
Abstract
Microbial enhanced oil recovery (MEOR) is a cost effective and efficient method for recovering residual oil. However, the presence of wax (paraffin) in residual oil can substantially reduce the efficiency of MEOR. Therefore, microbial dewaxing is a critical process in MEOR. In this study, a bacterial dewaxing agent of three spore-forming bacteria was developed. Among these bacteria, Bacillus subtilis GZ6 produced the biosurfactant surfactin. Replacing the promoter of the surfactin synthase gene cluster (srfA), increased the titer of surfactin in this strain from 0.33 g/L to 2.32 g/L. The genetically modified strain produced oil spreading rings with diameters increasing from 3.5 ± 0.1 to 4.1 ± 0.2 cm. The LadA F10L/N133R mutant was created by engineering an alkane monooxygenase (LadA) using site-directed mutagenesis in the Escherichia coli host. Compared to the wild-type enzyme, the resulting mutant exhibited an 11.7-fold increase in catalytic efficiency toward the substrate octadecane. When the mutant (pIMPpladA2mu) was expressed in Geobacillus stearothermophilus GZ178 cells, it exhibited a 2.0-fold increase in octadecane-degrading activity. Cultures of the two modified strains (B. subtilis GZ6 (pg3srfA) and G. stearothermophilus GZ178 (pIMPpladA2mu)) were mixed with the culture of Geobacillus thermodenitrificans GZ156 at a ratio of 5:80:15. The resulting composition increased the rate of wax removal by 35% compared to the composition composed of three native strains. This study successfully developed a multi-strain bacterial agent with enhanced oil wax removal capabilities by genetically engineering two bacterial strains.
Similar content being viewed by others


Background
Non-renewable fossil fuels currently account for approximately 80–90% of the global energy supply (Norouzi et al. 2020). According to a report by the Organization of Petroleum Exporting Countries, by the end of 2023, the global total oil reserves reached 1.56 trillion barrels. Modern oil production processes can recover around 65% of crude oil by using primary and secondary extraction methods (Shibulal et al. 2014). However, approximately 35% of the original oil remains as residual oil (RO) in depleted oil fields, which is difficult to recover because of its high viscosity and low fluidity (Hall et al. 2003). To recover RO, various techniques have been developed over the years, which can be classified into enhanced oil recovery and tertiary recovery methods. Currently, three main methods of enhanced oil recovery are used in practical oilfield applications: chemical flooding, thermal enhanced oil recovery, and microbial methods (Shibulal et al. 2014). Although the first two methods achieve high recovery efficiencies, they also have drawbacks (Murungi and Sulaimon 2022). For example, the addition of chemicals often leads to secondary pollution, and the application of heat over a large oilfield area substantially increases costs (Zhao et al. 2023).
To overcome these limitations, scientists have developed a non-toxic and eco-friendly technique for oil recovery using microbes. This method is known as microbial enhanced oil recovery (MEOR) and it is particularly noteworthy because of its low operating costs and high recovery rates (Shibulal et al. 2014). MEOR utilizes the hydrocarbon metabolism of microbes to alter the physical properties and composition of RO, thereby rendering RO more readily recoverable (Astuti et al. 2019). The primary target of the MEOR process is the heavy oil fraction, which constitutes approximately 15% of total RO (Zhang et al. 2020). Heavy oil is a complex mixture composed mainly of alkanes (paraffins), cycloalkanes (naphthenes), and aromatic hydrocarbons. The majority of heavy oil components are saturated alkanes, of which those with carbon chain lengths exceeding C18 are classified as waxes. At room temperature, this fraction of saturated alkane is present in the solid state (Zhang et al. 2020). During the extraction of crude oil from high-temperature underground oil wells, the decreasing temperature results in the precipitation of waxes. The result is the formation of wax crystals that adhere to oil wells and oil production equipment, thus considerably reducing oil recovery efficiency. The MEOR process mainly deals with waxes and is, therefore, actually a dewaxing process. The microorganisms involved in MEOR also act as dewaxing agents. Waxy crude oil is a common feedstock problem encountered by the petroleum industry. Statistical data show that waxy crude oils account for up to 90% of the total crude oil produced from Chinese oil fields. Most of these crude oils have wax contents above 20%, with certain samples containing up to 40% wax (Wang et al. 2013). Currently, microbial dewaxing technologies are used in about 65% of all oil fields in China (Bian et al. 2021).
The microbial dewaxing process enhances heavy oil recovery through two mechanisms: increasing the solubility of hydrophobic compounds and degrading complex compositions (Wu et al. 2022). Bacteria are the primary microorganisms used for microbial dewaxing and during the process, they produce several bioactive molecules, the most important of which are biosurfactants. These biosurfactants prevent wax crystals from aggregating by creating polar surfaces that are not conducive to crystal growth, thus keeping the wax in a highly dispersed state, and enabling its easy transport by oil flow (Wang et al. 2022). In addition, dewaxing bacteria are capable of degrading waxes, such as heavy hydrocarbons with 22 to 40 carbon atoms, as demonstrated by Pseudomonas species (Etoumi et al. 2008). Chen et al. (2017) reported that a bacterium belonging to the Dietzia genus can metabolize n-alkanes ranging from C14 to C31 and achieve high degradation rates at neutral and slightly alkaline pH levels. In conclusion, the production of biosurfactants and the degradation of hydrocarbons are crucial capabilities of dewaxing strains. To optimize this biotechnology, it is essential to isolate native microbes with superior capabilities in both aspects. Alternatively, microbial chassis cells can be modified through gene editing to enhance the yield of biosurfactants, or protein engineering can be employed to enhance the catalytic efficiency of relevant metabolic enzymes within hydrocarbon metabolic pathways.
This paper reports the development of a tri-strain microbial dewaxing agent consisting of a genomically modified Bacillus subtilis strain, an alkane monooxygenase-enhanced Geobacillus stearothermophilus strain, and a native Geobacillus thermodenitrificans strain. The dewaxing rate for this tri-strain compound was significantly higher than the formulation containing the native forms of these three strains. According to references, the vast majority of microbial dewaxing agents that have been developed so far are native microbial strains (Patel et al. 2015). In this study, genetic editing and protein engineering methods were employed to modify dewaxing strains. The performances of the single modified strains and the combined agent were greatly enhanced, indicating that these two approaches are effective tools in the development of microbial dewaxing agents. The agent developed is currently being field tested in several Chinese oil wells.
Methods
Chemicals, bacterial strains, plasmids, and culture media
Flavin mononucleotide (FMN), octadecane and surfactin were purchased from Acros (Beijing, China). Crude oil samples (alkane content, 19.4% ) were provided by the PetroChina Liaohe Petrochemical Refinery. Other reagents used were analytical grade and commercially available. Escherichia coli ) cultures were grown in Lysogeny Broth (LB) medium at 37 °C. E. coli TOP10 cells (TransGen Biotech, Beijing, China) were used for plasmid construction. The pET30a plasmid (Invitrogen, California, USA) was used for routine cloning and expression in E. coli BL21(DE3). Bacterial clones carrying the desired recombinant plasmids were selected by supplementing growth media with specific antibiotics.
