
Abstract
Background
Drug repurposing (i.e., finding novel uses for existing drugs) is essential for maximizing medicines’ therapeutic utility, but obtaining regulatory approval for new indications is costly. Policymakers have therefore created temporary indication-specific market exclusivities to incentivize drug innovators to run new clinical investigations. The effectiveness of these exclusivities is poorly understood.
Objective
To determine whether generic entry impacts the probability of new indication additions.
Methods
For a cohort of all new small-molecule drugs approved by the FDA between July 1997 and May 2020, we tracked new indications added for the subset of drugs that experienced generic entry during the observation period and then analyzed how the probability of a new indication changed with the number of years since/to generic entry.
Results
Of the 197 new drugs that subsequently experienced generic entry, only 64 (32%) had at least one new indication added. The probability of a new indication addition peaked above 4% between 7 and 8 years prior to generic entry and then to dropped to near zero 15 years after FDA approval. We show that the limited duration of exclusivity reduces the number of secondary indications significantly.
Conclusion
Status quo for most drug innovators is creating novel one-indication products. Despite indication-specific exclusivities, the imminence of generic entry still has a detectable impact on reducing the chances of new indication additions. There is much room for improvement when it comes to incentivizing clinical investigations for new uses and unlocking existing medicines’ full therapeutic potential.
Similar content being viewed by others


Key points
-
Drug repurposing is finding novel uses for existing drugs.
-
Indication-specific market exclusivities have been used to incentivize drug innovators to conduct the clinical trials preferred by drug regulators for label changes, but these are difficult to enforce because generic equivalents may still be approved and used off-label.
-
Our study finds that, despite indication-specific exclusivities, no new indications are added after FDA approval for two-thirds of drugs and that the probability of new indication additions drops to near zero after generic entry occurs.
-
As status quo for most drug innovators appears is creating novel one-indication products, there is much room for improving incentivizes for clinical investigations for new uses and for maximizing existing medicines’ full therapeutic potential.
Background
Drug repurposing (i.e., using an existing drug for a new use) is essential for making the most of our medical armamentarium. Many drugs have accrued new indications over a period of years: adalimumab (Humira) was approved by the FDA for sale in 2002 with one indication and now has 10; onabotulinumtoxinA (Botox) was approved in 1989 with two indications and now has 9. Berndt et al. examine the history of uses of histamine-2 antagonists, proton-pump inhibitors, and selective-serotonin reuptake inhibitors and find that about 75% of their use was ultimately for uses other than their first-approved indications [1]. Drug repurposing has become one of the key tools in the search for medicines in the early fight against Covid-19 [2, 3], with existing products remdesivir and dexamethasone both being found to show improvements in clinical endpoints among hospitalized patients [4].
The timing of new indication innovation varies considerably between drugs. For many drugs, repurposing happens during the development process. For example, remdesivir, initially explored as a treatment for hepatitis C virus infection, was tested unsuccessfully against Ebola, and eventually demonstrated efficacy in reducing the length of hospitalization for patients with COVID-19. While our focus in this paper is on new indications added after a primary indication has been approved, the fact that drugs may be tested for multiple indications before receiving any approval is testimony to the fact that researchers may not know all the possible uses for a drug when it is developed. Sometimes new uses are explored using high-throughput screening, genomic, transcriptomic or proteomic analysis, and analysis of administrative health data [5]. For other drugs, including famously sildenafil (Viagra), indications that become the basis for repurposing are discovered fortuitously through patient experience.
When new indications are discovered for already-approved drugs based on trials showing some evidence for effectiveness, physicians may then begin to use a drug for that purpose “off-label” [6,7,8,9,10,11,12]. However, many off-label uses are not based on high-quality clinical trials and in general any studies supporting an off-label use have not been reviewed by an expert regulator, or have been reviewed and found not to support a new indication, so they may be biased or may need to be weighed against other negative trials. As a result, the drug being used off-label may be ineffective or even unsafe, a point made clear by the recent widespread use of hydroxychloroquine for Covid-19 [13]. Off-label use is particularly common in children. A recent study of ambulatory pediatric visits in the US found that that 38% of prescriptions were off-label, with the majority being off-label by reason of indication rather than age [14,15,16,17]. The ubiquity of off-label use suggests that there is a need for more clinical trials to support new indications for approved drugs.
