
Abstract
The ability to quickly and accurately analyze Staphylococcus aureus (S. aureus) and isolate the bacteria in a simplified setting is crucial for the early identification and treatment of infectious illnesses. Here, we describe the development of a new aptamer-based detection and separation technique that combines Mg2+-dependent DNAzyme amplification cascades with catalytic hairpin assembly for enhanced sensitivity. This technique uses a rolling circle amplification procedure to build a detection scaffold with a repetitive functional hairpin structure that, upon identifying S. aureus, can launch a catalytic hairpin assembly-mediated DNAzyme-based cascade signal amplification. This allows S. aureus to be isolated using low-speed centrifugation and simultaneously quantified. The approach has a low limit of detection of 21 cfu/mL and a broad detection range of six orders of magnitude due to the inclusion of the catalytic hairpin assembly for signal amplification. In addition to high sensitivity, the method also demonstrates high selectivity for the identification and isolation of S. aureus, making it a useful instrument for reporting S. aureus infections.
Similar content being viewed by others

Introduction
Staphylococcus aureus (S. aureus) is a major bacterium responsible for nosocomial and community-acquired infections in humans, and these infections have persisted despite widespread prevention interventions (Becker and Bubeck Wardenburg 2015; Kadariya et al. 2014; Linzner et al. 2021). In addition to an increase in S. aureus infections in pregnant and postpartum women, as well as outbreaks in newborn nurseries, there have been reports of S. aureus colonization of the female vaginal tract (Deng et al. 2019; Lyon et al. 2023). S. aureus can produce toxins under various environmental conditions and can cause a variety of diseases, ranging from minor infections of the epidermis and soft tissues to life-threatening illnesses (Chen et al. 2022; Gherardi 2023). As a result, there is a pressing need to advance the state of public health protection and consumer safety by developing rapid and reliable technologies for detection and identification of infections.
Culture and colony counting, the standard method for bacterial detection, offers a high detection limit and a high degree of reproducibility (Locke et al. 2020; Rajapaksha et al. 2019). However, the process is very time-consuming as the outcome is not typically known for many days. Researchers have dedicated quite much time and energy to create methods for rapid bacterial identification, such as polymerase chain reaction (PCR)-based approaches and immunology-based methods (Alarcon et al. 2006; Brakstad et al. 1992; Hassan et al. 2022; Song et al. 2020; Wang et al. 2019), to compensate this shortcoming. Antibody production and purification remains a discouraging task in immunology-based approaches. PCR-based technologies are widely used because of their speed, consistency, and high specificity. These PCR-based assays are highly sensitive, but they are also difficult to perform, time-consuming, and fraught with problems inherent to PCR (such as the possibility of false-positive results, the need for rapid temperature cycling, and the requirement of thermostable DNA polymerases). Several methods, including fluorescence tests (Wang et al. 2022), the electrochemical method (Gill et al. 2019; Huang et al. 2021), and colorimetric procedures (Li et al. 2020; Suaifan et al. 2019), have been developed in recent years for detecting microorganisms. Rather than using antibodies, aptamers have been widely used to convert target signal to a nucleic acid signal. Aptamer-based methods are adaptable, sensitive, and cost-effective in comparison with more traditional methods (Li et al. 2021). For instance, DNA hybridization and fluorescence resonance energy transfer have been used to create a fluorescent aptasensor. The approach has a high selectivity and a broad detection range of 1.28 × 103 to 2.00 × 107 cfu/mL (Gao et al. 2018). Many electrochemical aptasensors, which typically consist of an electrode fixed with a molecular identification element, have also been developed in the meantime. There are still certain drawbacks to the above methods, despite the fact that they have made significant strides in S. aureus detection. These techniques, for instance, can only be used to detect and quantify target bacteria, but not to isolate microorganisms for further study. Xiao Xu et al. presented simultaneous individual glucose meter-assisted measurement of target bacteria after isolation via low-speed centrifugation (Xu et al. 2023). Although the method incorporated both bacterial quantification and isolation, it was not sensitive enough. Therefore, there is an urgent need to create a method that can detect and isolate target bacteria at the same time, ideally with detection sensitivity and specificity on par with or better than the current methods.
Using catalytic hairpin assembly (CHA)-mediated DNAzyme-based cascade signal amplification loading on the rolling circle amplification (RCA) product, this paper introduces a novel fluorescent sensing platform for ultrasensitive detection and low-speed centrifugation-based isolation of S. aureus. After identifying S. aureus, the suggested technique involves a catalytic hairpin assembly-mediated DNAzyme-based cascade signal amplification by the production of many single-stranded DNA (ssDNA) products having repetitive functional hairpin structure. The CHA product’s DNAzyme fragments can cleave reporter probes, hence releasing fluorescent signals. Ultrahigh sensitivity was demonstrated by the suggested approach, allowing for the detection and simultaneous low-centrifugation-based separation of S. aureus.
Materials and methods
Reagents and apparatus
Additional file 1 (Table S1, Oligonucleotide Sequences used in this research): Table S1 lists the oligonucleotide information of the sequences used in this approach. The oligonucleotides were produced and purified by the Shanghai Sangon Biological Science and Technology Co., Ltd. (Shanghai, China). Bacteria strains, including Pseudomonas aeruginosa (P aeruginosa, ATCC15442), Escherichia coli O157:H7 (E coli, ACTT25922), Staphylococcus aureus (S aureus, ATCC 26003), and Salmonella enterica (S enterica, ATCC 14028), were all obtained from the Institute of Microbiology Chinese Academy (Beijing, China). Both the DMSO and the BSA were provided by Beyotime Biotechnology Co., Ltd. (Shanghai, China). The enzymes, including DNA ligase T4 and the DNA polymerase phi29, were purchased from Genewiz Biotechnology Co., Ltd. (Suzhou, China).
Construction of the detection scaffold
The steps of rolling circle amplification that are involved in constructing the detection scaffold are as follows: In a tube containing 10 μL PBS buffer, 5 μL primer sequences and 5 μL padlock sequences were mixed. After 15 min (minutes) of incubation at room temperature, 1 μL of T4 DNA ligase was added. T4 DNA ligase was inactivated by heating the mixture to 70 C for 5 min after it had been incubated for 60 min. After that, 1 μL of phi29 enzyme was added, and the mixture was incubated at 30 °C for 2 h (hour). After heating to 90 °C for 10 min, the mixture was allowed to cool to room temperature to construct the detection scaffold.
Isolation and quantification of S. aureus
5 μL H1 probe, 5 μL H2 probe, 5 μL H3 probe and 5 μL S. aureus were mixed with 75 μL PBS buffer solution containing 5 μL constructed detection scaffold, and the mixture was incubated at 30 °C for 60 min. Afterward, the supernatant was collected to test the fluorescent signals. The recorded fluorescent signals were compared with the concentrations of S. aureus.
Results and discussion
Principle of sensitive detection and simultaneous low-centrifugation-based isolation of S. aureus
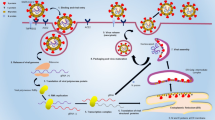
Figure 1 illustrates the basic concept behind the proposed method. The “1” component of the padlock is responsible for transcription of the aptamer sequence (The Protein A aptamer is a short oligonucleotide sequence discovered through in vitro screening that can bind to protein A with high affinity and specificity by forming a unique molecular conformation), and the “2” section is responsible for building of the detection scaffold. Primers and T4 DNA ligase work together to cyclize the padlock sequences. When phi29 polymerase is introduced, a ssDNA chain with a hairpin structure is formed. The aptamer sequence, which is transcribed from the padlock’s “1’” segment, and the “2’” sequence, which initiates further readout of data, form the hairpin structure in the ssDNA product. The aptamer sequence prevents the “2’” motif from activating signal amplification in the hairpin structure. Catalytic hairpin assembly-mediated DNAzyme-based cascade signal amplification begins when the “1’” sequence connects with protein A, revealing the “2’” sequence, upon detecting S. aureus.

