
Abstract
Background
Neurological injury is the primary cause of death after out-of-hospital cardiac arrest. There is a lack of studies investigating cerebral injury beyond the immediate post-resuscitation phase in a controlled cardiac arrest experimental setting.
Methods
The aim of this study was to investigate temporal changes in measures of cerebral injury and metabolism in a cardiac arrest pig model with clinically relevant post-cardiac arrest intensive care. A cardiac arrest group (n = 11) underwent 7 min of no-flow and was compared with a sham group (n = 6). Pigs underwent intensive care with 24 h of hypothermia at 33 °C. Blood markers of cerebral injury, cerebral microdialysis, and intracranial pressure (ICP) were measured. After 48 h, pigs underwent a cerebral MRI scan. Data are presented as median [25th; 75th percentiles].
Results
Return of spontaneous circulation was achieved in 7/11 pigs. Time to ROSC was 4.4 min [4.2; 10.9]. Both NSE and NfL increased over time (p < 0.001), and were higher in the cardiac arrest group at 48 h (NSE 4.2 µg/L [2.4; 6.1] vs 0.9 [0.7; 0.9], p < 0.001; NfL 63 ng/L [35; 232] vs 29 [21; 34], p = 0.02). There was no difference in ICP at 48 h (17 mmHg [14; 24] vs 18 [13; 20], p = 0.44). The cerebral lactate/pyruvate ratio had secondary surges in 3/7 cardiac arrest pigs after successful resuscitation. Apparent diffusion coefficient was lower in the cardiac arrest group in white matter cortex (689 × 10–6 mm2/s [524; 765] vs 800 [799; 815], p = 0.04) and hippocampus (854 [834; 910] vs 1049 [964; 1180], p = 0.03). N-Acetylaspartate was lower on MR spectroscopy in the cardiac arrest group (− 17.2 log [− 17.4; − 17.0] vs − 16.9 [− 16.9; − 16.9], p = 0.03).
Conclusions
We have developed a clinically relevant cardiac arrest pig model that displays cerebral injury as marked by NSE and NfL elevations, signs of cerebral oedema, and reduced neuron viability. Overall, the burden of elevated ICP was low in the cardiac arrest group. A subset of pigs undergoing cardiac arrest had persisting metabolic disturbances after successful resuscitation.
Similar content being viewed by others


Introduction
The number of published studies including a clinically relevant cardiac arrest animal model is limited [1]. To address this, we have developed a cardiac arrest pig model with myocardial infarction and 48 h of post-resuscitation care [2]. In that model, we found significantly depressed cardiovascular function as well as signs of renal, hepatic, and pulmonary injury during the post-cardiac arrest phase.
Neurological injury is the primary cause of death after out-of-hospital cardiac arrest [3]. A multimodal approach to neuroprognostication, and non-invasive cerebral injury assessment, is preferred in the clinical setting [4]. This often includes magnetic resonance imaging (MRI) and biomarkers of neurological injury [5]. Few studies on post-cardiac arrest patients have used intracranial pressure (ICP), brain oxygenation (PTiO2), and microdialysis to assess cerebral injury [6,7,8,9]. However, invasive cerebral measurements are not common in clinical practice due to risk of bleeding and infection. Several cardiac arrest pig studies have utilized ICP, PTiO2, and microdialysis monitoring [10,11,12,13,14,15]. Common to most of these studies are measurements only being collected during the intra-arrest and early return of spontaneous circulation (ROSC) periods, with only two investigations having an observation period beyond 6 h [16, 17]. A multimodal description of long-term cerebral injury development in a controlled experimental setting is therefore warranted.
The aim of this study was to investigate temporal changes in measures of cerebral injury and metabolism in a cardiac arrest pig model with clinically relevant post-cardiac arrest intensive care.
Methods and materials
The study was approved by the Danish Animal Experiments Inspectorate (License number: 2019-15-0201-01647) and conducted and reported in accordance with the ARRIVE [18] and Utstein-Style [19] guidelines. This manuscript describes cerebral outcome data from a previously published experiment. Detailed methodological descriptions are described therein [2]. Animals were assigned to the groups at the discretion of the primary investigator in a non-randomized order. The investigators were not blinded to group allocation.
Animals
Female crossbred Landrace/Yorkshire/Duroc pigs (40 kg) were fasted overnight with free access to water.
Animal preparation
Esketamine (6.25 mg/kg), midazolam (0.625 mg/kg), and atropine (0.5 mg) were used to induce anaesthesia, which was maintained with propofol (4.0–5.5 mg/kg/h) and remifentanil (0.6–1.0 µg/kg/min). Pigs were intubated and ventilated at a tidal volume of 8 ml/kg with pressure-controlled volume guarantee settings (Datex Ohmeda S5, GE Healthcare, IL, USA). The ventilator at baseline was adjusted as follows: respiratory rate to control arterial partial CO2 pressure (PaCO2) between 4.7 and 6.0 kPa, fraction of inspired O2 (FiO2) to maintain arterial partial O2 pressure (PaO2) between 20–25 kPa, and positive end-expiratory pressure (PEEP) at 5 cmH2O. Temperature was regulated at baseline to normothermia (38.5 ± 0.5 °C).