Obtaining regulatory approval for new indications may be costly. To address this challenge, policymakers in the US and in some other countries have designed incentive programs that offer manufacturers that obtain a new indication for an existing drug a temporary market exclusivity in the use of the drug for the newly approved indication [18]. These exclusivities last for 7 years for drugs intended to treat rare diseases and 3 years for more common conditions in the US.
These exclusivities, however, are difficult to enforce, particularly if there is existing competition from generic versions of the same drug already available for ongoing off-label use by prescribers. Without the revenue that would arise from monopoly pricing during market exclusivity, manufacturers may not have sufficient market-driven incentive to make the upfront investment in clinical trials preferred by regulators to validate these new uses [19, 20]. In the cases of adalimumab and onabotulinumtoxinA, for example, numerous indications were tested after FDA approval during long exclusivity periods free of generic competition.
To investigate the possible impact of generic competition upon post-approval indication innovation, we sought to investigate the relationship between the timing of generic entry and the number of new post-approval indications added per drug. Our hypothesis was that once generic competition is present, fewer new indications are FDA-approved and that as generic entry approaches, the probability of new indications being added would diminish. As such, we expected that the highest probability of new indications would be in the years soon after drug approval because it would provide the manufacturer with the longest period of exclusivity during which to reap the benefits and that most exclusivities associated with new indications will expire before generic entry (since generic entry typically occurs about 12–14 years after FDA approval [21, 22]).
Methods
Design
For a cohort of all new small-molecule drugs (i.e., New Molecular Entities [NMEs]) approved by the FDA between July 1997 and May 2020 (23.4 years), we tracked new indication exclusivities granted, and for the subset of drugs that experienced generic entry during the observation period, we analyzed the relationship between the number of years since/to generic entry and the number of new indications granted.
Data sources
Our chief data source was electronic archives of the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (the “Orange Book”) covering July 1997 through May 2020 [23]. All new indications that have been added on the basis of a new clinical investigation by the drug’s manufacturer receive an exclusivity. These datasets included the timing and type of new indication exclusivity (i.e., a 3-year New Indication exclusivity or a 7-year Orphan Drug Exclusivity) as well as which products the exclusivities were protecting. The Orange Book also provides all small-molecule drugs’ active ingredients, trade names, manufacturers, FDA approval dates, and whether the products are brand name or generic. To distinguish which brand-name drugs were NMEs, we linked to the Drugs@FDA Data Files [24] so that all subsequently approved brand-name drugs (i.e., new formulations) could be grouped with their parent products.
New variables
To study the association between the timing of generic entry and the frequency of a new indication, we created a variable (i.e., “Age”) for the NME’s ages during each observation year by calculating the number of years since their respective FDA approval dates. We also generated variables with Age raised to the power 2, 3, and 4 to allow for a non-linear relationship between age and the frequency of new indications. For NMEs with an equivalent generic drug approved during the observation period, we calculated the number of years to/since the first FDA approval of a generic equivalent. We then constructed dummy variables categorizing time to generic as being more than 10 years, 5–10 years away, 0–5 years away, or post-generic entry. Using these variables, each outcome (i.e., the number of new indications added) could be observed in years according to the new drug’s FDA approval date and in time period since/to the first generic approval. Since our objective was to focus upon new indications added during the post-approval period and to isolate the effect of generic entry, we excluded any new indications added at the time of FDA approval (i.e., AGE = 0) as well as drugs that had no generic equivalents by May 2020. Finally, for each year observed since NME approval and to/since generic entry, we observed whether there was either zero or at least one new indication. There was one observation for each year following the approval of each parent drug until 2020, for a total of 3154 observations.
We also categorized drugs by disease category in which the drug was first introduced, and calculated the proportion of drugs within each disease category. (We applied the MeSH disease categories from the National Library of Medicine.)
Analysis
We first report basic descriptive statistics of interest, including the proportion of drugs with one or more post-approval indications added during the observation period, the proportions of new indications added for more common versus rare diseases, and the proportion of new indication exclusivities that expired before versus after generic entry.
