
Abstract
Background
Acute myeloid leukemia (AML) is an aggressive and mostly incurable hematological malignancy with frequent relapses after an initial response to standard chemotherapy. Therefore, novel therapies are urgently required to improve AML clinical outcomes. 4-Amino-2-trifluoromethyl-phenyl retinate (ATPR), a novel all-trans retinoic acid (ATRA) derivative designed and synthesized by our team, has been proven to show biological anti-tumor characteristics in our previous studies. However, its potential effect on leukemia remains unknown. The present research aims to investigate the underlying mechanism of treating leukemia with ATPR in vitro.
Methods
In this study, the AML cell lines NB4 and THP-1 were treated with ATPR. Cell proliferation was analyzed by the CCK-8 assay. Flow cytometry was used to measure the cell cycle distribution and cell differentiation. The expression levels of cell cycle and differentiation-related proteins were detected by western blotting and immunofluorescence staining. The NBT reduction assay was used to detect cell differentiation.
Results
ATPR inhibited cell proliferation, induced cell differentiation and arrested the cell cycle at the G0/G1 phase. Moreover, ATPR treatment induced a time-dependent release of reactive oxygen species (ROS). Additionally, the PTEN/PI3K/Akt pathway was downregulated 24 h after ATPR treatment, which might account for the anti-AML effects of ATPR that result from the ROS-mediated regulation of the PTEN/PI3K/AKT signaling pathway.
Conclusions
Our observations could help to develop new drugs targeting the ROS/PTEN/PI3K/Akt pathway for the treatment of AML.
Similar content being viewed by others

Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease that affects 3–4 out of every 100,000 people, and the median age of AML patients is 67 years. The 5-year survival rate is approximately 20% [1]. The progression of the disease depends on many factors, including cytogenetics, molecular genetics, comorbidity scores, and the age of the patient. As the understanding of AML pathogenesis has increased cytotoxic chemotherapy with or without subsequent hematopoietic cell transplantation has been established as the primary treatment for AML. Despite many efforts to identify treatments for AML, the prognosis has not improved significantly over the past decade, and this endeavor remains a challenge [2]. Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia (AML) characterized by the accumulation of immature promyelocytes in the peripheral blood and the bone marrow. For decades, APL has been considered the most malignant AML because of the occurrence of severe bleeding in the disease and its high early mortality rate [3, 4]. Currently, retinoic acid (RA) and arsenic trioxide (ATO) are two classic drugs used for the treatment of APL. Treatments for APL are associated with a number of issues, such as ATO or all-trans retinoic acid (ATRA) resistance, relapse, differentiation syndrome and adverse effects [5,6,7,8]. In addition, ATRA seems to be a poor treatment for non-APL. Therefore, it is necessary to identify other therapeutic strategies for AML, including APL (using NB4 cells) and non-AML (using THP-1 cells).
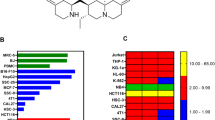
To overcome the side effects of ATRA, our team has altered the structure of ATRA to obtain a series of retinoic acid derivatives. After pre-pharmacodynamic screening, we found that 4-amino-2-trifluoromethyl-phenyl retinate (ATPR) (Fig. 1) has a favorable anti-tumor effect. ATPR shows better solubility than ATRA [9]. The anti-tumor effect has been studied in several types of solid tumors. Some studies have shown that ATPR can effectively inhibit growth and differentiation induction in breast cancer MCF-7 cells and gastric cancer SGC-7901 cells via the upregulation of retinoid receptor-induced gene-1 or retinoic acid receptors [10, 11]. These studies suggest that ATPR can exhibit strong anti-tumor effects and has potential as a cancer chemotherapeutic agent, but the molecular mechanism remains unclear.