The enrichment medium consisted of 0.2% crude oil, NaNO3 0.2%, (NH4)2SO4 0.1%, NaCl 0.5%, MgSO4 0.025%, KH2PO4 0.5%, K2HPO4 1%.
Medium 1(for bio-surfactant producing bacteria): sucrose 0.5%, NaNO3 1%, K2HPO4 0.3%, CaCl2 0.012%, MgSO4 0.024%, FeSO4 0.012%, Na2MoO4 0.008%, yeast extract 0.05%.
Medium 2 (for crude oil degrading bacteria): sucrose 0.5%, NaCl 0.3%, (NH4)2SO4 0.15%, MgSO4 0.02%, NaNO3 0.3%, KH2 PO4 0.01%, K2HPO4 0.05%, FeSO4 0.001%, yeast extract 0.05%.
Medium C18 (for pIMPpLad2Mu expression in Geobacillus stearothermophilus): octadecane 0.3%, NaCl 0.5%, (NH4)2SO4 0.15%, MgSO4 4.1%, KH2 PO4 4.0%, K2HPO4 4.1%, (NH4)SO4 4.1%.
Soil sample collection
Crude oil contaminated soil samples were collected from the Huaziping well at the Ansai Oilfield (Latitude 34°35’48"N, Longitude 108°95’35"E) located in North Shaanxi, Shaanxi Province, China. These samples were collected from a layer approximately 10 cm below the soil surface and placed into sterilized bags. Debris was removed from samples prior to screening experiments.
Bio-surfactant producing bacteria screening
A total of 50 mL of enrichment medium was inoculated with 10 g of crude oil-contaminated soil and cultured on a rotary shaker at 40 °C and 220 RPM for 48 h. After enrichment, cultures were diluted 10-fold with sterile water. Next, 0.1 mL aliquots were plated on Medium 1 plates and incubated for 48 h at 40 °C. Subsequently, individual colonies from these plates were inoculated into 5 mL of Medium 1 and grown on a rotary shaker at 40 °C and 220 RPM for 48 h. Finally, the culture supernatant was collected and used for oil spreading assays. Oil spreading assays were conducted following the method as described by Morikawa et al. (2000). Each experiment was replicated three times.
Screening crude oil degrading bacteria
A total of 50 mL of enrichment medium was inoculated with 10 g of crude oil-contaminated soil and cultured on a rotary shaker at 60 °C and 220 RPM for 48 h. After enrichment, cultures were diluted 10-fold with sterile water, and 0.1 mL of the diluted culture was plated onto Medium 2 plates. Plates were incubated at 60 °C for 48 h. Single colonies on these plates were then inoculated into 5 mL of Medium 2, supplemented with 1% crude oil, and grown on a rotary shaker for 48 h. The culture was then extracted with ethyl acetate and subjected to GC analysis.
Strain identification
The TIANamp Bacteria Kit (Tiangen, Beijing, China) was used to isolate bacterial chromosomal DNA following the manufacturer’s protocols. The isolated genomic DNA was then used as template to amplify the 16 S rRNA gene via PCR, using the primers listed in Table S1 (see Supplementary Materials). The resulting PCR products were ligated into pGM-Simple-T Fast Vector (Tiangen, Beijing, China), and the resulting plasmids were isolated using the TIANprep Mini Kit (Tiangen). The 16 S rRNA gene fragment from the plasmid was sequenced and analyzed using BLAST with the NCBI nucleotide database.
Preparation and analysis of surfactin using high-performance liquid chromatography
Surfactin was extracted from the broth of B. subtilis GZ6 and B. subtilis GZ6 (pg3srfA) as previously described (Li et al. 2015). High-performance liquid chromatography (HPLC) analysis was performed using an LC-20 A system (Shimadzu, Kyoto, Japan) equipped with a UV detector. The analytical column was a GL C18 column (25 mm × 4.6 mm, 5 μm; GL Sciences, Kyoto, Japan).
The mobile phase consisted of a mixture of acetonitrile, water, and trifluoroacetic acid (85:15:0.1) at a flow rate of 0.8 mL/min. Surfactin was detected at a wavelength of 205 nm and its concentration was quantified using a standard curve for surfactin.
Gas chromatography analysis and determination of the alkane degradation rate
Analysis was performed on an Agilent 7890 gas chromatography (GC) system with a flame ionization detector, (Agilent, Santa Clara, CA, USA) equipped with an HP-5 column (30 m × 0.25 μm; Agilent). GC conditions were as follows: injection and detection temperatures were set to 280 °C, the initial oven temperature was set to 50 °C, and increased to 300 °C at a rate of 9 °C/min. The hydrogen and air flow rates were 35 and 400 mL/min, respectively, and the split ratio was set to 10:1. The alkane degradation rate (ADR) was calculated using the following formula:
where P2 is the total peak area of the alkane control on GC, and P1 is the total alkane peak area of the alkane-degrading strain.
Gas chromatography-mass spectrometry analysis
Gas chromatography-mass spectrometry (GC–MS) analyses were performed on an Agilent GC–MS system (7890 A gas chromatograph and 5975 C mass-selective detector; Agilent Technologies, Santa Clara, CA, USA) equipped with a DB-WAX column (30 m × 0.25 μm, Agilent Technologies). The GC conditions were as follows: split injection (injector temperature 230 °C, split 1/5); oven temperature, programmed from 35 °C (held for 3 min) to 47 °C at 5 °C/min, then to 100 °C at 25 °C/min, then to 145 °C at 2.5 °C/min, and then to 200 °C (held for 5 min) at 25 °C/min; the post-injection dwell time was 0.04 min; carrier gas was He and the and flow rate was 1.0 mL/min; interface temperature was 160 °C; injection volume was 0.2 µL. The mass spectrometry was used in electron impact ionization mode, with an electron energy of 70 eV, an ion source temperature of 230 °C, and a quadrupole temperature of 150 °C. Data were acquired in full-scan (m/z 10–500) mode. Other GC–MS conditions were the same as described in Ai et al. (2014). Data were acquired and analyzed using Enhanced ChemStation (version E.02.00.493, Agilent Technologies). Peaks were identified by comparing volatile sample mass spectra with spectra in the NIST08 Mass Spectral Database of the National Institute of Standards and Technology.
Routine PCR amplification
PCR amplification was performed using Tsingke PCR Master Mix (Tsingke, China) according to specifications (PCR amplification speed, 1 kb/10 sec). The PCR cycling protocol included an initial denaturation step at 95 °C for 5 min, followed by 30 cycles at 95 °C for 1 min, 53 °C for 1 min, and 72 °C for a specified time (calculated via the DNA fragment length). A final extension step at 72 °C was then performed for 10 min.