Given that generic entry occurred at different times for different drugs (Additional file 1: Fig. 1), the variation in a new drug’s period on the market (“age”) at generic entry can be used to disentangle the effects of age and generic entry on the probability of new indication development. We used the following logistic regression model to study the effect of the first generic entry timing on the possibility of having a second indication controlling for the drug’s age:
where \(p\) is the probability of having a new indication; Age is observation year minus the new drug’s FDA approval date; and G is the years until generic entry (with \({G}_{\text{neg}}=1\) as observations following generic entry, \({G}_{\mathrm{0,5}}=1\) during the 5 years before generic entry, and \({G}_{\mathrm{5,10}}=1\) from 5 to 10 years before generic entry). The omitted category is all years more than 10 years before generic entry. Thus, the regression coefficients for the time to generic variables (G) in the model represent the predicted change in the logarithm of the odds ratio for each time-to-generic-entry category, compared to when generic entry is more than 10 years in the future. We use the polynomial values of Age up to \({\text{Age}}^{4}\) to ensure that we capture all the variation caused by years since the drug’s first approval.
Finally, while controlling for age, we estimated the counterfactual number of new indications, assuming the influence of generic entry were the same as when generic entry had already occurred, as when it is 0–5 years in the future, and as when it is 5–10 years into the future. To calculate the counterfactual number of second indications, we used the estimated results and replaced \({\beta }_{1}\) and \({\beta }_{2}\) with the estimated value of \({\beta }_{3}\) (i.e., generic entry is not to occur for another 5–10 years), adjusting appropriately for the frequency of observations with multiple new indications.
Results
During the 23-year observation period, 197 new drugs were approved and subsequently experienced generic entry. Of these, 64 (32%) were issued at least one post-approval new indication. As shown in Additional file 2: Table 3, 163 of these drugs had only one indication at the time of approval; 22 had two indications, and 12 had three or more indications.
Incidence of post-approval new indications and timing of their associated exclusivity expirations
There were 116 post-approval new indications added during the observation period for the 64 genericized drugs (Table 1). About one-quarter (27/116, 23%) new indications expired after generic entry had occurred. Expiration after generic entry was more common for the longer-lasting rare disease indications with 14 of 18 (78%), expiring an average of 4 years after. Expiration after generic entry was less frequent among common conditions with 13 of 98 (13%), expiring an average of 85 days after.
Probability of new indication exclusivities by NME age
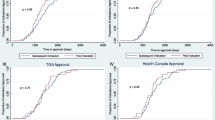
The probability that new indications would be developed changed according the age of the new drug. The probability peaked above 4% between 1–2 years after FDA approval and then dropped off (Fig. 1). By 15 years after FDA approval, the probability dropped to less than 1%, or about one-quarter of the peak.

Probability of new indication exclusivity granted by FDA for drugs approved, 1997–2020. The probability of a new indication addition is highest when NMEs have been relatively recently (within 1–2 years) and when generic entry is around 7–8 years into the future. The probability is lowest when an NME has been approved for more than 15 years and when generic entry has already occurred. “Age (in years)” = number of years since FDA approval
Probability of new indication exclusivities by year until/since generic entry
Similarly, the probability that a new indication would be developed also changed according to the number of years until the new drug experienced generic entry. The probability of a new indication peaked above 4% between 7 and 8 years prior to generic and then dropped off (Fig. 1). By 15 years after FDA approval, the probability dropped to near zero.
Counterfactual impact on the probability of new indication exclusivities controlling for NME age
When controlling for a new drug’s time on the market, regression results show that the probability of receiving a new indication exclusivity was independently conditional on the timing of generic entry (Fig. 2, Additional file 2: Table 1). The probability of new indication exclusivity dropped consistently from 5–10 years before generic entry, to 0–5 years before generic entry, and then especially after generic entry had already occurred.