Synthesis of ATPR by structural modification of ATRA
Reactive oxygen species (ROS) are primarily produced by NADPH oxidase (Nox), an important cellular signaling molecule involved in the progression of cancer cells, and are generally thought to be second messengers that augment inflammation by activating downstream signal cascades [12, 13]. Phosphatase and tensin homolog (PTEN) plays an important role in mature organisms as a tumor suppressor. The inactivation of PTEN genes by mutation or deletion is common in pediatric T-cell acute lymphoblastic leukemia (T-ALL) [14]. The major substrate of PTEN is phosphatidylinositol-3,4,5-triphosphate (PIP3), which is produced by the action of phosphoinositide-3-kinase (PI3K) [15]. The PI3K/AKT signaling pathway plays an important role in the development of anticancer therapies, and the inhibition of the PI3K/AKT signaling pathway may induce cycle arrest and differentiation in vitro.
Our results demonstrated that ATPR, a novel derivative of ATRA, inhibits the proliferation and induces the differentiation of acute myelocytic leukemia cells via the ROS-mediated regulation of the PTEN/PI3K/Akt signaling pathway. These findings suggest that ATPR may be a promising agent for acute myelocytic leukemia treatment.
Materials and methods
Chemicals and reagents
ATPR (purity: 99.66%) was synthesized by our laboratory (School of Pharmacy, Anhui Medical University). A 10−2 mol/l stock solution of ATPR was prepared in absolute alcohol and stored at − 20 °C. In addition, no effect of the solvent (alcohol) was found. Antibodies against cyclin A2, cyclin D3, CDK4, Rb (phosphatase), PTEN, AKT, phospho-Akt (Ser473), PI3K p85, CD11b (PE/CY5-conjugated anti-human CD11b) and CD14 (FITC-conjugated anti-human CD14) were obtained from Abcam (Danvers, MA, USA). β-Actin antibodies were purchased from Bioss. All of the antibodies used in the WB assay were diluted to 1:1000.
Cell lines and cell culture
The human leukemia cell lines NB4 and THP-1 were purchased from Gecko Gene (Shanghai, China). They were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, Gibco, USA), 100 U/ml penicillin and 100 μg/ml streptomycin in a 37 °C, 5% CO2 incubator. The medium was changed every 2 or 3 days. Cells in exponential growth were subjected to the following experiments.
Cell viability assay
The indicated concentrations of ATPR (0, 10−9, 10−8, 10−7, 10−6, or 10−5 mol/l) were used to treat the cells for 24, 48, and 72 h. After the addition of the Cell Counting Kit-8 (CCK-8) solution (10 μl per well), the cells were then incubated at 37 °C for 1 h. The optical density (OD) levels were measured at 450 nm using a BioTek ELx808 Microplate Reader. The results are presented as the viability rates. Cell viability was calculated according to the following formula: cell viability (%) = (OD treatment − OD blank)/(OD control − OD blank).
Cell cycle analysis
NB4 and THP-1 cells were seeded in six-well plates at a density of 2 × 105 cells per well. Then, the cells were treated with ATPR (10−6 mol/l) for 24 h, 48 h and 72 h at 37 °C. For cell cycle analysis, the cells were harvested, washed twice with PBS and fixed in 70% ethanol at 4 °C overnight. After a 15-min incubation period with 20 μl RNase A plus 400 μl propidium iodide (PI), the cells were subjected to cell cycle analysis using CytoFLEX (Becton–Dickinson, USA). The cell cycle distribution was analyzed by ModFIT software (BD Biosciences).
Differentiation marker analysis
Then, the cells were treated with ATPR (10−6 mol/l) for 24 h, 48 h and 72 h at 37 °C. After treatment, the cells were washed with PBS and then incubated with monoclonal antibodies against CD11b (CD11b-PE/CY5) and CD14 (CD14-FITC) for 30 min on ice away from light. The cells were then washed twice with ice-cold PBS and finally resuspended in 500 μl PBS for measurement. CD11b and CD14 expression levels were measured using CytoFLEX (Becton–Dickinson, USA). The data were processed using CytoFLEX.
Nitroblue tetrazolium (NBT) assay
For the NBT reduction analysis, NB4 and THP-1 cells (1 × 105 cells/ml) were seeded in a 6-well plate and treated with ATPR. A 10-μl aliquot of the NBT solution, composed of 10 mg/ml NBT (Sigma) and 2 μg/ml PMA (Sigma), was added to each well. Then, the cells were incubated for 30 min at 37 °C. Both the percentage of positive cells and the intensity of reduction when measured at a wavelength of 570 nm with a BioTek ELx808 Microplate Reader was evaluated in the NBT test.