Promoter replacement by single-cross homologous recombination
PCR was used to amplify a 2 kb sequence upstream of the srfA promoter region with primers P1 and P2 (provided in Table S1, see Supplementary Materials). An artificial PG3 promoter was introduced at the 5’ end of the 2 kb DNA fragment using primers P3–P6 (listed in Table S1, see Supplementary Materials) via Gibson assembly (Gibson 2011). The resulting hybrid gene construct (2.1 kb) was then ligated into the RepB site of the pMA5 plasmid (Novogen). The final plasmid construct was designated as pMA5cmPG3SrfA. B. subtilis GZ6 competent cells were prepared following a previously described method (Xue et al. 1999). The pMA5cmPG3SrfA recombinant plasmids were then electroporated into B. subtilis GZ6 competent cells using a MicroPulser Plus electroporator (Bio-Rad, Richmond, CA, USA) with settings of 2.5 kV voltage, two pulses, and 2.5 ms time constant. The electroporation was performed in 0.2 cm cuvettes. After electroporation, cells were plated on LB agar containing 30 µg/mL kanamycin. Colony PCR and sequencing were utilized to identify emerging clones. The confirmed strain was named B. subtilis GZ6 (pg3srfA).
Plasmid constructions and expression of the LadA gene in E. Coli or G. Stearothermophilus
The ladA gene (1.3 kb, GenBank: ABO68832) was synthesized by Genscript Co (Nanjing, China). The gene was ligated into the expression vector pET30a between the NdeI and XhoI sites by DNA assembly. This ligation resulted in the recombinant plasmid pETladA, which was then transformed into chemically competent E. coli BL21(DE3) cells. A colony of positive transformants was selected and inoculated into 5 mL of Luria-Bertani (LB) medium supplemented with kanamycin. The culture was incubated at 37 °C under shaking until the optical density at 600 nm (OD600) reached 0.8. Protein expression was induced by adding isopropyl-β-D-thiogalactoside to a final concentration of 1 mM. After 12 h of induction, cells were harvested by centrifugation at 8,000 RPM for 10 min.
The plasmid for expression in Geobacillus strains was constructed by amplifying and inserting a 100 bp PnppT12 constitutive promoter upstream of the ladA2mu gene (Xue et al. 1999). The resulting hybrid gene, PpladA2mu (1.4 kb), was then cloned into the HindIII site of the pIM1773 plasmid (Addgene, Cambridge, MA, USA) using primers P9–P12 and Gibson assembly. The final plasmid was designated as pIMPpladA2mu, which was then transformed into G. stearothermophilus competent cells as previously described (Xue et al. 1999). The cells were cultured on Medium 2 plates at 60 °C overnight. Single clones were then inoculated into C18 medium and incubated at 60 °C and 220 RPM for 36 h.
Molecular docking and in silico mutagenesis
The catalytic site of the LadA protein (PDB ID: 3B9O) was docked with an 18-carbon alkane molecule using Discovery Studio 2019 software (Accelrys, San Diego, CA, USA). The ligand was docked, and interaction energies were calculated using the CDOCKER docking algorithm. Protein and ligand structures were parameterized using the CHARMm force field. The ligand conformations that ranked highest were clustered using a 2.0 Å root-mean-square deviation cut-off and scored based on the CDOCKER interaction energy. The global structure with the lowest binding energy was chosen for further analysis.
Computational site-directed mutagenesis was performed using the Calculate Mutation Energy (Binding) protocol in Discovery Studio 2019 to examine the role of binding pocket residues in complex stabilization. The final docked complexes were energy-minimized using the CHARMm force field and the Smart Minimizer algorithm. In silico mutagenesis was performed by calculating the free binding energy of the docked complex. To estimate the effect of individual mutations on complex binding, each residue in the binding pocket was mutated to 19 different amino acids. The mutation binding energy was then calculated as ΔΔGmut = ΔΔGbind(mutant) - ΔΔGbind(wild-type), where ΔΔGmut represents the mutation energy and ΔΔGbind represents the difference in free energy between bound and unbound states.
Construction of NNK saturation mutant libraries and high-throughput screening of mutants
NNK saturation mutant libraries were created through NNK saturation mutagenesis (Kretz et al. 2004) using the primers listed in Table S2 (P13–P34, see Supplementary Materials). Mutations in clones were confirmed by DNA sequencing.
The library clones were transferred to 96-well plates, each containing 100 µL of LB medium supplemented with 50 µg/mL kanamycin, and incubated at 37 °C for 12 h under shaking at 200×g. Protein expression was induced by adding isopropyl-β-D-thiogalactoside to a final concentration of 1.0 mM. After a 12-h induction period, cells were harvested by centrifugation at 4,000×g for 10 min. The cell pellets were then suspended in 100 µL of chilled 50 mM Tris-HCl buffer (pH 7.5) containing 10 mg/mL lysozyme. Following 30-min incubation and subsequent centrifugation, the resulting supernatants were transferred to fresh 96-well plates for library screening. The reaction plate was prepared with 120 µL of 50 mM Tris-HCl buffer (pH 7.5), 1.5 mM octodecane, 1.5 mM NADPH, 1.5 mM MgSO4, and 0.015% (w/v) Triton X-100 in each well. Next, 30 µL of the LadA mutant library supernatant was added to each well. The plates were incubated at 60 °C for 10 min under shaking. NADPH consumption was determined by measuring the absorbance at 360 nm using a BioTek Synergy 2 Multi-Mode Microplate Reader (BioTek, Beijing, China). A sample without octodecane was used as background control.
Purification of his-tagged proteins on nickel-chelating columns
Cells grown in a 500 mL expression culture were suspended in 40 mL binding buffer (50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole at pH 8.0) and lysed by sonication in an ice bath for 20 min at 200 W. The supernatant was then applied to a 1 mL Novagen His-Bind gravity flow column equilibrated with 20 mL Ni-NTA binding buffer. The column was washed with 20 mL wash buffer (50 mM NaH2PO4, 300 mM NaCl, and 20 mM imidazole at pH 8.0). The His-tagged protein was eluted with 10 mL elution buffer (50 mM NaH2PO4, 300 mM NaCl, and 200 mM imidazole at pH 8.0). To remove imidazole from the elution buffer, the eluate was applied to a 5 mL HiTrap desalting column (GE Healthcare, USA) and eluted with 50 mM Tris-HCl buffer.
Activity assays of purified and crude LadA and determination of kinetic constants
The reaction mixture was composed of 1 mM octodecane, 0.015% (w/v) Triton X-100, and 10 mM NADPH in 50 mM Tris-HCl buffer (pH 8.0) with a volume of 200 µL. To ensure homogenization, the mixture was sonicated for 1 min. Subsequently, 100 µl of the same buffer containing 0.1 µg of purified enzyme was added. Reactions were initiated by incubation at 60 °C for 10–20 min and terminated by extraction with an equal volume of chloroform. NADPH consumption was determined following the method as described (Dubbels et al. 2007). Km and Vmax values were calculated using Lineweaver-Burk plotting plots. Reduced flavin mononucleotide (FMNH2) consumption was calculated by measuring the increase at A455 during alkane oxidation (Uetz et al. 1992) using a Shimadzu PharmaSpec UV-1700 UV-VIS spectrophotometer (Tokyo, Japan). A total of 10–30 µmol FMN was added to the reaction mixture and the initial reaction rate was measured spectrophotometrically. Michaelis-Menten analyses for FMN were then performed with initial velocities using GraphPad Prism v5.0 (GraphPad Software, Boston, MA, USA) to calculate the Michaelis-Menten constant (Km).