Estimated regression coefficients. The imminence of generic entry negatively impacts the chances of a new indication being developed for an NME. Holding the effect of age constant, generic entry’s influence upon reducing the chances of a new indication addition is strongest when generic has already occurred and weakest when generic entry is still 5–10 years into the future
Predicted probabilities of new indications if the impact of generic entry is applied to all drugs
In the counterfactual case that all drugs had the same probability of a new indication as when generic had already occurred, our results show that the chance of a new indication exclusivity was nearly zero, regardless of the new drug’s time on the market (Fig. 3). By contrast, in the counterfactual case that all drugs had the same probability as when generic entry is 5–10 years into the future, the probability peaked at nearly 4% after around 4 years after FDA approval and before declining and levelling out to around 1% after around 15 years after FDA approval. Our model predicts that if the effect of generic entry could be removed, the expected number of new indications for our cohort of NMEs would have been 134 rather than the 116 which occurred in actuality—in other words, a 16% increase was predicted (Table 2).

Predicted conditional probability of a new indication exclusivity approval. When we applied the strongest observed influence of generic entry (i.e., generic entry had already occurred [\({\beta }_{1}\)]) to all NMEs, shown in black is our model’s predicted probability of new indications additions, which is near zero, regardless of the age of the drug. Note that by law, generic entry cannot occur in the United States prior to 5 years after FDA approval when the product in question is designated as a small-molecule NME, and therefore, the black line begins only after 5 years accordingly. When we applied the weakest observed influence of generic entry (i.e., generic entry is 5–10 years into the future [\({\beta }_{3}\)]) to all NMEs, shown in red is our model’s predicted probability of new indications additions, which is substantially higher, regardless of the age of the drug
Therapeutic area differences
The proportion of drugs which received a secondary indication following approval varied noticeably across disease categories, as shown in Table 3. Secondary indications were most common for drugs categorized as treating neoplasms and skin and connective tissue disease.
Discussion
Our analysis shows that for two-thirds of all new drugs developed, no new indications are added during the post-approval period by drugs’ manufacturers. New uses are mostly added soon after FDA approval and long before generic entry, particularly for new indications for more common conditions (as opposed to rare diseases). Generic entry’s effect on reducing new indication innovation activity makes a detectable difference. Together, these findings may imply that there is much potential for improvement when it comes to providing incentives for clinical investigations that lead to regulatory authorization of new uses of approved drugs.
Our finding that two-thirds of drugs have no new indications added during the post-approval period can be explained in two (non-exclusive) ways: either those drugs are intrinsically highly specific to a disease area or the incentive to invest in clinical trials to obtain new indications is weak. To the extent that it is the latter reason, the failure to make full use of drug repurposing opportunities reduces the value of existing medicines: it reduces the effectiveness of our therapeutic toolkit and also puts patients at risk. Previous studies have demonstrated that once a drug is approved, off-label use is widespread (as high as 46% of all prescribing for some types of medications, such as cardiac medications and anticonvulsants) and that most (73%) off-label uses have little or no scientific basis [7]. In other words, a substantial share of prescribing is not validated by the FDA and is based on weak data. Historically, drug representatives have not been allowed to advertise off-label uses of their products to prescribers, although over the past two decades nearly all pharmaceutical manufacturers have been investigated for violating this rule [25]. Further, a number of practice guidelines and practice compendia, such as in psychiatry, include off-label uses [9]. It would be preferable if common uses of all drugs could be validated by the neutral experts at government regulators like the FDA based on high-quality clinical trials.
Most new indication market exclusivities that have been issued by the FDA expire before generic entry based on the original drug approval. This occurs because a drug’s patents last further than most post-approval exclusivities granted for new indications [26, 26]. Thus, these exclusivities seldom play a practical role in market protection. To increase incentives to obtain secondary indications, the 3-year exclusivity could be lengthened, or the exclusivity could be extended to all patents and other exclusivities that exist for that same product. This is currently done for clinical trials to test existing indications for adults in pediatric populations (i.e., a new patient population)—a 6-month extension is given to all patents and exclusivities [27]. This policy has motivated a large number of pediatric trials, many extended indications and a small number of new indications, though at a high cost: the return on investment for drug makers has been shown to be highly lucrative [28]. Shifting to an exclusivity and patent extension model (even by a period of 3 months, rather than the current 3-year exclusivity that expires before it can protect the market in practice) would have an impact, although the therapeutic benefits would have to be weighed against the financial costs of extended exclusivity.
