
Abstract
Objective
Bevacizumab is an important component in the treatment of various cancers, and despite guidelines recommending its use in both ovarian and cervical cancer, patient access to bevacizumab and other angiogenesis inhibitors is limited. Biosimilars are large, structurally complex molecules that are intended to be highly similar to, and treat the same condition(s) as, an existing licensed or approved (reference) biologic, with no clinically meaningful differences in purity, potency and safety. This article summarizes the role of bevacizumab in the treatment paradigm of ovarian and cervical cancer. We also discuss the potential role of biosimilars to bevacizumab, which may offer more affordable options in the future treatment of gynecologic cancers.
Methods
Literature searches of PubMed and ClinicalTrials.gov databases were conducted. Regulatory and individual pharmaceutical company web pages were also reviewed. Search terms included “biosimilar” and “bevacizumab,” and these were used to identify information regarding biosimilar development, reporting results of biosimilar studies or biosimilars in development.
Results
At present, four bevacizumab biosimilar candidates are undergoing comparative clinical assessment, with the potential to increase access and offer efficiencies across healthcare systems.
Conclusions
It is anticipated that biologics such as bevacizumab will continue to play a key role in the treatment of an array of gynecologic cancers. Biosimilars to bevacizumab are currently in development and have the potential to increase access to medicines in a variety of settings, including gynecologic cancers.
Similar content being viewed by others


Introduction
Bevacizumab (Avastin®) is a recombinant humanized monoclonal immunoglobulin G1 antibody that binds to the human vascular endothelial growth factor and blocks its activity and angiogenesis [1]. Bevacizumab is the only complex biologic therapy indicated for the treatment of patients with cervical, epithelial ovarian and fallopian tube cancer in the United States (Table 1) and Europe. Bevacizumab is also approved for the treatment of patients with metastatic colorectal cancer, non-small-cell lung cancer (NSCLC) and metastatic renal cell cancer [1, 2]. Additionally, it is indicated for the treatment of patients with glioblastoma in the United States [1] and for use in metastatic breast cancer in Europe [2].
It is expected that the use of bevacizumab in gynecologic cancers will increase, given the recent approval (in combination with carboplatin and gemcitabine) in platinum-sensitive ovarian cancer in the United States [3] and Canada [4]. However, patient access to bevacizumab and other angiogenesis inhibitors is limited [5]. This is due to several factors, including insurance coverage, drug availability, supply and manufacturing, and concerns regarding the cost-effectiveness of bevacizumab for some patients [5].
Biologics are large, structurally complex medicinal products. Their active ingredients are created by biological processes rather than chemical synthesis. Although biologics cannot be replicated, it is possible to create a version (termed “biosimilar”) that is highly similar to an already licensed or approved reference biologic in terms of purity, safety and efficacy [6, 7]. Biosimilars have the potential to increase patient access to biologic medicines, such as bevacizumab, and this may subsequently improve clinical outcomes.
This article reviews the role of bevacizumab in the treatment paradigm of ovarian and cervical cancer. We also discuss the potential role of biosimilars to bevacizumab, which may offer more affordable options in the future treatment of gynecologic cancers.
Review
Literature searches of PubMed and ClinicalTrials.gov databases were conducted. Regulatory and individual pharmaceutical company web pages were also reviewed. Search terms included “biosimilar” and “bevacizumab,” and these were used to identify information pertaining to biosimilar development, reporting results of biosimilar studies, or biosimilars in development.
Bevacizumab in the treatment of gynecologic cancers: an overview
Bevacizumab, in combination with chemotherapy, is an important component of treatment of ovarian cancer. Approval of combination bevacizumab for the treatment of platinum-resistant recurrent epithelial ovarian or fallopian tube cancer was based on the results of an international, open-label, randomized study, AURELIA (Avastin Use in Platinum-Resistant Epithelial Ovarian Cancer), in patients with measurable ovarian cancer that had progressed <6 months following platinum-based treatment [8]. Median progression-free survival (PFS) was 6.7 months with bevacizumab (10 mg/kg every 2 weeks or 15 mg/kg every 3 weeks) plus chemotherapy (weekly paclitaxel, pegylated liposomal doxorubicin or topotecan) vs 3.4 months with chemotherapy alone (P < 0.001). Objective response rate was 27.3% with bevacizumab plus chemotherapy vs 11.8% with chemotherapy alone (P = 0.001). No statistically significant difference in overall survival (OS) was observed between the two treatment regimens. Hypertension and proteinuria were common adverse events in patients treated with bevacizumab plus chemotherapy [8].