Invasive cerebral monitoring
The pig was placed in prone position and two burr holes were made in the skull 1.5 cm lateral to the sagittal suture and 1.5 cm caudal to the coronal suture. Both burr holes were fitted with a bolt (Bolt Kit, Raumedic AG, Helmbrechts, Germany). The pig was turned back to supine position and after puncture of the dura mater a catheter for measurement of intracranial pressure (ICP), brain tissue oxygenation (PTiO2), and temperature (NEUROVENT-PTO, Raumedic AG, Helmbrechts, Germany) was inserted in the right parietal lobe parenchyma and connected to a monitor (MPR2 logO Datalogger, Raumedic AG, Helmbrechts, Germany). Cerebral perfusion (CerPP) was calculated as MAP-ICP. ICP and PTiO2 data are reported as timepoint values. In the bolt over the left parietal lobe, a customized microdialysis catheter (CMA 20 Elite 10 mm, 20 kD cut-off, CMA CFM, Marseille, France) was inserted into the brain parenchyma. The microdialysis catheter was perfused with an artificial CNS fluid (Perfusion fluid CNS, CMA CFM) at a flow rate of 0.5 µL/min (CMA 402 Microdialysis Syringe Pump, CMA CFM). The dialysate samples were collected in microvials and stored at − 80 °C for later analysis (CMA 600 Microdialysis Analyzer, CMA CFM). The pre-cardiac arrest baseline sample was collected after 60 min of tissue recovery time for a sampling period of 60 min. After ROSC samples were collected every two hours. No dialysate samples were collected during cardiac arrest (sampling vials were exchanged 5 min after ROSC to collect the first post-resuscitation sample). The following metabolic markers were analysed: lactate, pyruvate, glucose, and glycerol (Reagent Set A, µdialysis, Stockholm, Sweden). Recovery rates of analytes in the dialysate were not calculated.
Experimental protocol
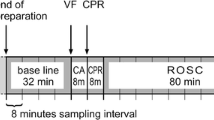
Induction of myocardial infarction, cardiac arrest, resuscitative efforts, and the post-cardiac arrest intensive care protocol for this experiment have been described in detail previously [2]. The earlier report included two different methods of myocardial infarction. One group had a 45 min continuous coronary occlusion starting 10 min prior to cardiac arrest induction and lasting throughout cardiac arrest, resuscitation, and the early post-ROSC phase. This resulted in severe cardiovascular instability during the post-cardiac arrest period (AMI-Cont group). The other method included an intermittent coronary reperfusion during cardiac arrest and resuscitation (see details below), which resulted in moderate cardiovascular instability during the post-cardiac arrest period (AMI-Int). This manuscript only reports neurological data from the AMI-Int group, as the AMI-Cont group only consisted of two animals surviving the entire protocol. Data from the AMI-Cont group are, however, presented in the supplemental material. In the main manuscript, the AMI-Int group is referred to as the cardiac arrest group.
In short, myocardial infarction was induced by endovascular balloon occlusion of the left anterior descending artery after the second diagonal branch. After 10 min of occlusion, the coronary artery was reperfused for 2 min, and then ventricular fibrillation (VF) was induced by a 9-V DC current delivered to the right ventricle. After a no-flow period of 7 min, mechanical chest compressions (LUCAS2) and ventilations were initiated. Five minutes after ROSC, the balloon catheter was re-inflated for another 35 min of coronary occlusion. Furthermore, goal-directed intensive care was initiated with special emphasis on cardiovascular stability after set treatment goals (for details see Additional file 1: Methods and Table S1). One hour after ROSC, targeted temperature management (TTM) was initiated to cool the pigs to 33 °C and maintain this temperature for 24 h before rewarming to normothermia (38.5 °C ± 0.5) by 0.5 °C per hour. For comparison, a group of sham animals was included and underwent the same procedures except for myocardial infarction, cardiac arrest, and subsequent resuscitative efforts.
Magnetic resonance imaging
After 48 h of intensive care the pigs were transported to an MRI scanner (Achieva DStream 1.5 T, Phillips, Best, The Netherlands). For details see Supplemental Methods. The perfusion scans were analysed by a semi-quantitative method, creating parametric maps of the upslope of the signal intensity–time curve (MIStar, Apollo, Melbourne, Australia). For the calculated apparent diffusion coefficient (ADC), blood oxygenation level dependent (BOLD) and relative perfusion maps, regions of interest (ROIs) included frontal cortex white matter and grey matter, thalamus, hippocampus, and cerebellum. Each cortical ROI were ~ 0.05 cm2 while non-cortical ROIs were ~ 0.1 cm2. An online histological/MRI atlas of the Göttingen minipig brain was used to guide ROI placement [20]. For the analysis of MRI scans Horos software version 3.3.5 (Annapolis, MD, USA) was used. All ROI placements were checked by an experienced neurosurgeon blinded to group allocation before analysis. All ROIs were drawn by free hand to best fit the anatomical area of interest (see Additional file 1: Fig. S1). For proton-spectroscopy a single whole brain voxel was applied.
Blood sampling and analysis
Serum samples were collected at baseline and throughout the post-resuscitation phase to measure neuron-specific enolase (NSE) and neurofilament light chain (NfL). After clotting for a minimum of 20 min, serum samples were centrifuged (1850 G, 4 °C, 9 min) and supernatants stored at − 80 °C for later analysis. NSE was analysed by an electrochemiluminescent immunoassay using Cobas 8000 e602 (Roche Diagnostics GmbH, Mannheim, Germany; analytical variation 11.3 µg/L: ± 10%). NfL was measured by the NF-light® assay using the ultrasensitive SimoaTM HD-1 platform (Quanterix©, Lexington, MA, USA; analytical variation 12.3 ng/L: ± 20%).