Alkane consumption was analyzed using GC, as described above. Enzyme activity was defined as the amount that oxidizes 1 µmol of NADPH or 1 µmol of octodecane per min. To measure expressed alkane monooxygenase activity in G. stearothermophilus GZ178, cells from a 50 mL expression culture were suspended in 4 mL of Tris-HCl buffer (50 mM, pH 8.0) and disrupted by sonication for 20 min at 200 W in an ice bath. The resulting supernatant (100 µL) was added to the same reaction mixture under identical conditions as described for purified LadA.
Determining the dewaxing rate by crude oil weight loss test
In a flask containing 40 g of crude oil (M1), 40 mL of microbial dewaxing agent culture was added, while in the control flask (M2), the culture was replaced by 40 mL of sterile water. The empty weights of the flasks were measured and recorded as M10h and M20h, respectively. The two flasks were incubated at 60 °C and 120 RPM for 72 h. After incubation, they were placed in an incubator at 35 °C for 90 min to allow the oil to solidify. The flasks were then inverted and incubated at 40 °C for 60 min. Finally, the total weights of flasks were measured again and recorded as M172h and M272h, respectively. Each experiment was replicated three times. The dewaxing rate (DWR) was calculated using the following formula.
where ∆ M1 = M172h - M10h; ∆ M2 = M272h - M20h.
Optimizing the composition of the microbial dewaxing agent
Individual colonies from strain-preserving plates were inoculated into 5 mL of medium and grown on a rotary shaker at 40–60 °C and 220 RPM for 24 h. The cultures were then transferred into fresh 50 mL medium and further grown under the same conditions for 48 h. B. subtilis GZ6 (pg3srfA) was cultured in Medium 1 at 40 °C, while G. thermodenitrificans GZ156 and G. stearothermophilus GZ178 (pIMPpladA2mu) were cultured in Medium 2 at 60 °C. The resulting cultures were mixed at a specific ratio and subjected to crude oil weight loss test (Yi 2015).
Results and discussion
Bacterial screening and strain identification

Positive strains in the oil spreading test. Average diameters of oil discharge rings: Kocuria carniphila GZ64, 1.7 ± 0.1 cm, Pseudomonas aeruginosa GZ37, 1.9 ± 0.1 cm, and Bacillus subtilis GZ6, 3.5 ± 0.2 cm; Each experiment was performed in triplicate
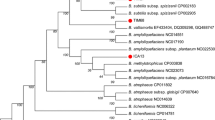
Oil wells are typically located at depths of around 2000 m, resulting in temperatures of approximately 60–70 °C. Therefore, initial screenings for bacteria capable of producing biosurfactants and degrading crude oil were conducted at 60 °C. However, no biosurfactant-producing strains were identified through oil spreading tests at this temperature. Subsequently, screening was conducted at gradually decreasing temperatures, and biosurfactant production was observed in three strains when cultured at 40 °C. These three bacteria that produced biosurfactants were identified as B. subtilis, Pseudomonas aeruginosa, and Kocuria carniphila through 16 S rRNA gene sequencing (Table 1). Among these three strains, B. subtilis GZ6 exhibited the most effective performance in oil spreading assays (Fig. 1). Currently, the most commonly used microbial species for biosurfactant production in microbial dewaxing agents is Pseudomonas aeruginosa (Soares Dos Santos et al. 2016). This bacterium is known for its high-yield production of rhamnolipids, and several strategies have been proposed to further enhance rhamnolipid production in this strain (She et al. 2011). However, despite being obtained in the initial round of screening, this strain was ultimately excluded from further experimentation because of its potential risk to public health and safety. The screening process in this study did not yield any thermophilic microorganisms capable of biosurfactant synthesis.
During the screening process for crude oil degrading bacteria, six strains capable of degrading alkanes were obtained and identified through sequencing of their 16 S rRNA gene (Table 1). GC–MS analysis was used to determine the substrate specificities and degradation rates of these strains, with G. thermodenitrificans GZ156 and G. stearothermophilus GZ178 exhibiting the highest rates at approximately 47.6% and 30.1%, respectively (Fig. 2 and Table S3). These two strains have different alkane substrate specificities: G. thermodenitrificans GZ156 degrades alkanes with low boiling points, such as dodecane, tetradecane, and pentadecane, while G. stearothermophilus GZ178 prefers alkanes with high boiling points, such as heptacosane and octacosane (Fig. 2 and Table S3).

Gas chromatography-mass spectrometry (GC–MS) analysis of alkane degradation. The degradation experiment was performed in triplicate and no significant differences were observed between the obtained GC–MS chromatograms. Blue line, G. thermodenitrificans GZ156 degradation, Red line, G. stearothermophilus GZ178 degradation
Enhancing B. subtilis biosurfactant productivity by promoter replacement

Enhancing the B. subtilis GZ6 surfactin productivity by promoter replacement. A, PCR amplification of the srfA gene cluster from the B. subtilis GZ6 genome. B, Surfactin produced by B. subtilis GZ6 (blue); and B. subtilis GZ6 (pg3srfA) (green). Four peaks (1, 2, 3, and 4) were observed for the surfactin standard (red) when analyzed by HPLC. C, Oil spreading tests for B. subtilis GZ6 and B. subtilis GZ6 (pg3srfA). Average diameters of oil spreading rings (each experiment was performed with three replicates), B. subtilis GZ6, 3.5 ± 0.1 cm; B. subtilis GZ6 (pg3srfA), 4.1 ± 0.2 cm
HPLC analysis using purchased standards (Fig. 3B) identified the biosurfactant produced by B. subtilis GZ6 as surfactin (Hsieh et al. 2008). Surfactin is typically composed of four cyclic lipopeptides, with chain lengths ranging from C13 to C16, that differ slightly in their amino acid sequences. The proportion of these four cyclic lipopeptides in the surfactin mixture varies among different B. subtilis strains (Jiao et al. 2017).
Native B. subtilis strains typically have low surfactin titers. To increase surfactin yield in these strains, several strategies were employed, including promoter replacement, which was identified as the most effective and simple approach (Li et al. 2015; Jiao et al. 2017). Surfactin is produced by surfactin synthase (SrfA), which is encoded by the seven-module non-ribosomal peptide synthetase gene cluster (srfA). When using the native srfA promoter, the transcription levels of srfA were low. However, the transcription of srfA in B. subtilis THY-7 could be initiated effectively by using a modified hybrid Pg3 promoter (Jiao et al. 2017). The B. subtilis GZ6 srfA gene cluster identified by PCR amplification and sequencing showed 99.8% homology to the B. subtilis THY-7 srfA gene cluster (Fig. 3A). Subsequently, the srfA promoter was replaced with the Pg3 promoter through the recombination of single-cross homologous (Jiao et al. 2017). The genetically modified B. subtilis GZ6 strain was confirmed through PCR and sequencing.