Measurement of intracellular ROS
Intracellular ROS were analyzed by 2′-7′-dichlorofluorescein diacetate (DCFH-DA; Beyotime Biotechnology). After treatment, the cells were washed with PBS and incubated with 20 μM DCFH-DA diluted in RPMI 1640 medium for 30 min at 37 °C in the dark. The cells were then washed with PBS and analyzed using CytoFLEX (Becton–Dickinson, USA). For the flow cytometry assay, the cells were analyzed using CytoFLEX. The ROS inhibitors NAC and Tiron were purchased from TagerMol.
Western blot analysis
NB4 and THP-1 cells were washed twice with prechilled PBS after treatment with different concentrations of ATPR (0, 10−9, 10−8, 10−7, 10−6, or 10−5 mol/l) for 72 h or with 10−6 mol/l ATPR at different time points (24, 48, and 72 h). Total protein was extracted using RIPA lysis buffer plus a protease inhibitor cocktail and then quantified by a BCA Protein Assay Kit (Beyotime). Equal amounts of protein (30 μg/well) were subjected to 10% or 12% SDS-PAGE and then electrotransferred onto 0.45 μm PVDF membranes (Millipore Corp). The membranes were blocked with 5% nonfat milk or bovine serum albumin for 2 h at room temperature followed by overnight incubation at 4 °C in primary antibodies as described above. After washing with TBST, the membranes were incubated with the indicated HRP-conjugated secondary antibodies for 1 h at room temperature. The blots were visualized with an ECL kit (ECL-plus; Thermo Fisher Scientific). ImageJ software was used to quantify the band intensity. For each sample, the signal intensity for each protein was normalized to that of β-actin and is expressed as the fold-increase relative to the control.
Immunofluorescence staining
NB4 and THP-1 cells were plated on 35-mm glass-bottom culture dishes. After 24 h and 72 h of treatment with ATPR (10−6 mol/l), the cells were fixed with 4% paraformaldehyde for 15 min. The fixed cells were permeabilized with 0.5% Triton X-100 for 10 min and blocked with 10% normal goat serum for 30 min. Then, the cells were incubated with a primary antibody against p-AKT (1:200) at 4 °C overnight. After rinsing with PBS three times for 5 min each time, the cells were incubated with a fluorescent-labeled secondary antibody in the dark for 1 h. Finally, the nuclei were counterstained with DAPI and examined and imaged using a NIKON fluorescence microscope.
Statistical analysis
Each experiment was repeated at least three times, and the data are shown as the mean ± standard deviation (SD). Statistical analyses were performed using SPSS software version 20.0 (SPSS Inc., Chicago, IL, USA). Between-group differences were estimated with one-way analysis of variance (ANOVA). p < 0.05 was considered to be statistically significant. All experiments were repeated three times.
Results
ATPR inhibits cell proliferation in human leukemia cells
In order to explore the proliferative effect of ATPR on AML cells, the CCK8 assay was used. As shown in Fig. 2a, b, ATPR inhibited the viability of NB4 and THP-1 cells in a time- and dose-dependent manner. The IC50 value of ATPR in NB4 and THP-1 cells at 24, 48, and 72 h was 10−6 M. As shown in Fig. 3a, d, a time-dependent accumulation of cells in G0/G1 phase was observed after ATPR treatment. However, the percentage of cells in S phase was reduced after ATPR treatment. These results indicate that NB4 and THP-1 cells were arrested in G0/G1 phase by ATPR. Moreover, Fig. 3b, c, e, and f shows that ATPR downregulated the expression of cyclin D3, cyclin A2, CDK4 and p-Rb, which are marker proteins of G0/G1 phase. These results also indicate that ATPR inhibited NB4 and THP-1 cell proliferation in a time- and concentration-dependent manner.

