
Abstract
Background
Perioperative acute kidney injury (AKI) caused by ischemia–reperfusion (IR) is a significant contributor to mortality and morbidity after major surgery. Furosemide is commonly used in postoperative patients to promote diuresis and reduce tissue edema. However, the effects of furosemide on renal microcirculation, oxygenation and function are poorly understood during perioperative period following ischemic insult. Herein, we investigated the effects of furosemide in rats subjected IR insult.
Methods
24 Wistar albino rats were divided into 4 groups, with 6 in each; Sham-operated Control (C), Control + Furosemide (C + F), ischemia/reperfusion (IR), and IR + F. After induction of anesthesia (BL), supra-aortic occlusion was applied to IR and IR + F groups for 45 min followed by ongoing reperfusion for 15 min (T1) and 2 h (T2). Furosemide infusion was initiated simultaneously in the intervention groups after ischemia. Renal blood flow (RBF), vascular resistance (RVR), oxygen delivery (DO2ren) and consumption (VO2ren), sodium reabsorption (TNa+), oxygen utilization efficiency (VO2/TNa+), cortical (CμO2) and medullary (MμO2) microvascular oxygen pressures, urine output (UO) and creatinine clearance (Ccr) were measured. Biomarkers of inflammation, oxidative and nitrosative stress were measured and kidneys were harvested for histological analysis.
Results
IR significantly decreased RBF, mainly by increasing RVR, which was exacerbated in the IR + F group at T2 (2198 ± 879 vs 4233 ± 2636 dyne/s/cm5, p = 0.07). CμO2 (61.6 ± 6.8 vs 86 ± 6.6 mmHg) and MμO2 (51.1 ± 4.1 vs 68.7 ± 4.9 mmHg, p < 0.05) were both reduced after IR and did not improve by furosemide. Moreover, VO2/TNa+ increased in the IR + F group at T2 with respect to the IR group (IR: 3.3 ± 2 vs IR + F: 8.2 ± 10 p = 0.07) suggesting a possible deterioration of oxygen utilization. Ccr did not change, but plasma creatinine increased significantly in IR + F groups. Histopathology revealed widespread damage both in the cortex and medulla in IR, IR + F and C + F groups.
Conclusion
Renal microvascular oxygenation, renal function, renal vascular resistance, oxygen utilization and damage were not improved by furosemide administration after IR insult. Our study suggests that furosemide may cause additional structural and functional impairment to the kidney following ischemic injury and should be used with caution.
Similar content being viewed by others
Background
Renal damage and loss of function can frequently occur during and after abdominal and cardiovascular surgery, transplantation, and renal arterial reconstruction surgery due to ischemia/reperfusion (IR) induced acute kidney injury (AKI) [1,2,3,4].
The kidney is vulnerable both to ischemic and hypoxic injury, due to its high energy expenditure and workload. The renal blood flow is continuously adjusted with strict autoregulatory mechanisms in physiological states to ensure a balance between oxygen delivery and consumption [5]. However, this balance can be impaired after IR injury. Renal tubular damage is triggered by the imbalance between oxygen delivery and consumption, hypoxia, inflammation, and it is further exacerbated by hyperoxia and oxidative stress during reperfusion [6].
Furosemide is a diuretic drug which inhibits the electrolyte transport receptors in the thick ascending limb (TAL) of the Henle. Furosemide can increase the excretion of Na+ and water by inhibiting Na–K–2Cl co-transporter in TAL, therefore it reduces Na–K-ATPase-mediated oxygen consumption. It is well known that fluid overload in patients with AKI is independently associated with mortality and poor outcome [7, 8]. Therefore, furosemide is commonly used to promote diuresis in the postoperative and critically ill patients with fluid overload, regardless of whether the patient has AKI or not. However, human and animal studies have shown that furosemide is not effective in preventing AKI in these settings [9, 10]. Furthermore, it was recently demonstrated that diuretic use was independently associated with higher AKI risk in patients hospitalized with Covid-19, which is a hyperinflammatory disease per se [11]. Several mechanisms including increased oxidative stress, relative hypovolemia, and higher renal vascular resistance are suggested as factors causing AKI following furosemide use [12, 13].
We hypothesized that the use of furosemide in perioperative phase may have some beneficial effect to reduce renal damage triggered by oxidative stress and hypoxia because its mechanism of action to reduce the medullary oxygen consumption as a result of increasing Na+ and water excretion. However, the existing research fails to resolve the impact of furosemide on IR-induced AKI in perioperative period. Here, we believed that understanding the effects of furosemide on the kidney, which is already damaged, may provide a new insight to use of furosemide in postoperative and ICU patients with renal injury. Therefore, we aimed to investigate the effects of furosemide on the renal cortical and medullary microvascular oxygenation, function, inflammation and extent of histopathological damage following a model of supra-aortic occlusion induced AKI in rats.
Methods
The current study uses an experimental design to simulate IR-induced AKI after cardiovascular or abdominal surgery. Oxygen phosphorescence technique employed in this study offers the advantage of the measurement of individual oxygen concentration both in the renal cortex and the medulla.
Animals
All experiments described in this study were approved by the Institutional Animal Experimentation Committee of the Academic Medical Centre of the University of Amsterdam (DFL83). Care and handling of the animals were following the guidelines for Institutional and Animal Care and Use Committees. The study has been carried out according to the Declaration of Helsinki. Experiments were performed on 24 male Wistar albino rats (Charles River, The Netherlands) with a mean ± SD bodyweight of 340 ± 26 g.
Surgical preparation
All the animals were anesthetized with an intraperitoneal injection of a mixture of 90 mg/kg ketamine (Nimatek, Eurovet, Bladel, The Netherlands), 0.5 mg/kg dexmedetomidine (Dexdomitor, Pfizer Animal Health BV, Capelle aan den IJssel, The Netherlands), and 0.05 mg/kg atropine-sulfate (Centrafarm Pharmaceuticals BV, Etten-Leur, The Netherlands). After preparing a tracheotomy, the animals were mechanically ventilated with a mixture of air and oxygen with a FiO2 of 0.4. Body temperature was maintained at 37 ± 0.5 °C during the entire experiment by an external thermal heating pad. Ventilator settings were adjusted to maintain end-tidal pCO2 between 30 and 35 mm Hg and arterial pCO2 between 35 mm Hg and 40 mm Hg.