The United States Food and Drug Administration (FDA) recently granted approval of bevacizumab for the treatment of platinum-sensitive recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer [3]. Approval was based on the results of two randomized Phase 3 studies. The Gynecologic Oncology Group (GOG) -0213 study showed that median OS was 42.6 months with bevacizumab plus chemotherapy vs 37.3 months with chemotherapy alone (P = 0.056). The GOG-0213 study also showed improvement in PFS with bevacizumab plus chemotherapy (13.8 months) compared with chemotherapy alone (10.4 months; P < 0.0001). In the OCEANS (Ovarian Cancer Study Comparing Efficacy and Safety of Chemotherapy and Anti-Angiogenic Therapy in Platinum-Sensitive Recurrent Disease) study, median PFS was 12.4 months with bevacizumab plus chemotherapy vs 8.4 months with chemotherapy plus placebo (P < 0.0001). However, no statistically significant difference in OS was observed between the two treatment groups. The adverse events associated with bevacizumab in the GOG-0213 and OCEANS studies were consistent with those observed in previous studies, and included fatigue, low white blood cell count with fever, low sodium, pain in extremity, low platelet count, elevated protein in the urine, high blood pressure and headache [3]. Bevacizumab, in combination with a chemotherapy backbone, is also a key component in the treatment of cervical cancer. Approval of combination bevacizumab for the treatment of persistent, recurrent or metastatic cervical cancer was granted on the results of an international Phase 2 randomized trial [9]. Results from this study showed that bevacizumab plus chemotherapy (cisplatin plus paclitaxel or topotecan) was associated with increased OS (17.0 months) compared with chemotherapy alone (13.3 months) (P = 0.004). Significantly higher response rates were observed with bevacizumab plus chemotherapy (48%) compared with chemotherapy alone (36%) (P = 0.008). In addition, bevacizumab plus chemotherapy was associated with a higher frequency of hypertension, thromboembolic events and gastrointestinal fistulas, compared with chemotherapy alone [9]. In summary, bevacizumab in combination with chemotherapy is the mainstay of treatment for a variety of gynecologic cancers.
Challenges and barriers to the use of bevacizumab in clinical practice
A recent retrospective population-based study using the Surveillance, Epidemiology, and End Results (SEER)-Medicare database of 9491 women with epithelial ovarian cancer showed that, despite strong evidence of improved survival associated with therapy recommended by National Comprehensive Cancer Network (NCCN) guidelines, [10] over 70% of women receiving initial treatment for epithelial ovarian cancer did not receive treatment consistent with NCCN recommendations [11]. Ultimately, this may adversely affect patient care and is a serious global concern.
Despite the clinical success of bevacizumab in cancers with a large global incidence, such as lung and colorectal cancers, and clear guidelines recommending its use in both ovarian [10] and cervical cancer, [12] there is a notable lack of patient access to bevacizumab. Disparities in access to bevacizumab and other targeted therapies have been reported in Europe, with some countries reporting only occasional access to bevacizumab, or access for only 50% of patients with ovarian cancer [5]. Therefore, it is important to identify areas of inefficiencies and to understand barriers to patient access.
Several factors, including healthcare system infrastructure, stage at diagnosis, population health and lifestyle and availability of anticancer agents, can influence access to cancer therapy. Issues related to insurance coverage, treatment cost, drug availability, supply and manufacturing may create barriers to the use of bevacizumab in many countries worldwide. A survey conducted by the European Society of Medical Oncology (ESMO) Consortium reported budget and affordability issues, and problems with the manufacture and supply of bevacizumab as the most common factors leading to suboptimal access to bevacizumab in a variety of cancers [5].
Clinical trials demonstrated bevacizumab improves PFS in patients with ovarian cancer [8] and OS in cervical cancer [9]. Although bevacizumab with chemotherapy is more effective with regard to PFS than chemotherapy alone, it is not a cost-effective, front-line treatment regimen in the overall population of patients with ovarian cancer (Table 2) [13]. Furthermore, approximately three-quarters of US oncologists do not consider bevacizumab a “good value” treatment option [14]. However, a recent analysis utilized results from the AURELIA study of bevacizumab plus chemotherapy versus chemotherapy alone in patients with platinum-resistant recurrent ovarian cancer [8]. This analysis concluded that bevacizumab was cost-effective in this setting [15]. Taken together, it is clear that further studies are needed to determine the cost-effectiveness of bevacizumab in the real-world setting.
Bevacizumab will continue to remain an important component in the treatment of gynecologic cancers as well as other settings. In light of the limited access to bevacizumab worldwide, additional treatment options for gynecologic cancers are eagerly awaited.