Statistics
Data in figures are presented as median with corresponding 25% and 75% quartiles. ICP, MAP, PaO2, and NSE from the same experiments have previously been reported [2], but to sufficiently interpret the dataset of this manuscript these variables have been included. Statistical significance was defined as a two-sided p-value < 0.05. Study data were collected using REDCap electronic data capture tools hosted at Aarhus University [21]. Data analysis and figures were produced in Stata 17.0 (StataCorp, TX, USA). No sample size calculation was performed due to the descriptive nature of the study.
Invasive cerebral, cardiopulmonary, and metabolic data
Data are presented and interpreted using descriptive statistics and a between-group test at baseline and 48 h after resuscitation. If data were approximately normally distributed and had equal variance, an unpaired t-test was applied. If data were right-skewed, it was log transformed. Normality was evaluated by qq-plots and equal variance tested by Bartlett’s test. If assumptions for parametric tests were not met a Mann–Whitney U test was applied. A Spearman correlation analysis was conducted between PTiO2 and PaO2. Microdialysis data were only analysed descriptively because of the numerous repeated measurements and no statistical tests were applied.
Blood samples
NSE and NfL were analysed with a linear mixed effects model with time (as a categorical value), group and their interaction as fixed effects and individual animals as random effects. Residual errors were structured from an exponential autocorrelation function to allow for unequal spaced categorical time. For NSE, variance differed between the two groups, and this was incorporated into the model. The model was used for checking differences in development over time in the two groups and between-group differences at baseline and 48 h post-resuscitation. Both NSE and NfL were analysed on log-scale, but group differences are presented on normal scale as ratios of geometric means with 95% confidence intervals. The models were checked by qq-plots of residuals and scatter plots of residuals vs. fitted values. Finally, a Spearman correlation analysis was carried out between NSE and NfL measurements from the cardiac arrest group.
MR data
Statistical differences between groups in ADC, BOLD sequence, and perfusion maps were tested by unpaired t-test or Mann–Whitney U test for each anatomical location. Proton spectroscopy data were analysed with LCModel (Stephen Provencher, Oakville, ON, Canada) version 6.3–1 M. Concentrations of lactate, N-acetylaspartate (NAA), choline, and glutamate/glutamine are presented in arbitrary units on log10 scale.
Results
As previously reported, 17 pigs were included in this study and divided in the two groups (11 cardiac arrest and 6 sham pigs) [2]. Seven pigs in the cardiac arrest group achieved ROSC (baseline data from all pigs are included in analyses). Time to ROSC was 4.4 min [4.2; 10.9]. One pig from the cardiac arrest group died from a tension pneumothorax at 37 h post-resuscitation, another pig from the cardiac arrest group died from fatal arrhythmia in the MRI scanner (only missing perfusion-weighted scan), and one pig from the sham group had an accidental bleeding from arterial catheter after 5 h. Data from all three animals are included up until time of death. In the AMI Cont cardiac arrest group, seven pigs were included. Three achieved ROSC, but one died just before MRI scan due to cardiovascular failure. Data from this group are presented in supplemental material (Additional file 1: Figs. S2–S6; Table S2).
Intracranial pressure
ICP, CerPP, and MAP are presented in Fig. 1. No statistically significant differences were seen between groups at baseline for ICP (cardiac arrest: 15 mmHg [12; 17] vs sham: 17 [15; 18], p = 0.12) or CerPP (cardiac arrest: 69 mmHg [59; 80] vs sham: 60 [59; 62], p = 0.06). In one animal in the cardiac arrest group ICP increased up to 46 mmHg at 48 h after resuscitation, but on visual inspection, ICP otherwise developed similarly between groups and no difference was found between groups at the end of the protocol (cardiac arrest: 17 mmHg [14; 24] vs sham: 18 [13; 20], p = 0.44). Accordingly, the CerPP followed the development of the arterial pressure, with elevated levels during the maintenance phase of TTM, and no statistically significant difference was found between groups for CerPP 48 h after resuscitation (cardiac arrest: 60 mmHg [49; 62] vs sham: 73 [54; 83], p = 0.25).

Intracranial, mean arterial, and cerebral perfusion pressure in the cardiac arrest and sham groups. Data are presented as median [25%; 75%] with overlaid scatter representing each animal. Lighter blue shading indicates induction/rewarming phase, respectively, while darker blue shading indicates maintenance phase of targeted temperature management. BL baseline before induction of myocardial infarction and cardiac arrest
Cerebral oxygenation
PTiO2 and PaO2 are presented in Fig. 2. There was no statistically significant difference between groups at baseline for PTiO2 (cardiac arrest: 7 mmHg [3; 17] vs sham: 13 [3; 25], p = 0.50). By inspection of scatter plot, large between-animal variations existed in PTiO2 in both groups during the first 12 h of recording, but PTiO2 overall developed similarly between groups. The amplitudes of all PTiO2 measurements were above 400. No statistically significant difference was seen at 48 h (cardiac arrest: 9 mmHg [7; 14] vs sham: 15 [12; 16], p = 0.28). The Spearman correlation coefficient between PaO2 and PTiO2 was − 0.02 (95%CI − 0.16 to 0.13, p = 0.84, see Additional file 1: Fig. S6A).