The production of surfactin by B. subtilis GZ6 (pg3srfA) increased to 2.32 g/L, compared to the production of 0.33 g/L of the wild-type strain. The modified strain of B. subtilis GZ6 (pg3srfA) produced significantly larger spreading rings in oil spreading assays (Fig. 3C). Additionally, B. subtilis GZ6 (pg3srfA) exhibited a growth curve comparable to that of B. subtilis GZ6, indicating that the replacement of the promoter did not affect cell growth (Fig. S2, A, Supplementary Materials).
LadA expression and engineering in E. Coli

Molecular docking (A) and residues of the LadA binding pocket (B). A, The distance between the oxygen atom of flavin mononucleotide (FMNOO) and the alkane terminal carbon is indicated by yellow dashed lines, while Pi-Alkyl interactions are indicated by orange dashed lines. B, Residues constituting the LadA binding pocket are represented by lines; C18 alkane and FMNOO are displayed as sticks
The high viscosity of RO is mainly the result of long-chain alkanes, also known as wax, with a carbon chain length of 18 or longer. The microbial dewaxing process targets these long-chain alkanes as they pose the primary obstacle to heavy crude oil recovery (Adlan et al. 2020). Based on preliminary screening results (Table S3), G. thermodenitrificans GZ156 did not degrade octadecane, while G. stearothermophilus GZ178 showed poor ADR on the substrate. Therefore, octadecane was selected as the target substrate for this research.
A previous study by Feng et al. (2007) illustrated the metabolic pathway for long-chain alkanes in G. thermodenitrificans. The pathway involves the utilization of an alkane monooxygenase (LadA), which activates alkanes to corresponding primary alcohols. LadA can degrade alkanes ranging from C15 to C36, with hexadecane being the optimal substrate and octadecane being the sub-optimal substrate. The first enzyme in a metabolic cascade usually catalyzes the rate-limiting step (Chen et al. 2008). It is worth noting that alkane monooxygenases generally have lower catalytic efficiencies than enzymes such as hydrolases. Increasing the copy number of the ladA gene in G. thermodenitrificans GZ156, particularly a version of LadA that was enhanced through protein engineering, may have the potential to improve the alkane metabolic cascade of the strain, especially in octadecane degradation, and subsequently enhance the wax removal rate.
Initial PCR amplification of the ladA gene from the total DNA of G. thermodenitrificans GZ156 or G. stearothermophilus GZ178 was unsuccessful. As previously reported, the ladA gene is located on a native plasmid (Feng et al. 2007), and it is likely that this plasmid is absent in these two strains. Despite this absence, their ability to degrade alkanes suggests the presence of an additional, as yet unidentified alkane hydroxylase system that also produces alcohol intermediates (Smits et al. 2002). To improve the alkane degrading capacity of these strains, the introduction of an improved LadA enzyme is a more promising option. However, protein engineering of LadA requires efficient gene manipulation and effective expression systems, that are lacking in Geobacillus species. The ladA gene was well expressed in E. coli BL21(DE3) under the T7 promoter (Fig. S1). Therefore, the initial engineering for LadA was conducted in the E. coli host.
Although the structure of LadA has been resolved, its catalytic mechanism has not yet been identified. However, LadA belongs to the SsuD subfamily within the bacterial luciferase family (Chen et al. 2008), and the catalytic mechanism of this subfamily has been well elucidated (Armacost et al. 2014). Therefore, the SsuD catalytic hypothesis was applied to propose a catalytic initiation mechanism for LadA. According to this hypothesis, the alkane terminal carbon is attacked by the distal oxygen atom of the C4a-hydroperoxyflavin of flavin mononucleotide. Protein-ligand docking was performed based on this hypothesis. The analysis focused on the lowest energy configuration among docking poses. In this configuration, the flavin mononucleotide distal oxygen atom was 2.8 Å away from the alkane terminal carbon. The terminal carbon was stabilized by two pi-alkyl interactions (Ozawa et al. 2008) with the imidazole rings of His17 and His311 (Fig. 4A).
According to the protein-ligand docking mechanism, the binding energy of the substrate ligand is typically positively correlated with catalytic efficiency (Londhe et al. 2019). To enhance substrate binding, virtual saturation mutagenesis was conducted and 25 residues within 5 Å of the docked substrate were analyzed. The changes in free energy (ΔG) were compared, showing that the Ala57His mutant presented the most pronounced reduction in binding energy (-5.0 kJ/mol). Mutations at the Phe10, His17, Ala57, Asp58, Val59, Tyr63, Gln79, Asn133, Val135, Ala227, and His311 sites resulted in ΔG values ranging from − 5.0 to -2.3 kJ/mol (Fig. 4B, Table S2, Supplementary Materials), suggesting that these mutations were beneficial. Mutants of other sites showed ΔG values ranging from − 2.3 to 2.9 kJ/mol, indicating either neutral or damaging effects (Spassov and Yan 2013).
The residues listed in Table S2 (Supplementary Materials) were applied for NNK saturation mutagenesis. The resulting mutant libraries were screened using high-throughput methods. A mutant with increased activity, namely Phe10Leu, was verified through GC analysis. NNK saturation mutagenesis was then performed on Phe10Leu at the other sites listed in Table S2. After screening and verification, the double mutant Phe10Leu/Asn133Arg (harboring pETladA2mu) was identified. The catalytic efficiency of this mutant was 11.7 times higher than that of the wild-type enzyme (Table 2). Table 2 shows that both single and double mutants had significantly lower Km values for the substrate compared to the wild-type strain, indicating an increased affinity between enzyme and substrate. The affinity between the single mutant and FMN remained unchanged, while the affinity between the double mutant and FMN decreased slightly. Overall, the mutation of the enzyme at these two sites has a minimal effect on the binding of coenzyme FMN to the enzyme. However, the mutation significantly increased the affinity between substrate and enzyme, thereby greatly improving the catalytic efficiency of the enzyme.
As previously reported, LadA was engineered through random and site-directed mutagenesis with the goal to enhance hexadecane oxidation (Dong et al. 2012). However, the best mutant obtained (F146N/N376I) differed significantly from the Phe10Leu/Asn133Arg mutant. This difference may be the result of the use of different substrates (hexadecane versus octadecane) for screening mutant libraries, resulting in variations in the binding of active sites and substrates. The use of random mutagenesis or virtual mutagenesis may have led to two different directed evolutionary pathways for LadA.