Development of biosimilars and their potential benefits
Patents for bevacizumab will shortly expire in the United States and Europe [16]. Biosimilars are large, structurally complex molecules that are intended to be highly similar to, and treat the same condition(s) as, an existing licensed or approved (reference) biologic [6, 7]. Biosimilars may offer increased treatment options for patients and physicians and have the potential to optimize efficiencies across healthcare systems worldwide. Additionally, biosimilars may provide lower cost alternatives and therefore increase access to biologics and allow greater use of biologic therapies, which may facilitate improved clinical outcomes.
The aim of biosimilar development is not to re-establish efficacy and safety, but to demonstrate similarity to the reference biologic in terms of quality, safety and efficacy (Fig. 1) [6, 7]. The development of biosimilars involves biochemical, biophysical and functional comparative studies, and detailed characterization of the potential biosimilar. Together with comparative nonclinical, pharmacokinetic (PK), and comparative clinical trials, these data comprise the “totality of the evidence” [6].

Development pathways for originator biologics and biosimilars: a different way of thinking. Adapted from Kozlowski et al., 2012. PD: pharmacodynamics, PK: pharmacokinetics
Biosimilars must have an identical primary amino acid sequence and the same route of administration, strength and type of administration as the reference biologic [6, 7]. Biosimilars are manufactured through a process of reverse engineering and must undergo extensive comparative structural and functional characterization using state-of-the-art technology and highly specialized techniques to identify any differences between the proposed biosimilar and the reference biologic, particularly those that may alter the mechanism of action [17].
A series of analytical similarity assessments are conducted to confirm identical amino acid sequences, similar post-translational modifications and highly similar biologic activity between the proposed biosimilar and the reference biologic. Analytical similarity forms the foundation for similarity in safety and efficacy. In addition, a comprehensive assessment of the structural and functional similarity of the potential biosimilar and the reference biologic is conducted using state-of-the-art techniques, physicochemical methods and functional assays [17].
Regulatory agencies do not typically require extensive nonclinical studies for the approval of biosimilars, although this is assessed on a case-by-case basis [6, 7]. A comparative clinical study is generally conducted in one therapeutic indication to demonstrate that there are no clinically meaningful differences in PK, pharmacodynamics (PD), efficacy or safety, including immunogenicity, between the potential biosimilar and the reference biologic. The goal of a comparative clinical study is to address any residual uncertainty between the proposed biosimilar and the reference biologic [6, 7]. Because all biologics, including biosimilars, have the potential to trigger an immunogenic response, which may alter the PK, efficacy or safety properties, [18] the formation of antidrug antibodies is carefully monitored throughout development and during postmarketing surveillance.
Biosimilars and the scientific basis of extrapolation across indications
Extrapolation is a scientific and regulatory principle that describes the approval of a biosimilar for use in an indication held by the reference biologic, which is not directly studied in a comparative clinical trial with a biosimilar. Extrapolation is key to the concept of biosimilarity and is based on establishing a similar mechanism of action for the biosimilar in various disease indications [6]. As well as reducing or eliminating the need for studies in multiple indications, extrapolation can potentially allow greater access to biosimilars, with minimal delays in treatment. The concept of extrapolation is supported by the US FDA and the European Medicines Agency (EMA) regulatory guidelines [6, 7]. However, the decision to extrapolate data from one indication to another is made on a case-by-case basis, with strong scientific justification and the totality of evidence.
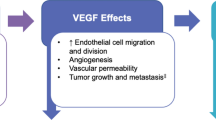
The mechanism of action of bevacizumab involves the inhibition of vascular endothelial growth factor (VEGF), which has an important role in tumor angiogenesis and vascularization [1, 2]. Bevacizumab is an effective treatment for a number of tumors and its mechanism of action is independent of tumor site [1]. This forms the basis of the scientific rationale for extrapolation of similarity data across indications and may support the approval of bevacizumab biosimilars in indications held by the reference biologic without clinical studies in gynecologic indications.