For drug and fluid administration and hemodynamic monitoring, vessels were cannulated with polyethylene catheters with an outer diameter of 0.9 mm (Braun, Melsungen, Germany). A catheter in the right carotid artery was connected to a pressure transducer to monitor mean arterial blood pressure (MAP). The right jugular vein was cannulated for continuous infusion of Ringer’s Lactate (Baxter, Utrecht, The Netherlands) at a rate of 15 ml/kg/h to continuous fluid resuscitation and 50 mg/kg/h ketamine dissolved in 5 ml Ringer’s Lactate for the maintenance of anesthesia. The right femoral artery was cannulated for withdrawing blood samples and the right femoral vein for drug administration.
The left kidney was exposed, decapsulated, and immobilized in a Lucite kidney cup (K. Effenbergerite, Pfaffingen, Germany) via a 4-cm left flank incision of each animal. Renal vessels were carefully separated from surrounding tissue, nerves, and adrenal gland. A perivascular ultrasonic transient time flow probe was placed around the left renal artery (type 0.7 RB Transonic Systems Inc, Ithaca, NY) (T206, Transonic Systems Inc, Ithaca, NY) to simultaneously measure RBF. The left ureter was isolated, ligated, and cannulated with a polyethylene catheter for urine collection.
After surgical preparation, an optical fiber was placed 1 mm above the decapsulated kidney and another optical fiber was placed 1 mm on the renal vein to measure renal microvascular and venous oxygen pressures using phosphorimetry, respectively. Oxyphor G2, a two-layer glutamate dendrimer of tetra-(4-carboxy-phenyl) benzoporphyrin (Oxygen Enterprises Ltd, Philadelphia, PA) was subsequently infused (6 mg/kg IV over 5 min) followed by 30 min of stabilization time. The surgical field was covered with a humidified gauze compress throughout the entire experiment to prevent drying of the exposed tissues.
Experimental protocol
The rats were randomly divided into four groups (n = 6 per group determined by the power analysis (nQuery advisor, GraphPad Software DBA Statistical Solutions, San Diego): a sham-operated time control group (C), a group subjected to renal ischemia for 45 min by supra-aortic occlusion with a custom-made vascular occluder placed in between the left renal artery and superior mesenteric artery followed by 2 h of reperfusion (IR), a group subjected to renal ischemia for 45 min and followed by 2 h of reperfusion in which furosemide 50 µγ/kg /h was continuously administered after the release of the clamp (IR + F), and finally a sham-operated group in which furosemide was administered to the control group (C + F). At the end of the experiments, renal tissue samples were harvested and stored in both %4 formaldehyde solution for histological analysis and – 80 °C for a marker of oxidative stress, inflammation, and nitric oxide levels.
Blood and plasma variables
Arterial blood samples (0.25 ml) were drawn from the femoral artery at three-time points: before aortic occlusion (baseline, BL); 15 min after reperfusion (initial reperfusion phase, T1); and 120 min after reperfusion (late reperfusion phase T2). The blood samples were replaced by the same volume of HES 130/0.4 (Voluven, 6% HES 130/0.4; Fresenius Kabi Schelle, Belgium). The samples were used for the determination of blood gas values, as well as for the determination of the hemoglobin concentration, hemoglobin oxygen saturation, and electrolytes concentrations (ABL80 Flex Blood Gas Analyzer, Radiometer, Copenhagen, Denmark). The plasma and urine samples were used for the measurement of creatinine levels.
Renal microvascular and venous oxygenation
Microvascular oxygen tension in the renal cortex (CµPO2), outer medulla (MµPO2), and renal venous oxygen tension (PrvO2) were measured by oxygen-dependent quenching of phosphorescence lifetimes of the systematically infused albumin targeted (therefore circulation-confined) phosphorescent dye Oxyphor G2 [14]. Oxygen measurements based on phosphorescence lifetime techniques rely on the principle that phosphorescence can be quenched by energy transfer to oxygen resulting in the shortening of the phosphorescence lifetime. A linear relationship between reciprocal phosphorescence lifetime and oxygen tension (i.e., Stern–Volmer relation) allows quantitative measurement of PO2 [15].
Calculation of derivative oxygenation parameters and renal vascular resistance
Arterial oxygen content (AOC) was calculated by the following equation: (1.31 × hemoglobin × SaO2) + (0.003 × PaO2), where SaO2 is arterial oxygen saturation and PaO2 is the arterial partial pressure of oxygen. Renal venous oxygen content (RVOC) was calculated as (1.31 × hemoglobin × SrvO2) + (0.003 × PrvO2), where SrvO2 is venous oxygen saturation and PrvO2 is renal vein partial pressure of oxygen (measured using phosphorimetry [14]. The SrvO2ren was calculated using the Hill equation with P50 = 37 Torr (4.9 kPa) and Hill coefficient = 2.7. Renal oxygen delivery was calculated as DO2ren (ml/min) = RBF x AOC. Renal oxygen consumption was calculated as VO2ren (ml/min) = RBF (AOC-RVOC). An estimation of the renal vascular resistance (RVR) was made as: RVR (dynes/s/cm5) = (MAP/RBF) × 100.
Assessment of kidney function
The high plasma creatinine level was accepted as a short-term AKI definition based on the KDIGO criteria. Creatinine clearance (Clearcrea(ml/min)) was measured as an index of the glomerular filtration rate and calculated with the following formula: Clearcrea = (Uurea × V)/Pcrea, where Ucrea was the concentration of creatinine in the urine, V is the urine volume per unit time, and Pcrea was the concentration of creatinine in plasma. Additionally, excretion fraction of Na+ [EFNa (%)] was calculated and used as a marker of tubular function in the following formula: EFNa = (UNa × Pcrea)/ (PNa+ × Ucrea) × 100, where UNa was Na+ concentration in urine and PNa was the Na+ concentration in plasma. The renal oxygen extraction ratio was calculated as O2ERren (%) = VO2ren/DO2ren × 100. Clearcrea and EFNa were determined at all time points. Furthermore, the renal energy efficiency for sodium transport (VO2ren/TNa) was assessed using a ratio portrayed by the total amount of VO2ren over the total amount of sodium reabsorbed (TNa, mmol/min) that was calculated according to: (Clearcrea × PNa) – UNa × V. This parameter can be regarded as an important variable reflecting tubular cell function and oxygen utilization.