The working mechanism of the method for S. aureus detection
H1 is unfolded by the “2’” sequence, resulting in an H1/ “2’” hybrid, and another toehold region of H1 is exposed at the same time. This foothold region of H1 is bound by H2 to form an H1/H2 duplex and liberate the “2’” sequence. The liberated “2’” sequence then re-hybridizes with H1, continuously setting off CHA that results in an abundance of Mg2+-dependent DNAzyme-containing H1/H2 duplexes. The H3 probe was labeled with Cy3 and quenching moiety BHQ (black hole quencher) on 3’ terminal and 5’ terminal, respectively. In addition, the DNAzyme recognizing site and cleavage site (rA) was in the loop section of H3 probe. The H3 hairpin signal probe is recognized and cleaved by the active DNAzyme, allowing Cy3 fluorescent signals to be released.
Construction of the detection scaffold and feasibility of the platform
A fluorescent assay was carried out, capitalizing on SYBR Green I’s specific characterization of the stem section in hairpin structure, to verify the RCA process and to test for the creation of the repeating hairpin structure in the detection scaffold. SYBR Green I is a fluorescent dye that specifically binds to the grooves in the double helix of dsDNA (double-stranded DNA). The SYBR Green I signal is low in its free state but significantly increased when bound to dsDNA. As a result, we monitored the SYBR signal at various incubation periods. As can be seen in Fig. 2A, B , a large quantity of dsDNA (stem) was generated in the detection scaffold group, as evidenced by the SYBR Green I signal being noticeably greater in this group. In the RCA process, a significant enhancement of SYBR Green I signal was observed only in the detection scaffold indicated the successful performance of RCA product (Additional file 1: Fig. S1, The SYBR Green I signals during the RCA process.). Dissociation of the stem part of the hairpin structure may account for the dramatic reduction in the SYBR signal upon addition of S. aureus. We put the entire procedure to the test to ensure it was workable. Figure 2C shows that the presence of the H1, H2, and H3 probes considerably amplified the fluorescence signals.