Brain oxygenation and partial pressure of O2 in arterial blood in the cardiac arrest and sham groups. Data are presented as median [25%;75%] with overlaid scatter representing each animal. Lighter blue shading indicates induction/rewarming phase, respectively, while darker blue shading indicates maintenance phase of targeted temperature management. *p < 0.01 between groups at 48 h. BL baseline before induction of myocardial infarction and cardiac arrest
Microdialysis
Cerebral microdialysis data are presented in Fig. 3. Median glucose levels rose from baseline in both groups during induction of TTM, however, to a higher level in the sham group compared with the cardiac arrest group. Glucose levels remained lower in the cardiac arrest group throughout the maintenance phase of TTM. Glucose levels in the sham group slowly declined during induction and maintenance phase and from the rewarming phase until end-of-protocol glucose levels were similar between groups. Median lactate levels were slightly higher in the cardiac arrest group during most of the protocol after cardiac arrest. Of interest, there was an increase in median lactate levels during the rewarming phase of TTM in the cardiac arrest group, which was not observed in the sham group. Pyruvate levels were overall similar between the two groups up until the rewarming phase. An increase in pyruvate was observed in both groups at the beginning of rewarming. The lactate/pyruvate (L/P) ratio was higher in the cardiac arrest group throughout the first 10 h after resuscitation and during the last part of the rewarming phase. Glycerol levels were higher at baseline in the sham group compared with the cardiac arrest group. Despite this, glycerol levels increased more in the cardiac arrest group two hours after resuscitation. Both groups had similarly declining glycerol levels throughout the remainder of the protocol. Individual scatter plots for lactate, pyruvate and their ratio are presented for the cardiac arrest group in Additional file 1: Fig. S7.

Cerebral microdialysis analytes in the cardiac arrest and sham groups. Data are presented as median [25%; 75%]. Lighter blue shading indicates induction or rewarming phase, while darker blue shading indicates maintenance phase of targeted temperature management
Blood markers of cerebral injury
NSE and NfL levels are depicted in Fig. 4. The repeated measurements analysis showed that the NSE increased significantly over time (p < 0.0001), and at 48 h after resuscitation the relative between-group difference was 5.19 (95%CI 3.08–8.74, p < 0.001) with values of 4.2 µg/L [2.4; 6.1] vs 0.9 [0.7; 0.9]. NfL likewise increased over time in the cardiac arrest group (p = 0.0001) reaching a larger relative values of 2.68 (95%CI 1.22–5.90, p = 0.02) at 48 h after resuscitation with values of 63 ng/L [35; 232] vs 29 [21; 34]. The Spearman correlation analysis of NSE and NfL in the cardiac arrest group revealed a coefficient of 0.74 (95%CI 0.62–0.83, p < 0.0001, see Additional file 1: Fig. S6B).

Level of neuron-specific enolase and neurofilament light chain over time in the cardiac arrest and sham groups. Data are presented as median [25%; 75%] with overlaid scatter plots representing each animal. Lighter blue shading indicates induction or rewarming phase, while darker blue shading indicates maintenance phase of targeted temperature management
MRI and MR spectroscopy
ADC, BOLD imaging, and upslope of the signal intensity–time curve results are presented in Fig. 5. In the cardiac arrest group, significantly lower ADC values were seen in white matter frontal cortex (689 × 10–6 mm2/s [524; 765] vs 800 [799; 815], p = 0.04) and hippocampus (854 × 10–6 mm2/s [834; 910] vs 1049 [965; 1180] p = 0.03), but no statistical significant difference was observed in grey matter (801 × 10–6 mm2/s [717; 826] vs 791 [781; 801], p = 0.93), thalamus (801 × 10–6 mm2/s [796; 805] vs 834 [824; 863], p = 0.18), or cerebellum (715 × 10–6 mm2/s [579; 802] vs 828 [798; 841], p = 0.05). The BOLD sequence showed that relaxation time, as a measure of deoxygenated blood levels, in the cardiac arrest group was statistical significantly lower in grey matter cortex (66 ms [59; 85] vs 91 [84; 92], p = 0.03). Lower values without statistical significance was seen in the cardiac arrest group in white matter cortex (73 ms [59; 79] vs 81 [80; 85], p = 0.52), thalamus (78 ms [71; 84] vs 80 [79;83], p = 0.54), hippocampus (77 ms [66; 90] vs 93 [88; 97], p = 0.18), and cerebellum (75 ms [68; 81] vs 80 [78; 87], p = 0.33). For the upslope of the signal intensity–time curve as a measure of perfusion, no differences were seen at any of the anatomical locations. MR spectroscopy metabolite levels are presented in Fig. 6. NAA was lower in the cardiac arrest group (− 17.2 log [− 17.4; − 17.0] vs − 16.9 [− 16.9; − 16.9], p = 0.03). No between-group difference (cardiac arrest vs sham) was observed for lactate (− 19.0 log [− 19.3; − 17.8] vs − 18.7 [− 19.0; − 18.7], p = 0.79), choline (− 18.7 log [− 18.9; − 18.5] vs − 18.5 [− 18.6; − 18.5], p = 0.14), or glutamate/glutamine (− 17.1 log [− 17.4; − 16.5] vs − 16.9 [− 17.2; − 16.8], p = 0.85) peaks in the spectroscopy data.

Apparent diffusion coefficient values, blood oxygenation level dependent (BOLD) imaging, and upslope of signal intensity curve of perfusion-weighted imaging in five different anatomical locations in the brain. Data are presented as median [25–75%] with overlaid scatter plots representing each animal. Ms milliseconds, a.u. arbitrary unit

MR spectroscopy with concentrations (in arbitrary units) for four metabolites from a whole brain voxel. Y-axis is presented on log 10 scale. Data are presented as median [25–75%] with overlaid scatter plots representing each animal. NAA N-acetylaspartate, a.u. arbitrary units
Discussion
Overall, our pig model of cardiac arrest demonstrated neuronal injury by elevated blood markers. Furthermore, signs of post-ischaemic cytotoxic oedema were shown on MRI ADC maps and decreased neuronal viability by NAA on proton-spectroscopy. Microdialysis data showed impaired cerebral metabolism in the cardiac arrest group, which was most pronounced during the first 10 h after resuscitation.