Measurement of inflammatory cytokines and glycocalyx component
Inflammatory cytokines TNF-α, IL-6, and hyaluronan (HA) (Rat TNF-a ELISA kit, DY510; Rat IL-6 ELISA kit, DY506 and Rat Hyaluronan Duoset ELISA kit, DY3614, R&D System Inc, Minneapolis, Minn) were determined by ELISA from renal frozen tissue samples. Malondialdehyde (MDA) was quantified using a Quattro Premier XE tandem mass spectrometer (MS/MS, Waters, Milford, Mass) with an Acquity sample manager and an Acquity binary solvent manager [16]. Nitric oxide levels were measured with chemiluminescence method by the Sievers NO analyzer [17]. The level of cytokines, MDA, NO and hyaluronan was expressed as per gram of protein (Bradford assay).
Histological analysis
Kidney tissues were fixed in 4% formalin and embedded in paraffin. Kidney sections (4 µm) were deparaffinized with xylene and rehydrated with decreasing percentages of ethanol and finally with water. The kidney sections were stained with periodic acid–Schiff reagent (PAS) and hematoxylin–eosin. Histologic changes in the cortex and medulla were assessed by semi-quantitative grading score of tubular damage, necrosis and inflammation. The degree of tubular dilatation in medulla and cortex were estimated at 400× magnification using 10 randomly selected fields by the three-point scoring system: 1 indicates < 25% of tubules show dilatation; 2, dilatation covering 25% to 50% of tubules; 3, covering > 50% of tubules. Tubular necrosis was classified the four-point scoring system as 1 = less that 25% tubular necrosis, 2 = 25–50% necrosis, 3 = 50–75% necrosis and 4 = more than 75% tubular necrosis. Inflammatory score was analyzed by the area of infiltrating cell as 1, none; 2, less that 25% of area; 3, 25–50% of area; 4, 50–75% of area and 5, more than 75% of area (adapted from [18]).
Statistical analysis
Data analysis and presentation were performed using GraphPad Prism 8 (GraphPad Software, San Diego, Calif). Shapiro–Wilk normality test was used for the Gaussian distribution of data. Values are reported as the mean ± SD. Two-way ANOVA for repeated measurements with a Tukey multiple comparison tests were used for comparative analysis of inter- and intra-group variations. The repeated-measures analysis of variance (one-way ANOVA with a Tukey multiple comparison test) was used for comparative analysis between the groups if baseline values differed distinctively. Statistical analysis of histological results (values are reported mean ± SE) was performed by one-way analysis of variance with Tukey multiple comparison test. p-value of < 0.05 was considered statistically significant.
Results
Systemic and renal hemodynamic variables
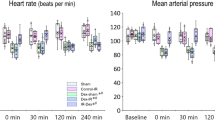
IR had a deleterious effect on all systemic and renal hemodynamic variables (Fig. 1). MAP and RBF were significantly reduced in the IR (MAP: 89.6 ± 8 mmHg at BL; 93.4 ± 16.6 mmHg at T1; 61.8 ± 6.9 mmHg at T2) (RBF: 5.5 ± 1.3 ml/min at BL; 2.1 ± 1 ml/min at T1 and 3.1 ± 0.9 ml/min at T2) and IR + F (MAP: 91.3 ± 9.6 mmHg at BL; 100 ± 29.3 mmHg at T1; 56.5 ± 8.6 mmHg at T2) (RBF: 5.4 ± 1 ml min at BL; 2.3 ± 1 ml/min at T1 and 1.8 ± 1 ml/min T2) groups at T2 in comparison to the BL and T1 (p < 0.05). Furosemide administration improved neither MAP nor RBF. RVR significantly increased in IR group at T1 (1678 ± 382 dyne/s/cm5 at BL vs 5338 ± 2860 dyne/s/cm5 at T1, p < 0.05) and IR + F group at T1 (1745 ± 452 dyne/s/cm5 at BL vs 5123 ± 2517 dyne/s/cm5 at T1, p < 0.05) and T2 (1745 ± 452 dyne/s/cm5 at BL vs 4223 ± 2636 dyne/s/cm5 at T2, p < 0.05) with respect to BL. RVR reduced in the IR group at T2 with respect to T1 (5338 ± 2860 dyne/s/cm5 at T1 vs 2198 dyne/s/cm5 at T2, p < 0.05) but in IR + F group, it remained higher at T2 than at BL (1745 ± 452 dyne/s/cm5 at BL vs 4223 ± 2636 dyne/s/cm5 at T2, p < 0.05). RVR in IR + F group at T2 was also higher than in the same time point of the C and C + F groups (4223 ± 2636 dyne/s/cm5 in IR + F vs 1645 ± 507 dyne/s/cm5 in C and 2198 ± 879 dyne/s/cm5 in C + F, p < 0.05). No significant changes were observed in the C and C + F groups.

Alteration of mean arterial pressure (MAP) (A), renal blood flow (RBF) (B) and renal vascular resistance (RVR) (C) between the experimental groups and time points. Values are represented as mean ± SD, *p < 0.05 vs. BL, +p < 0.05 vs. T1, #p < 0.05 vs. at same time point of Control (C) group, & < 0.05 vs. at same time point of Control + Furosemide (C + F) group
Renal oxygenation variables
CµPO2 and MµPO2 are shown in Fig. 2. IR caused a significant decrease in CµPO2 and MµPO2 at T2 compared to their BL values (CµPO2; 61.6 ± 6.8 vs 86 ± 6.6 mmHg and MµPO2; 51.1 ± 4.1 vs 68.7 ± 4.9 mmHg, p < 0.05). Furosemide administration did not alleviate this reduction in the IR + F group at T2 in compared to BL (45.6 ± 3 vs 60.2 ± 3.6 mmHg, p < 0.05). Additionally, furosemide without IR insult also decreased both CµPO2 and MµPO2 at T2 compared to the C group (CµPO2; 64.4 ± 4.6 vs 74.8 ± 5.6 mmHg and MµPO2; 46.1 ± 2.3 vs 53.3 ± 0.7 mmHg, p < 0.05).

