part conducted at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility.
Funding
We have acknowledged all relevant funding agencies in the Acknowledgements section.
Author information
Authors and Affiliations
Contributions
OSO, AO, SJ, SVK, AYB and STP conceived the project and wrote the manuscript. OSO and AO performed the data analysis. STEM data for the analysis was provided by BMH, SZY, ARL, MFC and WZ. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ovchinnikov, O.S., O’Hara, A., Jesse, S. et al. Detection of defects in atomic-resolution images of materials using cycle analysis. Adv Struct Chem Imag 6, 3 (2020). https://doi.org/10.1186/s40679-020-00070-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40679-020-00070-x