Expression of ladA2mu in G. Stearothermophilus GZ178 and octodecane transformation
After obtaining the ladA2mu gene from the E. coli host, attempts were made to express ladA2mu in G. thermodenitrificans GZ156 and G. stearothermophilus GZ178. However, no plasmid could be employed to express this gene in G. thermodenitrificans GZ156. The ladA2mu gene was successfully expressed in G. stearothermophilus GZ178, which was harboring plasmid pIMPpladA2mu. The relevant protein band was not detected in the sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis, suggesting low expression levels (Fig. S1, Supplementary Materials). However, the cells expressing the plasmid showed a significant increase in octodecane conversion compared to cells carrying the empty plasmid (from 25.0 to 75.3%, Fig. S3, Supplementary Materials). In addition, G. stearothermophilus GZ178 (pIMPpladA2mu) cells exhibited an expanded substrate spectrum toward short-chain alkanes, such as pentadecane and hexadecane (Table S3, Supplementary Materials). The growth curve of G. stearothermophilus GZ178 (pIMPpladA2mu) was comparable to that of G. stearothermophilus GZ178 (Fig. S2, B, Supplementary Materials). These data suggest that plasmid expression did not affect cell growth.
Optimization of the constitution of the microbial dewaxing agent
This study initially used the two strains of B. subtilis GZ6 (pg3srfA) and G. thermodenitrificans GZ156 in the agents. As shown in Fig. 5A, the DWR of the single strain agent (G. thermodenitrificans GZ156 alone) was only 50% compared to that of the double strain agent; therefore, the biosurfactant played a critical role in wax removal. Previous reports have shown that G. thermodenitrificans and G. stearothermophilus have different substrate preferences for carbon chain length (Lin et al. 2019). While G. thermodenitrificans prefers short-chain alkanes, such as dodecane, G. stearothermophilus prefers long-chain alkanes, such as octacosane (Fig. 2 and Table S3). It is proposed that the dewaxing rate could be improved by broadening the substrate spectrum of the microbial dewaxing agent. Therefore, G. stearothermophilus GZ178 (pIMPpladA2mu) was also introduced into the dewaxing agent. Compared to the wild-type strain, G. stearothermophilus GZ178 (pIMPpladA2mu) cells have an extended substrate spectrum (Table S3, Supplementary Material). The optimal microbial agent had the highest DWR when all three strains were mixed at a ratio of 5:80:15 (Fig. 5A). Figure 5B shows that the increase in surfactin production for B. subtilis GZ6 and the expression of ladA2mu in G. stearothermophilus GZ178 both contributed to the increase in DWR. DWR decreased when either modified strain was replaced by its wild-type strain. The complex composed of the native strains showed a DWR that was only 65% of that of the optimized composition (Fig. 5B).

Optimization of the constitution for microbial dewaxing agents. Each experiment was performed in triplicate. A, Bs, B. subtilis GZ6 (pg3srfA); Gt, G. thermodenitrificans GZ156; Gs, G. stearothermophilus GZ178 (pIMPpladA2mu). The 100% relative wax removal rate is 60.3 ± 3.2%. The OD580 of the cultures are 8.0 ± 0.1 for Bs, 3.0 ± 0.2 for Gt, and 6.0 ± 0.1 for Gs. B, All strain ratios are the same as 5/80/15 (Bs/Gt/Gs). 1, Same strain constitution ratio as the 5/80/15 agent in A; 2, Gs wild type strain (OD580 = 6.0) in the agent; 3, Bs wild type strain (OD580 = 8.0) in the agent; 4, All strains are wild type strains (Bs, OD580 = 8.0, Gt OD580 = 3.0, Gs OD580 = 6.0)
The microbial dewaxing agent operates through two main mechanisms: inhibiting the growth of wax crystals and degrading the existing wax. These dual functionalities rely on the biosurfactant production and wax-degrading activities of microbial strains. In this microbial dewaxing agent, strain B. subtilis GZ6 (pg3srfA) functions as biosurfactant producer while the other two strains degrade wax. The biosurfactant produced by B. subtilis GZ6 was surfactin, which remained stable even at a temperature of ~ 140 °C (Jiao et al. 2017). It is proposed that, during the sedimentation process following inoculation into oil wells, B. subtilis in the microbial agent proliferates within the appropriate temperature range at certain depths and continues to produce biosurfactants.
The introduction of G. stearothermophilus GZ178 (pIMPpladA2mu) into the agent significantly improved DWR for two reasons: First, the substrate spectrum of G. stearothermophilus GZ178 (pIMPpladA2mu) in alkane degradation was significantly extended by the introduction of a heterologous ladA2mu gene. When the strain was incorporated into the microbial agent, the DWR was increased by approximately 35%. Furthermore, although both Geobacillus species belong to the same genus, they have different alkane metabolic pathways. G. thermodenitrificans metabolizes long chain alkanes by the primary oxidation pathway, in which the C atom at the end of the alkane is first oxidized to alkanol by alkane oxidase, then sequentially converted to aldehyde and fatty acid, and finally enters β-oxidation (Feng et al. 2007). However, G. stearothermophilus metabolizes alkanes through secondary oxidation, in which long chain alkanes are oxidized to secondary alcohols and acetic acid. These secondary alcohols are further oxidized to ketones and esters, which then enter the same pathway as in G. thermodenitrificans (Liu et al. 2008). This diversity of pathways can effectively mitigate inhibition by intermediate metabolites during the alkane metabolism, resulting in significantly improved wax removal efficiency.
Biosafety and ethical considerations of microbial dewaxing agents in environmental applications
The use of genetically engineered microbes in environmental applications presents a complex set of challenges and imposes ethical considerations. The microbial dewaxing agent in this study has high biosafety. First, the agent was applied in oil fields with relatively harsh environments and relatively small microbial populations, thus minimizing horizontal transfer of resistance genes. Second, the genetically modified hyperthermophilic bacteria in this agent cannot multiply at room temperature, thus limiting their gene spread to mesophilic microbes. Third, Bacillus subtilis is considered a GRAS strain (i.e., Generally Recognized As Safe) and both native and gene modified strains are widely used in food, pharmaceutical, and other fermentation industries (Ejaz et al. 2022).
Research status and future research directions for microbial dewaxing agents
Currently, most microbial dewaxing agents are composed of multiple bacteria, while only few are composed of single bacteria (Patel et al. 2015). Certain agents may contain non-biological security microorganisms, such as Bacillus anthracis (Liu et al. 2014) and Pseudomonas aeruginosa (Câmara et al. 2019). The DWRs of these agents vary widely, ranging from 12.3 to 68.2%. Comparing the DWR values of these agents alone does not provide meaningful insight. Crude oil components have different geographic characteristics, especially regarding the amounts of heavy oil and wax contents. Therefore, it is necessary to verify the effectiveness of dewaxing agents through field experiments in oil wells. The microbial dewaxing agent developed in this study has a high dewaxing rate of 60.2% and is environmentally safe. Field tests conducted in several oil wells have presented positive results in terms of dewaxing and increased production (data not shown).