Alterations of the renal cortex (A) and medulla (B) partial oxygen pressure between the experimental groups and time points. Values are represented as mean ± SD, *p < 0.05 vs. BL, +p < 0.05 vs. T1, #p < 0.05 vs. at same tome point of Control (C) group, & < 0.05 vs. at same time point of Control + Furosemide (C + F) group
DO2ren, VO2ren and ERO2ren values are shown in Fig. 3. DO2ren was significantly reduced in the IR and IR + F groups at T1 and T2 in comparison to their BL (IR; 43.5 ± 20.4 vs 122.3 ± 33.9 and IR + F; 48.4 ± 20.1 vs. 115.5 ± 20.9) (IR; 55.8 ± 17 vs 122.3 ± 33.9 and IR + F; 34.4 ± 20.3 vs 115.5 ± 20.9) (p < 0.05). It did not significantly change in the C + F group. Also, DO2ren was significantly reduced in the IR and IR + F groups in compared with the C and C + F at both T1 (IR; 43.5 ± 20.4 and IR + F; 48.4 ± 20.1 vs. C; 90.3 ± 23.2 and C + F; 102.7 ± 27.7) and T2 (IR; 55.8 ± 17.1 and IR + F; 34.4 ± 20.3 vs C: 88.7 ± 25.7 and C + F: 94.8 ± 34 (p < 0.05) (Fig. 3A). VO2ren was significantly increased in the C group at T2 compared to their BL (C; 47.5 ± 18.2 vs 17.9 ± 3.7 and C + F; 45.3 ± 19.7 vs 18.6 ± 7.5, p < 0.05). It did not significantly change in IR and IR + F groups at T2 in comparison to their BL. It was significantly lower in IR and IR + F groups at T2 in compared with the C and C + F (IR; 26 ± 12.2 and IR + F; 20.7 ± 14.1 vs C; 47.5 ± 18.2 and C + F; 45.3 ± 19.7) (p < 0.05) (Fig. 3B). There was not any statistically significant difference between the IR and IR + F at T2. ERO2ren was significantly increased in all of the groups at T2 compared to their BL (C; 53.8 ± 12.6 vs 15.2 ± 3:

Renal oxygen deliver (DO2ren) (A), oxygen consumption (VO2ren) (B) and oxygen extraction ratio (ERO2ren) (C) between the experimental groups and time points. Values are represented as mean ± SD, *p < 0.05 vs. BL, +p < 0.05 vs. T1, #p < 0.05 vs. at same tome point of Control (C) group, & < 0.05 vs. at same time point of Control + Furosemide (C + F) group
C + F; 47.4 ± 11.6 vs 16.1 ± 4.8: IR; 4.57 ± 17 vs 17.6 ± 5.1: IR + F; 56 ± 12 vs 19.9 ± 6.2) (p < 0.05). There was not any significant difference between groups at any timepoint (Fig. 3C).
Renal function parameters and urine output
Renal function parameters are shown in Figs. 4, 5. There was no significant alteration of urine output percentage between the C (% 144 [86–440]), C + F (% 180 [106–468]), IR (% 23 [− 22 to 74]) and IR + F (% 47 [15–227]) groups from BL to T2 (Fig. 4A). IR caused a significant reduction in UO at T1 compared to the C group (− 8 ± 33 vs. 498 ± 207% p < 0.05) (Fig. 4B). While UO increased in IR group from T1 to T2 in comparison to the C (45 ± 40 vs. − 53 ± 40% p < 0.05), it was significantly reduced by furosemide administration after IR from T1 to T2 (+ 45 ± 40% vs. − 9 ± 32%, p < 0.05) (Fig. 4C).

The percentage changes of urine output from BL-T2 (A), BL-T1 (B) and T1-T2 (C). Values represented as mean ± SD, #p < 0.05 vs. control (C)

Alterations of plasma creatinine (A) and creatinine clearance (B) between the experimental groups and time points. Values are represented as mean ± SD, *p < 0.05 vs. BL, +p < 0.05 vs. T1, #p < 0.05 vs. at same tome point of Control (C) group, &p < 0.05 vs. at same time point of Control + Furosemide (C + F) group, $p < 0.05 vs. at same time point of ischemia/reperfusion (IR) group
Plasma creatinine was increased significantly at T2 compared to BL. The addition of furosemide did not improve creatinine clearance and, in contrast, it further increased plasma creatinine at T2, in the IR + F in compared to the IR only (54.83 ± 5.88 vs 37.17 ± 3.13 mmol/l, p < 0.05) (Fig. 5A). Creatinine clearance was significantly lower in the IR (0.04 ± 0.01 ml/min) and IR + F (0.05 ± 0.01 ml/min) groups in compared with the C (0.13 ± 0.02 ml/min) and C + F (0.12 ± 0.03 ml/min) groups at T1 (p < 0.05). This significance also persisted at T2 (0.06 ± 0.01 ml/min in the IR and 0.05 ± 0.01 ml/min in the IR + F vs 0.13 ± 0.01 ml/min in the C 0.13 ± 0.04 ml/min in C + F) (p < 0.05) (Fig. 5B).