ATPR inhibits cell proliferation in human NB4 and THP-1 cells. a, b Dose- and time-dependent effects of ATPR on NB4 and THP-1 cells. NB4 and THP-1 cells were exposed to various concentrations (0, 10−9, 10−8, 10−7, 10−6, or 10−5 M) of ATPR for 24–72 h, followed by the determination of cell viability using the CCK8 assay. Values are presented as the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 versus control group

ATPR triggers cell cycle arrest at G0/G1 phase in human NB4 and THP-1 cells. a NB4 cells were treated with ATPR (10−6 M) for different durations (0–72 h). The cell cycle distribution was analyzed by flow cytometry. b NB4 cells were treated with an ATPR concentration gradient (10−5–10−9 M) for 72 h. Then, the protein expression of cyclin A2, CDK4, cyclin D3 and p-Rb was assessed by western blot. c NB4 cells were treated with ATPR (10−6 M) for different duration (0–72 h). Then, the protein expression of cyclin A2, CDK4, cyclin D3 and p-Rb was assessed by western blot. d THP-1 cells were treated with ATPR (10−6 M) for different durations (0–72 h). The cell cycle distribution was analyzed by flow cytometry. e THP-1 cells were treated with an ATPR concentration gradient (10−5–10−9 M) for 72 h. Then, the protein expression of cyclin A2, CDK4, cyclin D3 and p-Rb was assessed by western blot. f THP-1 cells were treated with ATPR (10−6 M) at different time points (0–72 h). Then, the protein expression of cyclin A2, CDK4, cyclin D3 and p-Rb was assessed by western blot. *p < 0.05, **p < 0.01 versus control group
ATPR induces differentiation in NB4 and THP-1 cells
To investigate whether ATPR induces differentiation in NB4 and THP-1 cells, we measured CD11b and CD14 surface antigen expression in the treated cells. Flow cytometry showed that ATPR remarkably increased CD14 and CD11b expression levels in a time-dependent manner (Fig. 4a, b). Furthermore, western blot analysis also showed that CD11b expression was remarkably higher in ATPR-treated cells than in the control group (Fig. 4c). Finally, we found that ATPR-treated cells exhibited increased nitroblue tetrazolium (NBT) reduction (Fig. 4d). Altogether, these findings strongly suggest that ATPR induced leukemia cell differentiation.

ATPR induces differentiation in human NB4 and THP-1 cells. a NB4 cells were treated with ATPR (10−6 M) for different durations (0–72 h). CD11b and CD14 expression was analyzed by flow cytometry. b THP-1 cells were treated with ATPR (10−6 M) for different durations (0–72 h). CD11b and CD1 expression was analyzed by flow cytometry. c NB4 and THP-1 cells were treated with ATPR (10−6 M) for different durations (0–72 h). Then, the protein expression of CD11b was assessed by western blot. d NB4 and THP-1 cells were treated with ATPR (10−6 M) for 72 h, and the NBT reduction assay detected cell differentiation. *p < 0.05, **p < 0.01 versus control group
ATPR can regulate the PTEN/PI3K/AKT signaling pathway
ATPR increased PTEN expression in a time- and concentration-dependent manner (Fig. 5a–d). Next, we assessed the changes in PI3K/AKT-related marker proteins, e.g., PI3K, AKT, and p-AKT. The cells were exposed to ATPR at a concentration of 10−6 M for the indicated time points (0, 24, 48 and 72 h). As shown in Fig. 5a, c, p-Akt (Ser473) and PI3K expression was induced when the cells were treated with 10−8 M ATPR and decreased upon exposure to increasing ATPR concentrations, up to 10−5 M. As shown in Fig. 5b, d. p-Akt (Ser473) and PI3K expression was induced when the cells were treated with ATPR for 24 h and decreased upon exposure to ATPR for increasing durations, up to 72 h. Furthermore, the decreased expression of p-Akt (Ser473) was confirmed both in the cytoplasm and nucleus by immunofluorescence staining after 24 h of treatment with ATPR (Fig. 5e). Therefore, our data demonstrate that ATPR regulates the PTEN/PI3K/Akt pathway in NB4 and THP-1 cells.