Spore-forming bacteria, such as Bacillus, Geobacillus, and Clostridium, have been utilized as microbial dewaxing agents (Preeti et al. 2019; Zhou et al. 2016). These bacteria are frequently found in deep oil reservoirs and can withstand the prevalent harsh conditions, including nutrient deprivation, extreme temperatures, and high salt concentrations. The microbial dewaxing agent in this study was also developed using spore-producing microorganisms, which allows for a long-lasting effect because of the spores’ resistance to adverse environments. Therefore, bacterial strains capable of forming endospores and degrading petroleum or synthesizing biosurfactants are potential targets for further screening and isolation of microorganisms that remove wax. Similarly, extremophilic microbes have also been examined for microbial dewaxing applications because of their ability to withstand extreme environments. These microbes are either thermophilic, halophilic, or barophilic (Varjani and Upasani 2016; Lin et al. 2019). In addition, genetically engineered microbes have been used for microbial engineered wax removal through techniques such as protoplast fusion, gene knockout, and genome editing. These technologies have the potential to improve the temperature adaptability of microbes (Sun et al. 2017) and increase their biosurfactant productivity (Jiao et al. 2017).
Microbial dewaxing technology reduces reliance on conventional chemicals and lowers the risk of environmental contamination. It uses naturally occurring microorganisms to enhance the oil recovery process, thus reducing the need for harmful chemicals and helping to protect soil and water quality. The use of microbial dewaxing agents in oil recovery can not only reduce resource waste, but also extend the productive life stage of oil fields. Research and practical applications of microbial dewaxing agents have increased because of their potential economic benefits for oil extraction companies as well as for the green and sustainable development of energy extraction technology.
In summary, the ideal microbial dewaxing agent should possess thermophilic, halophilic, and barophilic properties. Such microbes can be isolated from the environment of deep-sea hydrothermal vents (Jin et al. 2019). However, native microbes with superior dewaxing performance are rarely obtained in such harsh reservoir conditions. To overcome this challenge, several methods can be employed. One such method is to customize microbial strains by designing or assembling novel synthetic metabolic pathways for the production of biosurfactants or wax-degrading pathways. Another method is to engineer and express enzymes with strong wax-degrading capabilities in a suitable host. The first step in achieving these goals is to create the appropriate genetic operating system and gene editing tools. The application of synthetic biology technology for microbial dewaxing, which is also the scope of microbial engineered wax removal, has the potential to significantly improve its efficiency.
Conclusion
In this study, three spore-forming bacterial strains were obtained via screening crude oil-contaminated soil samples. The surfactin titer of strain B. subtilis GZ6 was increased by promoter replacement of the surfactin synthase gene cluster (srfA). The enhanced alkane monooxygenase double mutant LadAF10L/N133R was generated by site-directed mutagenesis and then expressed in G. stearothermophilus GZ178. The final microbial dewaxing agent, consisting of two modified strains (B. subtilis GZ6 (pg3SrfA) and G. stearothermophilus GZ178 (pIMPpladA2mu)) and the native strain G. thermodenitrificans GZ156, achieved a 35% higher DWR compared to a composition consisting of three native strains.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Adlan NA, Sabri S, Masomian M, Ali MSM, Rahman R (2020) Microbial Biodegradation of Paraffin Wax in Malaysian crude oil mediated by Degradative Enzymes. Front Microbiol 11:565608
Ai G, Sun T, Dong X (2014) Evaluation of hydrolysis and alcoholysis reactions in gas chromatography/mass spectrometry inlets. J Chromatogr A 1356:283–288
Armacost K, Musila J, Gathiaka S, Ellis HR, Acevedo O (2014) Exploring the catalytic mechanism of alkanesulfonate monooxygenase using molecular dynamics. Biochemistry 53:3308–3317
Astuti DI, Purwasena IA, Putri RE, Amaniyah M, Sugai Y (2019) Screening and characterization of biosurfactant produced by Pseudoxanthomonas sp. G3 and its applicability for enhanced oil recovery. J Pet Explor Prod Te 9:2279–2289
Bian ZW, Zhang XC, Wu YY, Wei LS, Wu YF, Wu HN (2021) Review on microbial enhanced oil recovery in China: mechanisms, potential disadvantages, and application of genetic engineering. Energ Source Part A
Câmara, J.M.D.A., Sousa, M.A.S.B., Barros Neto, E.L., Oliveira, M.C.A.J.J.o.P.E. and Technologies, P. (2019) Application of rhamnolipid biosurfactant produced by Pseudomonas aeruginosa in microbial-enhanced oil recovery (MEOR)
Chen J, Shi X, Padmanabhan R, Wang Q, Wu Z, Stevenson SC, Hild M, Garza D, Li H (2008) Identification of novel modulators of mitochondrial function by a genome-wide RNAi screen in Drosophila melanogaster. Genome Res 18:123–136
Chen WW, Li JD, Sun XN, Min J, Hu XK (2017) High efficiency degradation of alkanes and crude oil by a salt-tolerant bacterium Dietzia species CN-3. Int Biodeter Biodegr 118:110–118
Dong Y, Yan J, Du H, Chen M, Ma T, Feng L (2012) Engineering of LadA for enhanced hexadecane oxidation using random- and site-directed mutagenesis. Appl Microbiol Biotechnol 94:1019–1029
Dubbels BL, Sayavedra-Soto LA, Arp DJ (2007) Butane monooxygenase of ‘Pseudomonas Butanovora’: purification and biochemical characterization of a terminal-alkane hydroxylating diiron monooxygenase. Microbiol (Reading) 153:1808–1816
Ejaz S, Khan H, Sarwar N, Aqeel MS, Al-Adeeb A, Liu S (2022) A review on recent Advancement in expression strategies used in Bacillus subtilis. Protein&Peptide Lett 29:733–743
Etoumi A, El Musrati I, El Gammoudi B, Behlil E, M (2008) The reduction of wax precipitation in waxy crude oils by Pseudomonas species. J Ind Microbiol Biotechnol 35:1241–1245
Feng L, Wang W, Cheng J, Ren Y, Zhao G, Gao C, Tang Y, Liu X, Han W, Peng X, Liu R, Wang L (2007) Genome and proteome of long-chain alkane degrading Geobacillus thermodenitrificans NG80-2 isolated from a deep-subsurface oil reservoir. Proc Natl Acad Sci USA 104:5602–5607
Gibson DG (2011) Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498:349–361
Hall C, Tharakan P, Hallock J, Cleveland C, Jefferson M (2003) Hydrocarbons and the evolution of human culture. Nature 426:318–322