Sodium handling is shown in Fig. 6. IR caused a significant reduction in TNa+. Furosemide administration did not affect TNa+ after IR. TNa+ was significantly reduced in the IR and IR + F at T2 in compared with the C and C + F groups (8.4 ± 1.8 and 4.2 ± 2.5 mmol/min vs 18.9 ± 3 and 15.8 ± 3.8 mmol/min, respectively) (p < 0.05) (Fig. 6A). IR alone did not cause a significant increase in EFNa+. However, EFNa+ was significantly increased in the IR + F group at T1 (46 ± 25 mmol/min) and T2 (41 ± 31 mmol/min) in comparison to the C (9.6 ± 7.4 and 4.5 ± 2.8 mmol/min), C + F (10.3 ± 3 and 19.3 ± 11.2 mmol/min) and the IR (21 ± 19.4 and 6 ± 2.3 mmol/min) groups (p < 0.05) (Fig. 6B). VO2/TNa+ ratio increased in the IR + F group at T2 compared to BL (1.9 ± 1.3 at BL vs 8.2 ± 10 at T2, p < 0.05), it was also higher in the IR + F group than in the C and C + F groups (2.5 ± 0.9 in the C and 2.8 ± 1.3 in C + F vs 8.2 ± 10 in IR + F group, p = 0.07), but this rise did not reach statistical significance in comparison to the IR group (3.3 ± 2 in IR vs 8.2 ± 10 in IR + F, p = 0.07) (Fig. 6C).

Renal sodium reabsorption (TNa+) (A), fractional sodium extraction (EFNa+) (B), and renal energy efficiency (VO2ren/TNa+) (C) between the experimental groups and the time points. Values are represented as Mean ± SD, *p < 0.05 vs. BL, +p < 0.05 vs. T1, #p < 0.05 vs. at same tome point of Control (C) group, & < 0.05 vs. at same time point of Control + Furosemide (C + F) group, $p < 0.05 vs. at same time point of ischemia/reperfusion (IR) group
Biochemical results
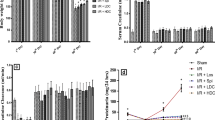
There was not any change in plasma anion gap, pH, PaO2, and the Hct between groups (Table 1). MDA, TNF-α, IL-6, and NO levels showed a non-significant alteration after IR compared with the (Fig. 7A–C, E). Hyaluronic acid levels increased in IR (p < 0.05) group compared with the C (p < 0.05). Hyaluronic acid also increased significantly in IR + F group compared with the C + F group (p < 0.05) (Fig. 7D).

Malondialdehyde (MDA) (A), tumor necrosis factor-alpha (TNF-α) (B), interleukine-6 (IL-6) (C), hyaluronic acid (D), and nitric oxide (E) levels between the experimental groups. Values are represented as mean ± SD, #p < 0.05 vs. Control (C), &p < 0.05 vs. Control + Furosemide (C + F) group
Renal histopathology
Renal cortical dilatation, necrosis and inflammatory cell migration were increased in the C + F, IR and IR + F groups in comparison to the C (p < 0.05) (Figs. 8, 9). While furosemide led to decrease the tubular dilatation and necrosis in the cortex of IR + F group (Fig. 8), increased the inflammatory cell migration with respect to the IR group (p < 0.05) (Fig. 9). In medulla, tubular necrosis, dilation and inflammatory score were evident in the IR, C + F and IR + F groups in comparison with the C group –(p < 0.05) (Figs. 10, 11).

Renal cortical tubular dilation and necrosis scores with representative figures. PAS– hematoxylin images: C; control, C+F; control + furosemide, I/R; ischemia/reperfusion, I/R+F; ischemia/reperfusion + furosemide. Values represented as mean ± SD, #p < 0.05 vs. control (C)), &p < 0.05 vs. control + furosemide (C + F), $p < 0.05 vs. ischemia/reperfusion (IR)

Renal cortical inflammatory scores and representative figures. Hematoxylin–eosin images: C; control, C+F; control + furosemide, I/R; ischemia/reperfusion, I/R+F; ischemia/reperfusion + furosemide. Values represented as mean ± SD, #p < 0.05 vs. control (C), &p < 0.05 vs. control + furosemide (C + F), $p < 0.05 vs. ischemia/reperfusion (IR)

Renal medullary tubular dilation and necrosis scores with representative figures. PAS–hematoxylin images: C; control, C+F; control + furosemide, I/R; ischermia/reperfusion, I/R+F; ischemia/reperfusion + furosemide. Values represented as mean ± SD, #p < 0.05 vs. control (C), &p < 0.05 vs. control + furosemide (C + F)

Renal medullary inflammatory scores and representative figures. Hematoxylin–eosin images: C control, C+F; control + furosemide, I/R; ischemia/reperfusion, I/R+F; ischemia/reperfusion + furosemide. Values represented as mean ± SD, #p < 0.05 vs. control (C), &p < 0.05 vs. control + furosemide (C + F)
Discussion
The previous studies evaluating the use of furosemide after kidney injury observed inconsistent results. It was hypothesized that furosemide administration following IR-induced AKI may have a beneficial role in renal tissue oxygenation, function and inflammation by means of facilitating oxygenation or reducing oxygen consumption. Our important finding is that furosemide did not improve either perfusion or oxygenation after IR-induced AKI in rats. Instead, furosemide aggravated the RVR and plasma lactate level. To complicate things further, furosemide exacerbated the oxygen utilization efficiency following IR insult. Moreover, furosemide administration augmented the renal injury and reduced the renal oxygenation even in the control condition.
Furosemide is commonly used to promote diuresis. It is also employed to prevent or limit kidney injury by uncoupling sodium absorption, thus conserving energy and oxygenation [19]. To date, however, there has been little definitive evidence that it is so [9, 20,21,22,23,24,25,26,27]. Additionally, diuretic use was independently associated with an increased risk of AKI among the hospitalized Covid-19 patients which is a disease characterized with widespread inflammation [11, 28, 29]. Besides, we demonstrated that furosemide disrupts kidney perfusion after IR insult without effecting the systemic hemodynamic parameters. Despite RBF was decreased in IR groups, much of the reduction in the perfusion was driven by an increased RVR following furosemide administration. As a result, we showed that furosemide has vasoconstrictive effects on the renal vascular bed after ischemia. Indeed, Castrop et al., demonstrated that furosemide increases renin secretion which subsequently activates renin–angiotensin–aldosterone system and causes afferent arteriole vasoconstriction [30]. Despite several studies indicating otherwise, such as improved medullary oxygenation without any increase in the medullary blood flow [31], we observed a reduction of oxygen availability in cortex and medulla after IR but only the medullary oxygenation was marginally decreased in IR groups received furosemide in compared to the Control group. An increased creatinine levels in IR group following furosemide administration injury indicates an aggravated renal dysfunction. Additionally, we also showed that furosemide has some negative effects of cortical and medullary oxygenation in the control condition without effecting the RVR. This may be explained by an increased renal oxygen shunting [6] in both IR and Control groups received furosemide. These results may also confirm that furosemide has an effect not only in the renal medullary perfusion and oxygenation, but also in the cortex.