ATPR regulates the PTEN/PI3K/Akt pathway in human NB4 and THP-1 cells. a NB4 cells were treated with an ATPR concentration gradient (10−5–10−9 M) for 72 h. Then, the protein expression of PTEN, AKT, p-AKT and PI3K was assessed by western blot. b NB4 cells were treated with ATPR (10−6 M) for different durations (0–72 h). Then, the protein expression of PTEN, AKT, p-AKT and PI3K was assessed by western blot. c THP-1 cells were treated with an ATPR concentration gradient (10−5–10−9 M) for 72 h. Then, the protein expression of PTEN, AKT, p-AKT and PI3K was assessed by western blot. d THP-1 cells were treated with ATPR (10−6 M) for different durations (0–72 h). Then, the protein expression of PTEN, AKT, p-AKT and PI3K was assessed by western blot. e NB4 cells were treated with ATPR (10−6 M) for 24 h and 72 h. Immunofluorescence was used to detect p-Akt. *p < 0.05, **p < 0.01 versus control group
ATPR promotes intracellular ROS accumulation, an important event in ATPR-induced cycle arrest and differentiation
As shown in Fig. 6a, b, we found that ATPR-treated NB4 cells had higher intracellular ROS levels than that of the control group cells. To further investigate whether ROS accumulation was involved in ATPR-induced cell cycle arrest and differentiation, CD11b and CD14 expression was assessed after ATPR treatment in the presence or absence of N-acetyl-l-cysteine (NAC) and Tiron. The results indicated that ATPR-induced ROS generation, cell cycle arrest and differentiation were markedly restrained by NAC and Tiron (Fig. 6c–f), and western blot analysis of CD11b expression revealed similar results (Fig. 6g).

ATPR induces the accumulation of intracellular ROS, leading to cell cycle arrest at G0/G1 and differentiation. a NB4 cells were treated with ATPR (10−6 M) for different durations (0–72 h). Intracellular ROS levels were assessed by flow cytometry. b Intracellular ROS levels were assessed by immunofluorescence staining after treatment with 10−6 M ATPR for 72 h. c–e NB4 cells were pretreated with NAC and Tiron for 1 h and then treated with ATPR (10−6 M) for 72 h. The intracellular ROS levels were measured by flow cytometry, and CD11b and CD14 expression was measured by flow cytometry. f NB4 cells were pretreated with NAC for 1 h and then treated with ATPR (10−6 M) for 72 h. CD11b levels were analyzed by western blot. *p < 0.05, **p < 0.01 versus control group; #p < 0.05, ##p < 0.01 versus ATPR group
ROS can regulate the PTEN/PI3K/AKT signaling pathway
Similarly, NAC was used to examine the relationship between the PTEN/PI3K/AKT signaling pathway and the generation of ROS. Interestingly, Fig. 7a shows that NAC inhibited the activation of the PI3K/AKT signaling pathway. Furthermore, NAC inhibited the induction of PTEN expression (Fig. 7b). ROS may play an important role in the ATPR-mediated induction of differentiation and proliferation inhibition of leukemia cells by regulating the PTEN/PI3K/AKT signaling pathway (Fig. 7c).

Assessment of PTEN/PI3K/AKT expression in NB4 cells after exposure to ATPR. a NB4 cells were pretreated with NAC for 1 h and then treated with ATPR (10−6 M) for 24 h. PI3K, AKT and P-AKT levels were analyzed by western blot. b NB4 cells were pretreated with NAC for 1 h and then treated with ATPR (10−6 M) for 72 h. b PTEN levels were analyzed by western blot. c A schematic of the proposed mechanism of ATPR-induced G0/G1 arrest and differentiation in human leukemia cells. The red arrows represent the inhibition of the subsequent signal, while the green arrows represent the promotion of the subsequent signal. *p < 0.05, **p < 0.01 versus control group; #p < 0.05, ##p < 0.01 versus ATPR group
Discussion