Bevacizumab biosimilar candidates in development
Four bevacizumab biosimilar candidates have completed preclinical assessments and, based on the totality of evidence, are currently undergoing comparative clinical assessments (Table 3). ABP 215 (Amgen) showed similar in vitro functional characteristics and equivalent human PK to bevacizumab [19] and demonstrated clinical equivalence and similar safety and immunogenic profiles as bevacizumab in patients with non-squamous NSCLC [20]. BCD-021 (Biocad) showed similar PK and safety to bevacizumab in patients with NSCLC [21]. BCD-021 also demonstrated similar efficacy, safety and immunogenicity to bevacizumab in patients with advanced non-squamous NSCLC [22]. A multicenter, randomized, double-blind clinical trial is ongoing to evaluate the efficacy and safety of BI 695502 (Boehringer Ingelheim) compared with bevacizumab (in combination with chemotherapy) in patients with advanced non-squamous NSCLC (ClinicalTrials.gov, NCT02272413). PF-06439535 (Pfizer) showed a similar structure and in vitro biological activity, and a similar in vivo toxicologic and toxicokinetic profile as bevacizumab [23, 24]. PF-06439535 also demonstrated PK similarity and comparable safety profiles to bevacizumab in healthy male volunteers [25]. A trial of PF-06439535 vs bevacizumab sourced in the EU in patients with advanced non-squamous NSCLC who have not received prior chemotherapy is ongoing (ClinicalTrials.gov, NCT02364999).
Conclusions
It is anticipated that biologics such as bevacizumab will continue to play a key role in the treatment of an array of gynecologic cancers. Limited access to bevacizumab and the lack of cost-effectiveness in some patients has driven the need to develop safe and effective biosimilars to bevacizumab, which have the potential to increase access to medicines and offer efficiencies across healthcare systems.
Abbreviations
- AURELIA:
-
Avastin Use in Platinum-Resistant Epithelial Ovarian Cancer
- EMA:
-
European Medicines Agency
- ESMO:
-
European Society of Medical Oncology
- FDA:
-
Food and Drug Administration
- GOG:
-
Gynecologic Oncology Group
- NCCN:
-
National Comprehensive Cancer Network
- NSCLC:
-
non-small-cell lung cancer
- OCEANS:
-
Ovarian Cancer Study Comparing Efficacy and Safety of Chemotherapy and Anti-Angiogenic Therapy in Platinum-Sensitive Recurrent Disease
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- SEER:
-
Surveillance, Epidemiology, and End Results
- VEGF:
-
Vascular endothelial growth factor.
References
Genentech Inc. Avastin (bevacizumab) injection prescribing information. 2004. http://www.gene.com/download/pdf/avastin_prescribing.pdf. Accessed 18 Oct 2016.
European Medicines Agency. Summary of product characteristics: Avastin (bevacizumab) 25 mg/ml concentrate for solution for infusion. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000582/WC500029271.pdf. Accessed 18 Oct 2016.
Wire B. FDA approves Genentech’s Avastin® (Bevacizumab) plus chemotherapy for a specific type of advanced ovarian cancer. 2016. http://www.businesswire.com/news/home/20161206006296/en/FDA-Approves-Genentech%E2%80%99s-Avastin%C2%AE-Bevacizumab-Chemotherapy-Specific. Accessed 17 Jan 2016.
Hoffmann-La Roche Limited. Avastin (bevacizumab) 100 mg and 400 mg vials (25 mg/mL solution for injection) Product monograph. 2016. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Avastin/Avastin_PM_E.pdf. Accessed 17 Jan 2016.
Cherny N, Sullivan R, Torode J, Saar M, Eniu A. ESMO European Consortium Study on the availability, out-of-pocket costs and accessibility of antineoplastic medicines in Europe. Ann Oncol. 2016;27:1423–43.
US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. 2015. http://www.fda.gov/downloads/DrugsGuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 18 Oct 2016.
European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf. Accessed 25 Aug 2015.
Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol. 2014;32:1302–8.
Tewari KS, Sill MW, Long 3rd HJ, Penson RT, Huang H, Ramondetta LM, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370:734–43.
National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: ovarian cancer. 2016. https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf. Accessed 18 Oct 2016.
Urban RR, He H, Alfonso-Cristancho R, Hardesty MM, Goff BA. The cost of initial care for Medicare patients with advanced ovarian cancer. J Natl Compr Canc Netw. 2016;14:429–37.
National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: cervical cancer. 2016. https://www.nccn.org/professionals/physician_gls/pdf/cervical.pdf. Accessed 18 Oct 2016.
Poonawalla IB, Parikh RC, Du XL, VonVille HM, Lairson DR. Cost effectiveness of chemotherapeutic agents and targeted biologics in ovarian cancer: a systematic review. Pharmacoeconomics. 2015;33:1155–85.
Nadler E, Eckert B, Neumann PJ. Do oncologists believe new cancer drugs offer good value? Oncologist. 2006;11:90–5.