It is assumed that nephron workload is the primary determinant of oxygen distribution in the kidney [32]. Renal medulla is the most susceptible part hypoxia due to its limited blood supply and high energy consumption in comparison to the cortex [33]. Therefore, reduced sodium delivery to distal parts of the nephron due to severely decreased glomerular filtration as a result of disrupted renal blood flow should simultaneously reduce the oxygen requirement of medullary tubules and increase its availability elsewhere [32]. Indeed, Ow et al. showed that renal tissue oxygenation is maintained by a reduction in oxygen consumption even 5 days after severe IR insult and cellular hypoxia was negligible in rats that did not receive furosemide [34]. However, perhaps the most striking finding in our study was reduced medullary oxygenation after furosemide and IR, which was shown previously in furosemide naïve rats [17, 34,35,36]. Inhibition of the electrolyte absorption in the TAL by furosemide exposes distal parts of the nephron to increased workload to compensate the electrolyte loss and ensure homeostasis [7]. This compensation is overwhelmed in the injured kidney as shown by disproportionately increased Na+ excretion compared to absorption despite reduced oxygen consumption in compared to the rats that did not receive furosemide after the IR insult. Additionally, VO2/TNa+ is increased following furosemide administration indicating impaired oxygen utilization efficiency per Na+, as also reported in the previous studies [37,38,39,40,41]. Altogether, these results indicate that furosemide may disrupt the adaptive responses against hypoxia in the injured kidney.
It is well known that IR generates reactive oxidant and nitrosative species forming peroxynitrite [42,43,44]. Peroxynitrite interaction triggers propagation of inflammation, loss of the local autoregulation of blood flow and subsequent tissue injury [33, 42,43,44]. Silbert and co-workers founded that use of furosemide caused an increase in oxidative stress depending on the severity of AKI [12]. In this study, we showed a distinct cortical and medullary injury in term of tubular dilatation, necrosis and inflammation caused by IR. In parallel to our findings, Chu et al., demonstrated that after 45 min renal arterial clamping resulted in severe tubular dilatation, necrosis, and high kidney injury molecule-1 (KIM-1) expression [18]. Moreover, they also showed that the use of empagliflozin as a sodium glucose cotransporter-2 (SGLT-2) inhibitor, which inhibits the SGLT-2 in proximal tubules to reduce Na+ and glucose re-uptake, in IR-induced AKI restored the renal tissue damage and KIM-1 expression [18]. In compared with SGLT-2 inhibitors, furosemide plays a role in TAL in Henle which has a higher oxygen requirement than the proximal tubules due to its function on great deal of salt and water reabsorption, and urine condensation. Thus, it seems that the inhibition of Na+ K+ and Cl− transport, and water reabsorption in TAL by furosemide results no beneficial effects on medullar damage as shown in this study. In fact, Rokuten et al., showed that use of the furosemide after myocardial infarction on rats were strongly associated with renal damage and mortality in compared to the placebo and the experimental group received angiotensin converting enzyme (ACE) inhibitor [45]. In present study, despite furosemide increased the inflammatory cell migration in renal cortex, it reduced the renal tubular dilatation and necrosis in comparison to the IR alone. However, furosemide showed no beneficial effects on the renal medullar dilation and necrosis after IR insult. This discrepancy in the effects of furosemide on cortex and medulla can be explained by the structural and functional differences of renal medulla which includes abundant of active furosemide receptor, limited blood supply, higher workload and oxygen consumption with respect to cortex. Importantly, furosemide also caused a distinct cortical and medullary damage in term of tubular dilation, cell necrosis and inflammatory cell migration in the control conditions. Therefore, this result indicates that furosemide can also injure the kidney without any pre-existing damage. These results may indicate that overwhelming effects of furosemide on medulla may be also overlapped by IR injury on its own because of the severity of AKI model we used in this study. However, surrogates of tissue inflammation and oxidative stress markers were not altered in our study. Plasma hyaluronic acid, a glycocalyx degradation biomarker, was significantly increased after IR and did not decrease after furosemide administration. Together with the renal oxygenation, plasma creatinine level, renal vascular resistance and sodium handling results, aggravation of hypoxia-induced tissue damage is the main factor causing extra renal injury following furosemide administration.
Conclusion
In conclusion, the present study was designed to determine the effect of furosemide on the kidneys after IR insult. Contrary to the expectations, our findings indicate that furosemide does not improve either the cortical and medullary microvascular oxygenation or renal function. Furthermore, it may even aggravate AKI by disrupting Na+ reabsorption, oxygen utilization efficiency, tissue perfusion and increasing hypoxic injury in the kidney. Taken together, our findings contribute in several ways to our understanding of AKI and provides a basis for future clinical studies which aims to prevent or limit the extent of renal injury following a hypoxic insult.
Despite its exploratory nature, our study offers insight into the relationship between furosemide and AKI. It is possible that these results may not fully apply to AKI in humans encountered after scenarios mentioned in the introduction. Therefore, there would be a certain need for clinical trials to better understand the possible link between hypoxia and oxygen utilization efficiency.
Availability of data and materials
Not applicable.