A high recurrence rate, a high progression rate and high mortality are characteristics of acute myelocytic leukemia (AML). There is an urgent need to identify the potential molecular mechanisms of and to develop a new anti-tumor treatment for acute myelocytic leukemia. According to previous reports, ATPR is a potential drug for the treatment of specific cancers [4]. The CCK-8 assay data in Fig. 1 of this study demonstrate that ATPR inhibited the proliferation of the human acute myelocytic leukemia cell lines NB4 and THP-1 in a time- and dose-dependent manner. The cell cycle proceeds sequentially through four phases, including G0/G1, S, G2, and M. Cell cycle dysregulation is one of the most common alterations in tumor development [19]. Blocking the cell cycle is often considered an effective strategy to eliminate cancer cells [20]. The results of previous studies indicate that ATPR arrests gastric cancer cells at G0/G1 stage of the cell cycle [11]. Our data show that ATPR induced G0/G1 arrest in NB4 and THP-1 cells in a time-dependent manner (Fig. 3a, d). ATPR-induced G0/G1 arrest may help NB4 and THP-1 cells properly repair DNA defects, thus preventing their transmission to the resulting daughter cells. The transition of the cell cycle from one phase to the next requires a regulatory mechanism called checkpoints. CDK/cyclin complexes are often involved in these checkpoints [16, 17]. Meanwhile, our results also show that ATPR reduced the expression of the G0/G1 cycle marker proteins cyclin D3, CDK4, p-Rb and cyclin A2 (Fig. 3b, c, e, f). Cells at the G0/G1 checkpoint are surveyed to determine whether their DNA replicates correctly, and these cells will not transition to the next phase until replication problems are solved. If DNA repair does not occur after some time, cells will undergo differentiation. Figure 4 of this study also shows that ATPR induced cell cycle arrest and inhibited DNA synthesis, subsequently leading to differentiation in NB4 and THP-1 cells. Increasing evidence shows that the PI3K/Akt pathway is involved in the pathogenesis of many cancers and regulates a variety of cellular processes, including differentiation, proliferation, metastasis and metabolism [18]. Therefore, we wanted to explore whether the anti-leukemia effect of ATPR is related to this pathway.
PI3Ks are divided into three categories (I, II, and III), and class I PI3K includes two subgroups, IA and IB. The widely studied class IA PI3K, which plays a key role in tumorigenesis, is a heterodimer composed of the p85 regulatory subunit and the p110 catalytic subunit [19]. The activation of PI3K converts PI (3, 4) P2 to PI (3, 4, 5) P3, a process that can be reversed by PTEN. PIP3, as a second messenger, recruits Akt to the plasma membrane where Akt is phosphorylated at Thr308 and Ser473 [20]. Activated Akt may promote cell cycle progression and tumor growth through its downstream p53 pathway and Bcl-2 family proteins. Akt phosphorylates mouse double minute 2 (MDM2), an E3 ubiquitin ligase that triggers p53 degradation [21]. The tumor suppressor gene p53 induces cell cycle arrest and apoptosis by increasing the expression of its two transcriptional targets, p21Waf1/Cip1 and Bax, respectively [22]. The PI3K/Akt pathway is considered to play a major role in acute myelocytic leukemia, but the specific mechanisms of its role remain to be investigated [23]. PTEN is an essential tumor suppressor that antagonizes the PI3K/AKT anti-apoptotic pathway [24, 25]. Our study found that ATPR promoted the upregulation of PTEN in a time- and concentration-dependent manner. Theoretically, PI3K/AKT should be inhibited when PTEN levels are increased. However, during the treatment of AML by ATPR, the relevant target proteins of the PI3K/AKT signaling pathway first increase and then decrease. This is a very interesting finding that has attracted our attention. It has been reported that ROS play a regulatory role in the PTEN/PI3K/AKT signaling pathway [26, 27]. It remains to be determined whether ROS play an important role in the ATPR-induced differentiation and proliferation inhibition of leukemia cells by regulating the PTEN/PI3K/AKT signaling pathway.
We observed that ATPR treatment significantly stimulates ROS generation (Fig. 6a, b). We also observed that the ROS scavengers NAC and Tiron can inhibit ATPR-induced differentiation (Fig. 6d, e, g) and G0/G1 cycle arrest (Fig. 6f). These results suggest that ATPR promotes intracellular ROS production, which is very important for the anti-leukemia effects of ATPR in cells. Initially, ATPR activates PI3K/Akt pathway signaling, but ROS inhibitors can reverse this behavior (Fig. 7a). As the levels of reactive oxygen species increase, PTEN is activated (Fig. 7b), which in turn inhibits the PI3K/AKT signaling pathway (Fig. 7c).