Intracranial pressure and cerebral perfusion
The CerPP was seemingly higher during the maintenance phase of TTM in the sham group. The interpretation of this difference is difficult because the intensive care protocol used, only ensured a MAP > 65 mmHg using vasopressor treatment. Hence, the difference in CerPP is caused by a higher MAP in the sham group. Interestingly, the ICP remained at similar levels in the cardiac arrest and sham groups during the intensive care period, disregarding the one cardiac arrest animal that developed a very high ICP. The ICP during cardiac arrest, resuscitation, and the immediate ROSC period is well described in animal models. ICP rises upon the cessation of cardiac function, and then gradually falls or stays elevated during resuscitation depending on the quality and mode of CPR [10, 14,15,16]. The increase during cardiac arrest and resuscitation is due to decreased venous return, as proven by decreased ICP values during CPR which incorporates active measures to decrease intrathoracic pressure [22]. Looking beyond the immediate post-resuscitation phase, case series of hypoxic–ischaemic brain injury patients have reported differentiation of poor vs. good neurological prognosis by looking at ICP during the first 5–6 days after ROSC [6, 9]. Other studies have shown a generally low prevalence of intracranial hypertension after cardiac arrest, although with a trend towards higher ICP values in non-survivors [23,24,25]. This picture of an overall low prevalence of intracranial hypertension is reproduced in our study. In some cases, high ICP levels indicate a higher degree of cerebral injury when also considering the two animals from the AMI-Cont group with intracranial hypertension and high NSE values. We did not see any major deviations in ICP during cooling or rewarming in contrast with previous investigations in pigs and humans [7, 26]. Although there is an apparent association between very high ICP values and increased cerebral injury, results from recent studies in both pigs and humans do agree that using ICP and blood pressure correlation analysis for indirect measurements of intracranial compliance or cerebral autoregulation shows stronger associations than ICP alone [9, 12, 23]. Unfortunately, due to the temporal granularity of our data, we were not able to conduct such analysis.
Brain oxygenation
The lower limit for brain normoxia is considered 20 mmHg and the PTiO2 measurements in our study were lower in both groups [8]. Of importance, PTiO2 increased with FiO2 elevations and fell accordingly during cardiac arrest indicating the probe was responsive to changes in oxygen levels [2]. Despite the low values, we can derive that the PTiO2 levels seemingly did not vary much between groups over the course of the intensive care period. A normal L/P ratio and normal biomarkers was seen throughout the protocol for sham pigs, which indicates the absence of brain hypoxia. Furthermore, the amplitude of the PTiO2 signal was above the level of the manufactures recommendation why we do not consider dampening of the signal as a factor. A large variance was seen during the first 6 h after resuscitation, due to probe stabilization, altered brain perfusion/metabolism in cardiac arrest animals, PaO2 titration determined by the protocol, and effect of TTM induction. The PTiO2 levels in this study were not associated with PaO2, which is in line with a previous investigation showing that PaO2 only describes a small portion of PTiO2 variance. [13]
Cerebral metabolism
To our knowledge, this is the first study of long-term cerebral metabolism after resuscitation with a high temporal solution in a controlled experimental setting. Markers of cerebral metabolism during cardiac arrest and resuscitation has been described with an immediate rise in lactate, glycerol, and L/P ratio, as well as a drop in glucose [10, 11, 27]. In our study, we saw a high peak in L/P ratio in the cardiac arrest group at 2 h after resuscitation. The microdialysis vials in our study were changed 5 min after successful resuscitation, so this early peak to some extent represents the no/low flow periods. Beyond 2 h, two different patterns emerged in the cardiac arrest group: (1) four animals had normalization of L/P ratio, and (2) three animals experienced secondary/tertiary surges in L/P ratio (see Additional file 1: Fig. S7). The exact mechanisms generating the secondary L/P ratio elevation could be manifold, e.g., microcirculatory dysfunction, neurovascular uncoupling, mitochondrial dysfunction, etc. In our data, secondary surges in L/P ratio were mainly accompanied by low pyruvate levels, indicating ischaemic/hypoxic conditions, and not mitochondrial dysfunction. Previous studies of cerebral metabolism have shown similar patterns as we observed: Yuan et al. have in two studies shown elevations in L/P ratio of around 50–100 between 6–16 h after resuscitation indicating secondary surges [28, 29], whereas Skåre et al. on the contrary showed early peak values of L/P, with a rapid near-normalization of L/P ratio during the first hour after resuscitation in a 10-min no-flow pig model [12]. The studies by Yuan et al. had low temporal granularity and the development over time is therefore difficult to interpret. The model by Skåre et al. utilized resuscitative efforts with high-flow cardiopulmonary bypass ensuring near-optimal macrocirculatory conditions prior to ROSC, which could have led to a faster recovery of cerebral metabolism. Further studies into the mechanisms of dysregulated cerebral metabolism, perhaps with focus on linking disturbances to vascular dysfunction, after successful resuscitation, are needed.
In a case series of 10 post-cardiac arrest patients, Hifumi et al. demonstrated increasing L/P ratios over the first four days after ROSC. Furthermore, they showed a correlation between increasing ICP levels and L/P ratio in a subgroup of patients with poor neurological outcome [25]. On this basis, they proposed a theory of brain oedema and likely ICP elevations as the reason for the cerebral metabolic disturbances. Our data showed downward trends in PTiO2 and cerebral metabolic disturbances prior to ICP elevations in pigs with intracranial hypertension (including the two animals from the AMI-Cont group, see Additional file 1: Fig. S2). This indicates reperfusion injury and/or secondary hypoxia as the instigating cause of cerebral oedema and ICP elevation and not vice versa.