Abbreviations
- AKI:
-
Acute kidney injury
- F:
-
Furosemide
- IR:
-
Ischemia/reperfusion
- MAP:
-
Mean arterial pressure
- RBF:
-
Renal blood flow
- RVR:
-
Vascular resistance
- DO2ren :
-
Renal oxygen delivery
- VO2ren :
-
Renal oxygen consumption
- TNa+ :
-
Sodium reabsorption
- VO2/TNa+ :
-
Oxygen utilization efficiency
- EFNa :
-
Excretion fraction of Na+
- O2ERren (%):
-
Renal oxygen extraction ratio
- CμO2 :
-
Cortical microvascular oxygen pressures,
- MμO2 :
-
Medullary microvascular oxygen pressures
- UO:
-
Urine output
- Ccr:
-
Creatinine clearance
- TAL:
-
Tick ascending limp
- PO2 :
-
Partial oxygen pressure
- FiO2 :
-
Inspired oxygen pressure
- PaO2 :
-
Arterial oxygen pressure
- PrvO2 :
-
Renal venous oxygen pressure
- SaO2 :
-
Arterial oxygen saturation
- SrvO2 :
-
Renal venous oxygen saturation
- AOC:
-
Arterial oxygen content
- RVOC:
-
Renal venous oxygen content
- mmHg:
-
Millimeter mercury
- kPa:
-
Kilo pascal
- TNF-α:
-
Tumor necrosis factor- alpha
- IL-6:
-
Interleukin-6
- HA:
-
Hyaluronic acid
- MDA:
-
Malondialdehyde
- NO:
-
Nitric oxide
References
Dovzhanskiy DI, Schwab S, Bischoff MS et al (2021) Extended intensive care correlates with worsening of surgical outcome after elective abdominal aortic reconstruction. J Cardiovasc Surg (Torino) 62:591–599. https://doi.org/10.23736/S0021-9509.21.11842-7
Liu X, Zhang J, Yang Y et al (2021) Analysis of risk factors of acute kidney injury in perioperative patients after lung transplantation. Ann Palliat Med 10:9841–9847. https://doi.org/10.21037/apm-21-2094
Prowle JR, Forni LG, Bell M et al (2021) Postoperative acute kidney injury in adult non-cardiac surgery: joint consensus report of the Acute Disease Quality Initiative and PeriOperative Quality Initiative. Nat Rev Nephrol 17:605–618. https://doi.org/10.1038/s41581-021-00418-2
Gumbert SD, Kork F, Jackson ML et al (2020) Perioperative Acute Kidney Injury. Anesthesiology 132:180–204. https://doi.org/10.1097/ALN.0000000000002968
Guerci P, Ergin B, Ince C (2017) The macro- and microcirculation of the kidney. Best Pract Res Clin Anaesthesiol 31:315–329. https://doi.org/10.1016/j.bpa.2017.10.002
Legrand M, Almac E, Mik EG et al (2009) L-NIL prevents renal microvascular hypoxia and increase of renal oxygen consumption after ischemia-reperfusion in rats. Am J Physiol-Ren Physiol 296:F1109–F1117
Bouchard J, Soroko SB, Chertow GM et al (2009) Fluid accumulation, survival and recovery of kidney function in critically ill patients with acute kidney injury. Kidney Int 76:422–427. https://doi.org/10.1038/ki.2009.159
Gillespie RS, Seidel K, Symons JM (2004) Effect of fluid overload and dose of replacement fluid on survival in hemofiltration. Pediatr Nephrol Berl Ger 19:1394–1399. https://doi.org/10.1007/s00467-004-1655-1
Bagshaw SM, Gibney RN, Kruger P et al (2017) The effect of low-dose furosemide in critically ill patients with early acute kidney injury: a pilot randomized blinded controlled trial (the SPARK study). J Crit Care 42:138–146
Bellomo R, Haase M, Ghali WA et al (2007) Loop diuretics in the management of acute renal failure: a systematic review and meta-analysis. Crit Care Resusc 9:60
Jewell PD, Bramham K, Galloway J et al (2021) COVID-19-related acute kidney injury; incidence, risk factors and outcomes in a large UK cohort. BMC Nephrol 22:359. https://doi.org/10.1186/s12882-021-02557-x
Silbert BI, Ho KM, Lipman J et al (2017) Does furosemide increase oxidative stress in acute kidney injury? Antioxid Redox Signal 26:221–226
O’Connor PM (2006) Renal oxygen delivery: matching delivery to metabolic demand. Clin Exp Pharmacol Physiol 33:961–967. https://doi.org/10.1111/j.1440-1681.2006.04475.x
Johannes T, Mik EG, Ince C (2006) Dual-wavelength phosphorimetry for determination of cortical and subcortical microvascular oxygenation in rat kidney. J Appl Physiol 100:1301–1310
Vanderkooi JM, Maniara G, Green TJ, Wilson DF (1987) An optical method for measurement of dioxygen concentration based upon quenching of phosphorescence. J Biol Chem 262:5476–5482
Ergin B, Bezemer R, Kandil A et al (2015) TEMPOL has limited protective effects on renal oxygenation and hemodynamics but reduces kidney damage and inflammation in a rat model of renal ischemia/reperfusion by aortic clamping. J Clin Transl Res 1:1
Ergin B, Zuurbier CJ, Kapucu A, Ince C (2018) Divergent effects of hypertonic fluid resuscitation on renal pathophysiological and structural parameters in rat model of lower body ischemia/reperfusion-induced sterile inflammation. Shock Augusta Ga 50:655–663. https://doi.org/10.1097/SHK.0000000000001096
Chu C, Delić D, Alber J et al (2022) Head-to-head comparison of two SGLT-2 inhibitors on AKI outcomes in a rat ischemia-reperfusion model. Biomed Pharmacother 153:113357. https://doi.org/10.1016/j.biopha.2022.113357
Haddock B, Larsson HB, Francis S, Andersen UB (2019) Human renal response to furosemide: Simultaneous oxygenation and perfusion measurements in cortex and medulla. Acta Physiol 227:e13292
Mahesh B, Yim B, Robson D et al (2008) Does furosemide prevent renal dysfunction in high-risk cardiac surgical patients? Results of a double-blinded prospective randomised trial. Eur J Cardiothorac Surg 33:370–376. https://doi.org/10.1016/j.ejcts.2007.12.030
Welch HK, Kellum JA, Kane-Gill SL (2018) Drug-associated acute kidney injury identified in the united states food and drug administration adverse event reporting system database. Pharmacother J Hum Pharmacol Drug Ther 38:785–793
Ng KT, Velayit A, Khoo DKY et al (2018) Continuous infusion versus intermittent bolus injection of furosemide in critically Ill patients: a systematic review and meta-analysis. J Cardiothorac Vasc Anesth 32:2303–2310