Other policy alternatives that are non-exclusivity-based are also possible. For example, priority review vouchers which can be sold or used to expedite FDA receive of new drugs (as is currently being done to incentivize research and development of neglected tropical diseases [29]), tax breaks (as is currently being done to incentivize treatments for rare diseases [26]), or greater upfront government funding are some examples which each have their own strengths and limitations. For example, priority review vouchers have attracted considerable criticism [30]. An important advantage of non-exclusivity-based incentives is that the timing of generic entry is unaffected, which is important for medicine accessibility, affordability, and public health impact.
Our study has certain limitations. Most importantly, the counterfactual estimate does not account for the fact that even when generic entry is distant, there may still be a disincentive to invest in the clinical trials necessary to establish a new indication, given that generic entry will eventually reduce the total possible sales of the drug in all indications. We have not been able to include biologics in this analysis, despite their growing importance, because of the small number of drugs with biosimilar competition. Moreover, the regulatory framework around biologics differs in numerous ways, which makes comparisons treacherous.
Conclusion
For two-thirds of new drugs approved in recent years by the FDA, drug manufacturers added no new indications during the post-approval period, suggesting that even when an off-label use becomes common, all too often no one invests in securing FDA approval for it. One way to address this issue would be to redesign current incentive structures such that firms can profit from investments into new clinical trials that meet FDA standards and could be submitted for inclusion on the official drug labeling. Such a move would help weed out secondary uses based on solid versus weak data and would help improve the dissemination of evidence-based prescribing practices, for the benefit of patients.
Availability of data and materials
This study is not reporting data from a trial. All raw data are publicly available from the United States Food and Drug Administration.
References
Berndt ER, Cockburn IM, Grépin KA. The impact of incremental innovation in biopharmaceuticals. PharmacoEconomics. 2006;24(2):69–86.
Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6(1):14.
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–71.
Thorlund K, Mills E, Mehta C. Global coronavirus COVID-19 clinical trial tracker. https://www.covid19-trials.org. 2020. Accessed 20 Jul 2020.
Cha Y, Erez T, Reynolds IJ, Kumar D, Ross J, Koytiger G, et al. Drug repurposing from the perspective of pharmaceutical companies. Br J Pharmacol. 2018;175(2):168–80.
Stafford RS. Regulating off-label drug use—rethinking the role of the FDA. N Engl J Med. 2008;358(14):1427–9.
Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021–6.
De Souza JA, Polite B, Perkins M, Meropol NJ, Ratain MJ, Newcomer LN, et al. Unsupported off-label chemotherapy in metastatic colon cancer. BMC Health Serv Res. 2012;12(1):481.
Paczynski RP, Alexander GC, Chinchilli VM, Kruszewski SP. Quality of evidence in drug compendia supporting off-label use of typical and atypical antipsychotic medications. Int J Risk Saf Med. 2012;24:137–46.
McKean A, Monasterio E. Off-label use of atypical antipsychotics. CNS Drugs. 2012;26(5):383–90.
Goločorbin-Kon S, Iličković I, Mikov M. Reasons for and frequency of off-label drug use. Med Pregl. 2015;68(1–2):35–40.
Gonçalves MG, Heineck I. Frequency of prescriptions of off-label drugs and drugs not approved for pediatric use in primary health care in a southern municipality of Brazil. Revista Paulista de Pediatria. 2016;34(1):11–7.
Ferner RE, Aronson JK. Chloroquine and hydroxychloroquine in covid-19. BMJ. 2020;369:m1432. https://doi.org/10.1136/bmj.m1432.
Allen HC, Garbe MC, Lees J, Aziz N, Chaaban H, Miller JL, et al. Off-label medication use in children, more common than we think: a systematic review of the literature. J Okla State Med Assoc. 2018;111(8):776–83.
Bazzano AT, Mangione-Smith R, Schonlau M, Suttorp MJ, Brook RH. Off-label prescribing to children in the United States outpatient setting. Acad Pediatr. 2009;9(2):81–8.