Chappell NP, Miller C, Barnett J, Fielden A. Is FDA approved bevacizumab cost-effective in the setting of platinum-resistant recurrent ovarian cancer? Obstet Gynecol. 2016;127:6–7.
Genetic Engineering & Biotechnology News. Biosimilars: 11 Drugs to watch. 2014. http://www.genengnews.com/insight-and-intelligence/biosimilars-11-drugs-to-watch/77900135. Accessed 14 Mar 2017.
Bui LA, Hurst S, Finch GL, Ingram B, Jacobs IA, Kirchhoff CF, et al. Key considerations in the preclinical development of biosimilars. Drug Discov Today. 2015;20(Suppl 1):3–15.
Bendtzen K. Anti-TNF-alpha biotherapies: perspectives for evidence-based personalized medicine. Immunotherapy. 2012;4:1169–79.
Born TL, Huynh Q, Mathur A, Velayudhan J, Canon J, Reynhardt K, et al. Functional similarity assessment results comparing bevacizumab to biosimilar candidate ABP 215. Ann Oncol. 2014;25(Suppl 4):v163.
Thatcher N, Thomas M, Paz-Ares Rodriguez L, Ostoros G, Pan J, Goldschmidt JH, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non squamous non-small-cell lung cancer. Paper presented at: 2016 Annual Meeting of the American Society of Clinical Oncology (ASCO); June 3–7, 2016; Chicago, IL, USA.
Orlov S, Burdaeva O, Nechaeva MP, Kopp MV, Kotiv BN, Sheveleva LP, et al. Pharmacokinetics and safety of BCD-021, bevacizumab biosimilar candidate, compared to Avastin in patients. J Thorac Oncol. 2014;32(Suppl):e13500.
Filon O, Orlov S, Burdaeva O, Kopp MV, Kotiv BN, Alekseev S, et al. Efficacy and safety of BCD-021, bevacizumab biosimilar candidate, compared to Avastin: Results of international multicenter randomized double blind phase III study in patients with advanced non-squamous NSCLC. J Clin Oncol. 2015;33(Suppl):8057.
Rule K, Peraza M, Shiue M, Finch G, Thibault S, Rosenberg JA, et al. Nonclinical development of PF 06439535, a potential biosimilar to bevacizumab. J Thorac Oncol. 2015;10:S485.
Peraza M, Shiue M, Phenix S. Comparative nonclinical assessment of the potential biosimilar PF-06439535 and bevacizumab. Paper presented at: 54th Annual Meeting and ToxExpo of Society of Toxicology (SOT); March 22–26, 2015; San Diego, CA, USA.
Knight B, Rassam D, Liao S, Ewesuedo R. A phase I pharmacokinetics study comparing PF-06439535 (a potential biosimilar) with bevacizumab in healthy male volunteers. Cancer Chemother Pharmacol. 2016;77:839–46.
Markus R, Born T, Chow V, Zhang N, Huynh Q, Maher G. Functional similarity and human pharmacokinetic (PK) equivalence of ABP 215 and bevacizumab. J Clin Oncol. 2015;33:15.
Acknowledgments
Medical writing support was provided by Neel Misra, MSc, of Engage Scientific Solutions and was funded by Pfizer.
Funding
This report is supported by Pfizer Inc.
Availability of data and materials
Not applicable
Authors’ contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be submitted for publication.
Authors’ information
Not applicable
Competing interests
Dr Monk discloses that St. Joseph’s Hospital institution has received research grants from Novartis, Amgen, Lilly, Genentech, Janssen/Johnson & Johnson, Array, TESARO and Morphotek. He has received honoraria for speaker bureaus from Roche/Genentech, AstraZeneca, Janssen/Johnson & Johnson, and Myriad. Additionally, Dr Monk has been a consultant for Roche/Genentech, Merck, TESARO, AstraZeneca, Gradalis, Advaxis, Cerulean, Amgen, Vemillion, ImmunoGen, Pfizer, Bayer, NuCana, Insys, GlaxoSmithKline, Verastem, Mateon (formerly OxiGENE), PPD and Clovis. Dr Warner K. Huh declared no conflicts of interest. Dr Julie Ann Rosenberg and Dr Ira Jacobs are full-time employees of Pfizer Inc.
Consent for publication
Not applicable
Ethics approval and consent to participate
Not applicable
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Monk, B.J., Huh, W.K., Rosenberg, J.A. et al. Will bevacizumab biosimilars impact the value of systemic therapy in gynecologic cancers?. gynaecol oncol res pract 4, 7 (2017). https://doi.org/10.1186/s40661-017-0045-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40661-017-0045-x