To our knowledge, this is the first report to demonstrate the anti-neoplastic effect of ATPR on acute myelocytic leukemia via the ROS-mediated regulation of the PTEN/PI3K/AKT pathway. However, it is worth noting that ATPR has good solubility in anhydrous ethanol but poor solubility in water, which poses a serious obstacle to its practical application as a drug for leukemia treatment. Injectable ATPR is being developed and may contribute to the wider use of ATPR for future research. Further studies on the characteristics of acute myelocytic leukemia are also needed to better understand the therapeutic potential of ATPR.
Conclusions
In summary, our observations show that ATPR inhibited the proliferation and induced the differentiation of AML cells and that this effect was related to the ROS-mediated regulation of the PTEN/PI3K/AKT signaling pathway. These studies provide a rationale for the application of ATPR in clinical therapies and in therapeutic regimens for AML patients.
Abbreviations
- AML:
-
acute myeloid leukemia
- ATPR:
-
4-amino-2-trifluoromethyl-phenyl retinate
- ATRA:
-
all-trans retinoic acid
- PML-RARα:
-
promyelocytic leukemia-retinoic acid receptor alpha
- RA:
-
retinoic acid
- ATO:
-
arsenic trioxide
- ROS:
-
reactive oxygen species
- PTEN:
-
phosphatases and tensin homolog
- T-ALL:
-
T cell acute lymphoblastic leukemia
- PI3K:
-
phosphoinositide-3-kinase
- CCK-8:
-
cell counting kit-8
- FBS:
-
fetal bovine serum
- OD:
-
optical density
- RIPA:
-
radio-immunoprecipitation assay
- NBT:
-
nitroblue tetrazolium
- PVDF:
-
polyvinylidene difluoride
- SDS:
-
sodium dodecyl sulfate
- PAGE:
-
polyacrylamide gel electrophoresis
- DAPI:
-
2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride
References
Kell J. Considerations and challenges for patients with refractory and relapsed acute myeloid leukaemia. Leuk Res. 2016;47:149–60.
Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, Pettenati MJ, Patil SR, Rao KW, Watson MS, Koduru PR, Moore JO, Stone RM, Mayer RJ, Feldman EJ, Davey FR, Schiffer CA, Larson RA, Bloomfield CD, Cancer, B Leukemia Group. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood. 2002;100(13):4325–36.
Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci USA. 1980;77(5):2936–40.
Friend C, Scher W, Holland JG, Sato T. Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc Natl Acad Sci USA. 1971;68(2):378–82.
Meyer PN, Roychowdhury S, Kini AR, Alkan S. HSP90 inhibitor 17AAG causes apoptosis in ATRA-resistant acute promyelocytic leukemia cells. Leuk Res. 2008;32(1):143–9.
Zuo L, Li J, Yang Y, Wang X, Shen T, Xu CM, Zhang ZN. Sodium selenite induces apoptosis in acute promyelocytic leukemia-derived NB4 cells by a caspase-3-dependent mechanism and a redox pathway different from that of arsenic trioxide. Ann Hematol. 2004;83(12):751–8.
Shen ZX, Shi ZZ, Fang J, Gu BW, Li JM, Zhu YM, Shi JY, Zheng PZ, Yan H, Liu YF, Chen Y, Shen Y, Wu W, Tang W, Waxman S, De The H, Wang ZY, Chen SJ, Chen Z. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA. 2004;101(15):5328–35.
Patatanian E, Thompson DF. Retinoic acid syndrome: a review. J Clin Pharm Ther. 2008;33(4):331–8.
Fan TT, Cheng Y, Wang YF, Gui SY, Chen FH, Zhou Q, Wang Y. A novel all-trans retinoid acid derivative N-(3-trifluoromethyl- phenyl)-retinamide inhibits lung adenocarcinoma A549 cell migration through down-regulating expression of myosin light chain kinase. Asian Pac J Cancer Prev. 2014;15(18):7687–92.
Wang N, Ge JF, Pan CX, Peng XQ, Chen HH, Wang XQ, Tang J, Hu W, Chen FH. Anti-tumor effect of 4-amino-2-trifluoromethyl-phenyl retinate on human breast cancer MCF-7 cells via up-regulation of retinoid receptor-induced gene-1. Biomed Pharmacother. 2013;67(8):687–92.