Blood markers
NSE is, as of now, the only recommended biomarker of cerebral injury to be used in prognostication of cardiac arrest survivors [30]. In recent retrospective observational studies NfL has been shown to be predictive of poor neurologic outcome as early as on hospital admission with peak values occurring around 48 h after admission [31,32,33]. To our knowledge this is the first study to investigate NfL in a pre-clinical model of cardiac arrest. Although NfL rose with the same kinetics as NSE the relative difference was smaller when compared with sham pigs.
MRI
For assessment of cerebral oedema by ADC maps in human studies, a threshold value in a proportion of the brain is often measured and growing evidence is supporting a threshold of < 650 × 10–6 mm2/s in > 10% of the brain [34]. For this study, although the ICP/PTiO2 probes in the pigs were removed before the scan, an MRI-sensible artefact was still present in the location of the catheter, which precluded a volume-ratio measurement. Therefore, we took the approach of locating ROIs in specific anatomical locations. Our findings with lower ADC values in the cardiac arrest group in white matter cortex and hippocampus (and lower in cerebellum but not significant) indicate that cerebral oedema formation did occur as a result of the ischaemia–reperfusion injury elicited by cardiac arrest. Diffusion-weighted imaging with ADC maps has the sensitivity to detect cytotoxic oedema [35]. In the cardiac arrest pigs with normal range ICP at the end of the protocol lower ADC values may therefore be indicative of cytotoxic oedema.
The cause of the relatively large ADC values in the hippocampi in the sham group is unknown. To exclude the possibility of cerebrospinal fluid from the lateral or third ventricles (which would otherwise increase ADC values) the hippocampal ROIs were minimized in a secondary analysis, but this yielded no change in ADC values.
The lower relaxation time in grey matter cortex from BOLD imaging, demonstrates a higher relative concentration of deoxyhemoglobin. Coinciding this finding with the normal perfusion measures indicates a larger metabolic demand than supply. When inspecting the relaxations times from all five regions there is a trend towards lower values in the cardiac arrest group in all regions. These results should be interpreted with caution due to a low number of animals undergoing MRI, but they could indicate a persisting mismatch in the demand and supply of oxygen in the vulnerable brain after cardiac arrest. Furthermore, although not significant, median PTiO2 levels did trend towards lower values in the cardiac arrest group. Reports in humans have shown secondary hypoxia as the cause of de novo neuronal damage. [8]
Limitations
Although the pig has many physiological and anatomical similarities with humans, establishing a direct link between the results from this study to the clinical setting is not possible. Furthermore, there were a relatively low number of animals in each group. Treatment of high ICP or low PTiO2 levels was not included in the study protocol, as either parameter could have affected other outcome measurements in this study (e.g., microdialysis and MRI). The microdialysis data only represent one area of the brain, and although the ischaemic insult after cardiac arrest is global, injury progression is different between anatomical locations.
Conclusion
We have developed a clinically relevant cardiac arrest pig model that displays cerebral injury as marked by NSE and NfL elevations, signs of cerebral oedema, and reduced neuron viability. Overall, the burden of elevated ICP was low in the cardiac arrest group. A subset of pigs undergoing cardiac arrest had persisting metabolic disturbances after successful resuscitation.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ADC:
-
Apparent diffusion coefficient
- AMI-Cont:
-
Cardiac arrest group with continuously myocardial infarction
- AMI-Int:
-
Cardiac arrest group with reperfusion of coronary artery prior to cardiac arrest and re-occlusion upon ROSC
- BOLD:
-
Blood oxygen level dependent
- CerPP:
-
Cerebral perfusion pressure
- FiO2 :
-
Fraction of inspired oxygen
- ICP:
-
Intracranial pressure
- L/P:
-
Lactate/pyruvate
- MAP:
-
Mean arterial blood pressure
- MRI:
-
Magnetic resonance imaging
- NAA:
-
N-Acetylaspartate
- NSE:
-
Neuron specific enolase
- NfL:
-
Neurofilament light chain
- PaCO2 :
-
Partial pressure of CO2 in arterial blood
- PaO2 :
-
Partial pressure of O2 in arterial blood
- PTiO2 :
-
Brain tissue oxygenation
- ROSC:
-
Return of spontaneous circulation
- SvO2 :
-
Mixed venous blood oxygen saturation
- TTM:
-
Targeted temperature management
References
Vognsen M, Fabian-Jessing BK, Secher N, Lofgren B, Dezfulian C, Andersen LW, Granfeldt A (2017) Contemporary animal models of cardiac arrest: a systematic review. Resuscitation 113:115–123
Vammen L, Munch Johannsen C, Magnussen A, Povlsen A, Riis Petersen S, Azizi A, Løfgren B, Andersen LW, Granfeldt A (2021) Cardiac arrest in pigs with 48 hours of post-resuscitation care induced by 2 methods of myocardial infarction: a methodological description. J Am Heart Assoc 2021:e022679