Bove T, Belletti A, Putzu A et al (2018) Intermittent furosemide administration in patients with or at risk for acute kidney injury: meta-analysis of randomized trials. PLoS ONE 13:e0196088
Shen Y, Wu M (2018) Loop diuretic use in patients with AKI: different severity, different response. Crit Care 22:202
Krzych LJ, Czempik PF (2019) Impact of furosemide on mortality and the requirement for renal replacement therapy in acute kidney injury: a systematic review and meta-analysis of randomised trials. Ann Intensive Care 9:85
Lee SJ, Kim CW, Lee MK et al (2017) Effect of high-dose furosemide on the prognosis of critically ill patients. J Crit Care 41:36–41
Bagshaw SM, Bellomo R, Kellum JA (2008) Oliguria, volume overload, and loop diuretics. Crit Care Med 36:S172–S178
Casas-Aparicio GA, León-Rodríguez I, Barrera CA et al (2021) Acute kidney injury in patients with severe COVID-19 in Mexico. PLoS ONE 16:e0246595. https://doi.org/10.1371/journal.pone.0246595
Dai Y, Liu Z, Du X et al (2021) Acute Kidney Injury in Hospitalized Patients Infected with COVID-19 from Wuhan, China: A Retrospective Study. BioMed Res Int 2021:6655185. https://doi.org/10.1155/2021/6655185
Castrop H, Lorenz JN, Hansen PB et al (2005) Contribution of the basolateral isoform of the Na-K-2Cl- cotransporter (NKCC1/BSC2) to renin secretion. Am J Physiol-Ren Physiol 289:F1185–F1192
Brezis M, Agmon Y, Epstein FH (1994) Determinants of intrarenal oxygenation. I. Effects of diuretics. Am J Physiol 267:F1059-1062. https://doi.org/10.1152/ajprenal.1994.267.6.F1059
Heyman SN, Khamaisi M, Zorbavel D et al (2019) Role of hypoxia in renal failure caused by nephrotoxins and hypertonic solutions. Semin Nephrol 39:530–542. https://doi.org/10.1016/j.semnephrol.2019.10.003
Ow CPC, Ngo JP, Ullah MM et al (2018) Renal hypoxia in kidney disease: Cause or consequence? Acta Physiol 222:e12999. https://doi.org/10.1111/apha.12999
Ow CPC, Ngo JP, Ullah MM et al (2018) Absence of renal hypoxia in the subacute phase of severe renal ischemia-reperfusion injury. Am J Physiol Ren Physiol 315:F1358–F1369. https://doi.org/10.1152/ajprenal.00249.2018
Ergin B, Heger M, Kandil A et al (2017) Mycophenolate mofetil improves renal haemodynamics, microvascular oxygenation, and inflammation in a rat model of supra-renal aortic clamping-mediated renal ischaemia reperfusion injury. Clin Exp Pharmacol Physiol 44:294–304. https://doi.org/10.1111/1440-1681.12687
Abdelkader A, Ho J, Ow CPC et al (2014) Renal oxygenation in acute renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 306:F1026-1038. https://doi.org/10.1152/ajprenal.00281.2013
Johannes T, Mik EG, Nohé B et al (2006) Influence of fluid resuscitation on renal microvascular PO2 in a normotensive rat model of endotoxemia. Crit Care 10:R88. https://doi.org/10.1186/cc4948
Heemskerk AE, Huisman E, van Lambalgen AA et al (1997) Renal function and oxygen consumption during bacteraemia and endotoxaemia in rats. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc Eur Ren Assoc 12:1586–1594. https://doi.org/10.1093/ndt/12.8.1586
Post EH, Kellum JA, Bellomo R, Vincent J-L (2017) Renal perfusion in sepsis: from macro- to microcirculation. Kidney Int 91:45–60. https://doi.org/10.1016/j.kint.2016.07.032
Evans RG, Harrop GK, Ngo JP et al (2014) Basal renal O2 consumption and the efficiency of O2 utilization for Na+ reabsorption. Am J Physiol Renal Physiol 306:F551-560. https://doi.org/10.1152/ajprenal.00473.2013
Redfors B, Bragadottir G, Sellgren J et al (2010) Acute renal failure is NOT an “acute renal success”—a clinical study on the renal oxygen supply/demand relationship in acute kidney injury. Crit Care Med 38:1695–1701. https://doi.org/10.1097/CCM.0b013e3181e61911
Szabó C, Módis K (2010) Pathophysiological roles of peroxynitrite in circulatory shock. Shock Augusta Ga 34:4
Walker LM, Walker PD, Imam SZ et al (2000) Evidence for Peroxynitrite Formation in Renal Ischemia-Reperfusion Injury: Studies with the Inducible Nitric Oxide Synthase Inhibitorl-N 6-(1-Iminoethyl) lysine. J Pharmacol Exp Ther 295:417–422
Paller MS, Weber K, Patten M (1998) Nitric oxide—mediated renal epithelial cell injury during hypoxia and reoxygenation. Ren Fail 20:459–469
Rokutan H, Suckow C, von Haehling S et al (2012) Furosemide induces mortality in a rat model of chronic heart failure. Int J Cardiol 160:20–25. https://doi.org/10.1016/j.ijcard.2011.03.005
Acknowledgements
Not applicable.
Funding
This study was carried out as a part of a project supported by the Dutch Kidney Foundation (Grant C 09.2290).
Author information
Authors and Affiliations
Contributions
OD, data analysis, writing he final version and review of manuscript. CI: writing he final version and review of manuscript. PH, design and manufacturing the phosphorimeters. AK, conducting the histological assessment. BE, study design, carried out experiments, data analysis, writing the first draft and review manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study is approved by the Institutional Animal Experimentation Committee of the Academic Medical Centre of the University of Amsterdam (DFL83). Care and handling of the animals were following the guidelines for Institutional and Animal Care and Use Committees. The study has been carried out according to the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors do not have any conflicts of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dilken, O., Ince, C., Kapucu, A. et al. Furosemide exacerbated the impairment of renal function, oxygenation and medullary damage in a rat model of renal ischemia/reperfusion induced AKI. ICMx 11, 25 (2023). https://doi.org/10.1186/s40635-023-00509-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40635-023-00509-3