Xia Q, Zhao Y, Wang J, Qiao W, Zhang D, Yin H, Xu D, Chen F. Proteomic analysis of cell cycle arrest and differentiation induction caused by ATPR, a derivative of all-trans retinoic acid, in human gastric cancer SGC-7901 cells. Proteomics Clin Appl. 2017;11:7–8.
Yang CS, Kim JJ, Lee SJ, Hwang JH, Lee CH, Lee MS, Jo EK. TLR3-triggered reactive oxygen species contribute to inflammatory responses by activating signal transducer and activator of transcription-1. J Immunol. 2013;190(12):6368–77.
Hong HY, Jeon WK, Kim BC. Up-regulation of heme oxygenase-1 expression through the Rac1/NADPH oxidase/ROS/p38 signaling cascade mediates the anti-inflammatory effect of 15-deoxy-delta 12,14-prostaglandin J2 in murine macrophages. FEBS Lett. 2008;582(6):861–8.
Jenkinson S, Kirkwood AA, Goulden N, Vora A, Linch DC, Gale RE. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia. 2016;30(1):39–47.
Kishimoto H, Hamada K, Saunders M, Backman S, Sasaki T, Nakano T, Mak TW, Suzuki A. Physiological functions of Pten in mouse tissues. Cell Struct Funct. 2003;28(1):11–21.
Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;138(2):255–71.
Barta T, Dolezalova D, Holubcova Z, Hampl A. Cell cycle regulation in human embryonic stem cells: links to adaptation to cell culture. Exp Biol Med. 2013;238(3):271–5.
Kamada S, Kikkawa U, Tsujimoto Y, Hunter T. Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-like protein(s). J Biol Chem. 2005;280(2):857–60.
Guo K, Huang P, Xu N, Xu P, Kaku H, Zheng S, Xu A, Matsuura E, Liu C, Kumon H. A combination of YM-155, a small molecule survivin inhibitor, and IL-2 potently suppresses renal cell carcinoma in murine model. Oncotarget. 2015;6(25):21137–47.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–19.
Ihle NT, Powis G. Inhibitors of phosphatidylinositol-3-kinase in cancer therapy. Mol Aspects Med. 2010;31(2):135–44.
Tu Y, Kim E, Gao Y, Rankin GO, Li B, Chen YC. Theaflavin-3, 3′-digallate induces apoptosis and G2 cell cycle arrest through the Akt/MDM2/p53 pathway in cisplatin-resistant ovarian cancer A2780/CP70 cells. Int J Oncol. 2016;48(6):2657–65.
Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, Gu W. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell. 2017;68(1):224–32.
Lim HJ, Crowe P, Yang JL. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J Cancer Res Clin Oncol. 2015;141(4):671–89.
Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. PTEN function: the long and the short of it. Trends Biochem Sci. 2014;39(4):183–90.
Luo H, Yang Y, Duan J, Wu P, Jiang Q, Xu C. PTEN-regulated AKT/FoxO3a/Bim signaling contributes to reactive oxygen species-mediated apoptosis in selenite-treated colorectal cancer cells. Cell Death Dis. 2013;4:e481.
Kim JH, Choi TG, Park S, Yun HR, Nguyen NNY, Jo YH, et al. Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ. 2018;25:1921–37.
Authors’ contributions
FYB planned and conducted the experiments, analyzed the data and wrote the text. FYB conducted the experiments and analyzed the data. HXX, NUW, WK, DY, SCJ and ZRP participated in the design of the experiment and contributed to discussing the data and text. CFH conceived of the study and participated in its design and coordination and the critical review of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not available.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not available.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This work was supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2011ZX09401) to Prof. Chen.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Feng, Y., Hua, X., Niu, R. et al. ROS play an important role in ATPR inducing differentiation and inhibiting proliferation of leukemia cells by regulating the PTEN/PI3K/AKT signaling pathway. Biol Res 52, 26 (2019). https://doi.org/10.1186/s40659-019-0232-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40659-019-0232-9