Palmaro A, Bissuel R, Renaud N, Durrieu G, Escourrou B, Oustric S, et al. Off-label prescribing in pediatric outpatients. Pediatrics. 2015;135(1):49–58.
Hoon D, Taylor MT, Kapadia P, Gerhard T, Strom BL, Horton DB. Trends in off-label drug use in ambulatory settings: 2006–2015. Pediatrics. 2019;144:4.
Federal Food, Dug, and Cosmetic Act 1938 s.505C.3.E and s.505F.J.5 (USA). https://ballotpedia.org/Federal_Food,_Drug,_and_Cosmetic_Act_of_1938. Accessed 1 Feb 2020.
Benjamin N. Roin. Solving the Problem of New Uses. Mich. St. L. Rev; 2013. 67 p. https://dash.harvard.edu/handle/1/11189865. Accessed 10 Jul 2020.
Eisenberg RS. The problem of new uses. Yale J Health Pol’y L & Ethics. 2005;5:717.
Wang B, Liu J, Kesselheim AS. Variations in time of market exclusivity among top-selling prescription drugs in the United States. JAMA Internal Med. 2015;175(4):635–7.
Grabowski H, Long G, Mortimer R, Boyo A. Updated trends in US brand-name and generic drug competition. J Med Econ. 2016;19(9):836–44.
Approved Drug Products with Therapeutic Equivalence Evaluations. The Orange Book. United States Food and Drug Administration. 2020. https://www.fda.gov/drugs/drug-approvals-and-databases/approved-drug-products-therapeutic-equivalence-evaluations-orange-book. Accessed 7 Feb 2020.
Drugs@FDA Data Files. Drugs Approvals and Databases. United States Food and Drug Administration. 2020. https://www.fda.gov/drugs/drug-approvals-and-databases/drugsfda-data-files. Accessed 15 Feb 2020.
Richardson E. Off-Label Drug Promotion. Health Affairs. 30 June 2016; doi: https://doi.org/10.1377/hpb20160630.920075
Copenhagen Economics. Study on the economic impact of supplementary protection certificates, pharmaceutical incentives and rewards in Europe: Final Report . Publications Office of the European Union. 2018. 386 p.
Sarpatwari A, Beall RF, Abdurrob A, He M, Kesselheim AS. Evaluating the impact of the orphan drug act’s seven-year market exclusivity period. Health Aff. 2018;37(5):732–7.
Patents and Exclusivity. FDA/CDER SBIA Chronicles. United States Food and Drug Administration. https://www.fda.gov/media/92548/download. Accessed 15 Apr 2020.
Sinha MS, Najafzadeh M, Rajasingh EK, Love J, Kesselheim AS. Labeling changes and costs for clinical trials performed under the US Food and Drug Administration pediatric exclusivity extension, 2007 to 2012. JAMA Internal Med. 2018;178(11):1458–66.
Ridley DB, Régnier SA. The commercial market for priority review vouchers. Health Aff. 2016;35(5):776–83.
Kesselheim AS. Drug development for neglected diseases—the trouble with FDA review vouchers. N Engl J Med. 2008;359(19):1981–3.
Acknowledgements
Dr. Beall’s work is funded by an O’Brien Institute for Public Health catalyst grant for equitable drug innovation and access. Dr. Kesselheim’s work is funded by Arnold Ventures and Harvard-MIT Center for Regulatory Science. Dr. Hollis’s work is funded by a grant from Canadian Institutes for Health Research.
Author information
Authors and Affiliations
Contributions
RB, ASK, and AH wrote the manuscript; BS, RB, ASK, and AH designed the research; BS, RB, and AH performed the research; BS, RB, and AH analyzed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
IRB approval was not necessary as all data used in this study are secondary, has been de-identified, published online, and is publicly available.
Patient involvement
This study required no direct patient or public involvement.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure A-1. Time between brand and first generic entry.
Additional file 2:
Appendices.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Sahragardjoonegani, B., Beall, R.F., Kesselheim, A.S. et al. Repurposing existing drugs for new uses: a cohort study of the frequency of FDA-granted new indication exclusivities since 1997. J of Pharm Policy and Pract 14, 3 (2021). https://doi.org/10.1186/s40545-020-00282-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40545-020-00282-8