Hsieh FC, Lin TC, Meng M, Kao SS (2008) Comparing methods for identifying Bacillus strains capable of producing the antifungal lipopeptide iturin A. Curr Microbiol 56:1–5
Jiao S, Li X, Yu H, Yang H, Shen Z (2017) In situ enhancement of surfactin biosynthesis in Bacillus subtilis using novel artificial inducible promoters. Biotechnol Bioeng 114:832–842
Jin M, Gai Y, Guo X, Hou Y, Zeng R (2019) Properties and Applications of Extremozymes from Deep-Sea Extremophilic Microorganisms: A Mini Review. Marine drugs 17
Kretz KA, Richardson TH, Gray KA, Robertson DE, Tan X, Short JM (2004) Gene site saturation mutagenesis: a comprehensive mutagenesis approach. Methods Enzymol 388:3–11
Li X, Yang H, Zhang D, Li X, Yu H, Shen Z (2015) Overexpression of specific proton motive force-dependent transporters facilitate the export of surfactin in Bacillus subtilis. J Ind Microbiol Biotechnol 42:93–103
Lin JH, Zhang KC, Tao WY, Wang D, Li S (2019) Geobacillus strains that have potential value in microbial enhanced oil recovery. Appl Microbiol Biotechnol 103:8339–8350
Liu QK, Wang J, Li GQ, Ma T, Liang FL, Liu RL (2008) Characterization of a thermophilic Geobacillus strain DM-2 degrading hydrocarbons. Huan Jing Ke Xue 29:3554–3560
Liu J, Chen Y, Xu R, Jia Y, Wang J, Lu Y, Sun H, Yuan (2014) Microbial paraffin-removal technology using paraffin-degrading and biosurfactant-producing strain. D J J o C 26:2957–2959
Londhe AM, Gadhe CG, Lim SM, Pae AN (2019) Investigation of Molecular Details of Keap1-Nrf2 Inhibitors Using Molecular Dynamics and Umbrella Sampling Techniques. Molecules 24
Morikawa M, Hirata Y, Imanaka T (2000) A study on the structure-function relationship of lipopeptide biosurfactants. Biochim Biophys Acta 1488:211–218
Murungi PI, Sulaimon AA (2022) Petroleum sludge treatment and disposal techniques: a review. Environ Sci Pollut Res Int 29:40358–40372
Norouzi N, Fani M, Ziarani ZK (2020) The fall of oil age:a scenario planning approach over the last peak oil of human history by 2040. J Petrol Sci Eng 188
Ozawa T, Tsuji E, Ozawa M, Handa C, Mukaiyama H, Nishimura T, Kobayashi S, Okazaki K (2008) The importance of CH/pi hydrogen bonds in rational drug design: an ab initio fragment molecular orbital study to leukocyte-specific protein tyrosine (LCK) kinase. Bioorg Med Chem 16:10311–10318
Patel J, Borgohain S, Kumar M, Rangarajan V, Somasundaran P, Sen R (2015) Recent developments in microbial enhanced oil recovery. Renew Sustain Energy Rev 52:1539–1558
Preeti, Arora P, Kshirsagar R, Dolly, Pal, Rana P, Colloids K, D.J. and, Biointerfaces SB (2019) Hyperthermophilic Clostridium sp. N-4 produced a glycoprotein biosurfactant that enhanced recovery of residual oil at 96°C in lab studies. Colloids Surf B 182:110372–110372
She YH, Zhang F, Xia JJ, Kong SQ, Wang ZL, Shu FC, Hu JM (2011) Investigation of biosurfactant-producing indigenous microorganisms that enhance residue oil recovery in an oil reservoir after polymer flooding. Appl Biochem Biotechnol 163:223–234
Shibulal B, Al-Bahry SN, Al-Wahaibi YM, Elshafie AE, Al-Bemani AS, Joshi SJ (2014) Microbial enhanced heavy oil recovery by the aid of inhabitant spore-forming bacteria: an insight review. TheScientificWorldJournal 2014, 309159
Smits TH, Balada SB, Witholt B, van Beilen JB (2002) Functional analysis of alkane hydroxylases from gram-negative and gram-positive bacteria. J Bacteriol 184:1733–1742
Soares Dos Santos A, Pereira N, Jr., Freire DM (2016) Strategies for improved rhamnolipid production by Pseudomonas aeruginosa PA1. PeerJ 4:e2078
Spassov VZ, Yan L (2013) pH-selective mutagenesis of protein-protein interfaces: in silico design of therapeutic antibodies with prolonged half-life. Proteins 81:704–714
Sun SS, Luo YJ, Zhou Y, Xiao M, Zhang ZY, Hou JR, Wei XF, Xu QS, Sha T, Dong H, Song H, Zhang ZZ (2017) Application of Bacillus spp. in Pilot Test of Microbial Huff and Puff to Improve Heavy Oil Recovery. Energ Fuel 31:13724–13732
Uetz T, Schneider R, Snozzi M, Egli T (1992) Purification and characterization of a 2-Component monooxygenase that hydroxylates Nitrilotriacetate from Chelatobacter Strain Atcc-29600. J Bacteriol 174:1179–1188
Varjani SJ, Upasani VN (2016) Core Flood study for enhanced oil recovery through ex-situ bioaugmentation with thermo- and halo-tolerant rhamnolipid produced by Pseudomonas aeruginosa NCIM 5514. Bioresource Technol 220:175–182
Wang ZW, Zhu LQ, Liu HC, Li WP (2013) A conversion coating on carbon steel with good anti-wax performance in crude oil. J Petrol Sci Eng 112:266–272
Wang WQ, He Y, Wang B, Dong M, Zhang HJ, Shen C (2022) Experimental study on wax removal and viscosity reduction of waxy crude oil by Ochrobactrum intermedium. J Petrol Sci Eng 213
Wu B, Xiu J, Yu L, Huang L, Yi L, Ma Y (2022) Research advances of microbial enhanced oil recovery. Heliyon 8, e11424
Xue GP, Johnson JS, Dalrymple BP (1999) High osmolarity improves the electro-transformation efficiency of the gram-positive bacteria Bacillus subtilis and Bacillus licheniformis. J Microbiol Methods 34:183–191
Yi SJ (2015) A method for determining the wax removing rate of microbiological wax-cleaning agents. China
Zhang J, Gao H, Xue Q (2020) Potential applications of microbial enhanced oil recovery to heavy oil. Crit Rev Biotechnol 40:459–474
Zhao G, Liang LH, Lv DF, Ji WJ, You Q, Dai CL (2023) A novel nanofluid of modified carbon black nanoparticles for enhanced oil recovery in low permeability reservoirs. Petrol Sci 20:1598–1607
Zhou JF, Li GQ, Xie JJ, Cui XY, Dai XH, Tian HM, Gao PK, Wu MM, Ma TJRA (2016) A novel bioemulsifier from Geobacillus stearothermophilus A-2 and its potential application in microbial enhanced oil recovery. RSC Adv 6:96347–96354
Acknowledgements
We greatly acknowledge the financial support from Scientific Research Program of Beijing Municipal Commission of Education (KM202110017009, granted to Xiaoyan Guo), Undergraduates Research Training Program of Beijing Institute of Petrochemical Technology (2022J00178, granted to Lizhu Li). Discovery Studio 4.5 software analysis was conducted by Dr. Ma Rui, Key Laboratory for Feed Biotechnology of the Ministry of Agriculture, Feed Research Institute, Chinese Academy of Agricultural Sciences, Beijing.
Author information
Authors and Affiliations
Contributions
JW and HJ conceived and designed research. XG and XZ conducted experiments. LL contributed analytical tools. JW analyzed data. JW wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors hereby declare that there is no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Guo, X., Zhao, X., Li, L. et al. Development of a microbial dewaxing agent using three spore forming bacteria. Bioresour. Bioprocess. 11, 80 (2024). https://doi.org/10.1186/s40643-024-00795-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-024-00795-z