Witten L, Gardner R, Holmberg MJ, Wiberg S, Moskowitz A, Mehta S, Grossestreuer AV, Yankama T, Donnino MW, Berg KM (2019) Reasons for death in patients successfully resuscitated from out-of-hospital and in-hospital cardiac arrest. Resuscitation 136:93–99
Sandroni C, D’Arrigo S, Nolan JP (2018) Prognostication after cardiac arrest. Crit Care 22(1):150
Sandroni C, D’Arrigo S, Cacciola S, Hoedemaekers CWE, Kamps MJA, Oddo M, Taccone FS, Di Rocco A, Meijer FJA, Westhall E et al (2020) Prediction of poor neurological outcome in comatose survivors of cardiac arrest: a systematic review. Intensive Care Med 46(10):1803–1851
Gueugniaud PY, Garcia-Darennes F, Gaussorgues P, Bancalari G, Petit P, Robert D (1991) Prognostic significance of early intracranial and cerebral perfusion pressures in post-cardiac arrest anoxic coma. Intensive Care Med 17(7):392–398
Naito H, Isotani E, Callaway CW, Hagioka S, Morimoto N (2016) Intracranial pressure increases during rewarming period after mild therapeutic hypothermia in postcardiac arrest patients. Ther Hypothermia Temp Manag 6(4):189–193
Hoiland RL, Ainslie PN, Wellington CL, Cooper J, Stukas S, Thiara S, Foster D, Fergusson NA, Conway EM, Menon DK et al (2021) Brain hypoxia is associated with neuroglial injury in humans post-cardiac arrest. Circ Res 129(5):583–597
Balu R, Rajagopalan S, Baghshomali S, Kirschen M, Amurthur A, Kofke WA, Abella BS (2021) Cerebrovascular pressure reactivity and intracranial pressure are associated with neurologic outcome after hypoxic-ischemic brain injury. Resuscitation
Putzer G, Martini J, Spraider P, Hornung R, Pinggera D, Abram J, Altaner N, Hell T, Glodny B, Helbok R et al (2020) Effects of different adrenaline doses on cerebral oxygenation and cerebral metabolism during cardiopulmonary resuscitation in pigs. Resuscitation
Bahlmann L, Klaus S, Baumeier W, Schmucker P, Raedler C, Schmittinger CA, Wenzel V, Voelckel W, Lindner KH (2003) Brain metabolism during cardiopulmonary resuscitation assessed with microdialysis. Resuscitation 59(2):255–260
Skåre C, Karlsen H, Strand-Amundsen RJ, Eriksen M, Skulberg VM, Sunde K, Tønnessen TI, Olasveengen TM (2021) Cerebral perfusion and metabolism with mean arterial pressure 90 vs. 60 mmHg in a porcine post cardiac arrest model with and without targeted temperature management. Resuscitation
Jung YH, Shamsiev K, Mamadjonov N, Jeung KW, Lee HY, Lee BK, Kang BS, Heo T, Min YI (2021) Relationship of common hemodynamic and respiratory target parameters with brain tissue oxygen tension in the absence of hypoxemia or hypotension after cardiac arrest: a post-hoc analysis of an experimental study using a pig model. PLoS ONE 16(2):e0245931
Cavus E, Bein B, Dorges V, Stadlbauer KH, Wenzel V, Steinfath M, Hanss R, Scholz J (2006) Brain tissue oxygen pressure and cerebral metabolism in an animal model of cardiac arrest and cardiopulmonary resuscitation. Resuscitation 71(1):97–106
Bein B, Cavus E, Stadlbauer KH, Tonner PH, Steinfath M, Scholz J, Dörges V (2006) Monitoring of cerebral oxygenation with near infrared spectroscopy and tissue oxygen partial pressure during cardiopulmonary resuscitation in pigs. Eur J Anaesthesiol 23(6):501–509
Elmer J, Flickinger KL, Anderson MW, Koller AC, Sundermann ML, Dezfulian C, Okonkwo DO, Shutter LA, Salcido DD, Callaway CW et al (2018) Effect of neuromonitor-guided titrated care on brain tissue hypoxia after opioid overdose cardiac arrest. Resuscitation 129:121–126
Zhou D, Li Z, Zhang S, Wu L, Li Y, Shi G, Zhou J (2020) Mild hypercapnia improves brain tissue oxygen tension but not diffusion limitation in asphyxial cardiac arrest: an experimental study in pigs. BMC Anesthesiol 20(1):252
Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U et al (2020) The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. J Physiol
Idris AH, Becker LB, Ornato JP, Hedges JR, Bircher NG, Chandra NC, Cummins RO, Dick W, Ebmeyer U, Halperin HR et al (1996) Utstein-style guidelines for uniform reporting of laboratory CPR research. A statement for healthcare professionals from a task force of the American Heart Association, the American College of Emergency Physicians, the American College of Cardiology, the European Resuscitation Council, the Heart and Stroke Foundation of Canada, the Institute of Critical Care Medicine, the Safar Center for Resuscitation Research, and the Society for Academic Emergency Medicine. Writing Group. Circulation 94(9):2324–2336
Orlowski D, Glud AN, Palomero-Gallagher N, Sørensen JCH, Bjarkam CR (2019) Online histological atlas of the Göttingen minipig brain. Heliyon 5(3):e01363
Harris PA, Taylor R, Minor BL, Elliott V, Fernandez M, O’Neal L, McLeod L, Delacqua G, Delacqua F, Kirby J et al (2019) The REDCap consortium: Building an international community of software platform partners. J Biomed Inform 95:103208
Yannopoulos D, Aufderheide TP, McKnite S, Kotsifas K, Charris R, Nadkarni V, Lurie KG (2006) Hemodynamic and respiratory effects of negative tracheal pressure during CPR in pigs. Resuscitation 69(3):487–494
Sekhon MS, Griesdale DE, Ainslie PN, Gooderham P, Foster D, Czosnyka M, Robba C, Cardim D (2019) Intracranial pressure and compliance in hypoxic ischemic brain injury patients after cardiac arrest. Resuscitation 141:96–103
Sakabe T, Tateishi A, Miyauchi Y, Maekawa T, Matsumoto M, Tsutsui T, Takeshita H (1987) Intracranial pressure following cardiopulmonary resuscitation. Intensive Care Med 13(4):256–259
Hifumi T, Kawakita K, Yoda T, Okazaki T, Kuroda Y (2017) Association of brain metabolites with blood lactate and glucose levels with respect to neurological outcomes after out-of-hospital cardiac arrest: a preliminary microdialysis study. Resuscitation 110:26–31
Covaciu L, Allers M, Lunderquist A, Rubertsson S (2010) Intranasal cooling with or without intravenous cold fluids during and after cardiac arrest in pigs. Acta Anaesthesiol Scand 54(4):494–501
Gowers SAN, Samper IC, Murray DRK, Smith GK, Jeyaprakash S, Rogers ML, Karlsson M, Olsen MH, Møller K, Boutelle MG (2020) Real-time neurochemical measurement of dynamic metabolic events during cardiac arrest and resuscitation in a porcine model. Analyst 145(5):1894–1902
Yuan W, Wu JY, Zhao YZ, Li J, Li JB, Li ZH, Li CS (2017) Comparison of early sequential hypothermia and delayed hypothermia on neurological function after resuscitation in a swine model. Am J Emerg Med 35(11):1645–1652
Yuan W, Wu JY, Zhao YZ, Li J, Li JB, Li ZH, Li CS (2018) Effects of mild hypothermia on cardiac and neurological function in piglets under pathological and physiological stress conditions. Ther Hypothermia Temp Manag
Nolan JP, Sandroni C, Böttiger BW, Cariou A, Cronberg T, Friberg H, Genbrugge C, Haywood K, Lilja G, Moulaert VRM et al (2021) European Resuscitation Council and European Society of Intensive Care Medicine Guidelines 2021: post-resuscitation care. Resuscitation 161:220–269
Hunziker S, Quinto A, Ramin-Wright M, Becker C, Beck K, Vincent A, Tisljar K, Disanto G, Benkert P, Leppert D et al (2021) Serum neurofilament measurement improves clinical risk scores for outcome prediction after cardiac arrest: results of a prospective study. Crit Care 25(1):32
Moseby-Knappe M, Mattsson N, Nielsen N, Zetterberg H, Blennow K, Dankiewicz J, Dragancea I, Friberg H, Lilja G, Insel PS et al (2019) Serum neurofilament light chain for prognosis of outcome after cardiac arrest. JAMA Neurol 76(1):64–71
Wihersaari L, Ashton NJ, Reinikainen M, Jakkula P, Pettilä V, Hästbacka J, Tiainen M, Loisa P, Friberg H, Cronberg T et al (2020) Neurofilament light as an outcome predictor after cardiac arrest: a post hoc analysis of the COMACARE trial. Intensive Care Med
Hirsch KG, Fischbein N, Mlynash M, Kemp S, Bammer R, Eyngorn I, Tong J, Moseley M, Venkatasubramanian C, Caulfield AF et al (2020) Prognostic value of diffusion-weighted MRI for post-cardiac arrest coma. Neurology 94(16):e1684–e1692
von Kummer R, Dzialowski I (2017) Imaging of cerebral ischemic edema and neuronal death. Neuroradiology 59(6):545–553
Acknowledgements
Not applicable.
Funding
This study was conducted with funding from Independent Research Fund Denmark, Aarhus University, Augustinus Foundation, Riisfort Foundation, and Hede Nielsen Family Foundation. Asger Granfeldt was supported by a grant from the Health Research Foundation of Central Denmark Region. None of the funding sources played any role in the design, data collection, analysis, interpretation, writing, or submission of the paper for publication.
Author information
Authors and Affiliations
Contributions
LV, LWA, BL and AG contributed to idea and design. LV, ARK, and AG contributed to the pilot studies and model setup. LV, CMJ, AM, AP, SRP and AA contributed to the conduction of the experiments and data collection. LV, SR and MP contributed to the acquisition and analysis of MR data. LV conducted the data analysis. LV drafted the manuscript and supplemental material. All authors contributed to editing and approval of final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Danish Animal Experiments Inspectorate (License number: 2019-15-0201-01647) and conducted and reported in accordance with the ARRIVE [18] guidelines.
Consent for publication
Not applicable.
Competing interests
AG reported receiving personal fees from Noorik Biopharmaceuticals outside the submitted work. Furthermore AG is co-inventor on a patent owned by Aarhus University claiming the use of senicapoc for ARDS caused by COVID-19. Other authors have none to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Post-cardiac arrest intensive care protocol. Table S2 Haemoglobin, blood lactate and pH. Figure S1 Regions of interest displayed on T1 weighted MRI scan. Figure S2 Pressure and oxygen levels for both cardiac arrest groups and sham controls. Figure S3 Microdialysis data for both cardiac arrest groups and sham controls. Figure S4 MRI and MR spectroscopy for both cardiac arrest groups and sham controls. Figure S5 Neuronal injury markers for both cardiac arrest groups and sham controls. Figure S6 Scatter plot of blood/brain oxygenation and NSE/NfL levels for each animal. Figure S7 Scatter plots of lactate, pyruvate, and their ratio from animals in AMI-Int cardiac arrest group.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vammen, L., Johannsen, C.M., Magnussen, A. et al. Cerebral monitoring in a pig model of cardiac arrest with 48 h of intensive care. ICMx 10, 45 (2022). https://doi.org/10.1186/s40635-022-00475-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40635-022-00475-2