Construction of detection scaffold and feasibility of the approach. A Schematic illustration of the detection scaffold when S. aureus existed or not. B SYBR Green I signals of detection scaffold when S. aureus existed or not. ***, P < 0.01. C Fluorescent spectrum of whole sensing system when H1 probe, H2 probe, and H3 probe existed or not
Optimization of experimental parameter
We subsequently adjusted experimental parameters including incubation time and centrifugation g values to get a desirable detection performance. For signal amplification by chain displacement, reaction time was also a critical factor. Figure 3A shows that after 60 min (minute) of incubation, the fluorescent signal had increased significantly. Isolation of the target bacteria was dependent on centrifugation rpm. 5000 rpm was chosen for further testing because it is shown in Fig. 3B to produce the highest fluorescence enhancement.

Optimization of experimental parameters. A Fluorescence intensities of the approach with different incubation times. B Fluorescence intensities of the approach with different centrifugal speeds
Analytical performance of the approach
Under optimal experimental settings, we monitored the impact of S. aureus concentration on fluorescence intensities to ascertain the detection sensitivity of our CHA-DNAzyme method. Figure 4A shows that as the S. aureus concentration in the sensing solution is increased from 100 to 1.00 × 107 cfu/mL, the associated fluorescence intensities at 560 nm also increase with time. According to the findings, the production of Mg2+-dependent DNAzyme that catalyze the fluorescent signal process is triggered by the release of “2’” sequences, which in turn depends on the concentration of targets. With a correlation coefficient of 0.9943, the corresponding equation is as follows: Y = Y = 476.9 * lgC − 342.5 (Fig. 4B). The limit of detection (LOD) is calculated to be 21 cfu/mL using the 3SD/slope rule, making it competitive with or superior to other well-established methods for detecting S. aureus while retaining the advantages of low-speed centrifugation-based isolation (Additional file 1: Table S2, A brief comparison of the approach with former ones).

Analytical performance of the approach. A Fluorescent spectrum of the approach when detecting different concentrations of S. aureus. B Correlation between the recorded fluorescence intensity and the concentrations of target. Inserted is the linear equation between the recorded fluorescence intensity and the logarithmic concentrations of S. aureus. C Fluorescence intensity of the approach when detecting S. aureus and interfering bacteria
Possible interfering microorganisms, such as P. aeruginosa, E. coli, and S. enterica, were evaluated to determine the selectivity of the created platform. Each bacterial strain was present at a concentration of 105 cfu/mL. The results shown in Fig. 4C indicated a high specificity of the platform, as the fluorescence intensity was greatly increased with the addition of S. aureus, much more so than that found with possibly interfering pathogens. The method’s great selectivity originates from the strong binding between the aptamer and the bacteria of interest.
Clinical application of the method
S. aureus was quantified in artificial clinical samples using the established approach and the conventional colony-counting method to examine the applicability. After diluting the target microorganisms with a solution containing uterine secretion, clinical samples were generated. The computed quantities of S. aureus using the two approaches were highly correlated, as shown in Fig. 5.

Correlation between the calculated S. aureus concentrations by the proposed approach and the colony-counting method
Conclusion
In conclusion, we used a functional RCA product as a detection scaffold to develop a sensing method for the selective and sensitive quantification and low-speed centrifugation-based isolation of S. aureus. The scaffold is made up of repetitive protein A aptamer sections, which bind to their intended bacteria and reveal “2’” sequences, which then trigger a cascade of signal amplification and facilitated DNAzyme activity. Our method’s developed detection scaffold maintains precise target recognition and aggregation, high sensitivity, and greatly simplified detection procedures when compared to previously published approaches for detecting S. aureus. Our method also has a low detection limit because of the considerable signal amplification it employs, allowing it to be used for quantitative analysis of S. aureus in diluted clinical samples. The new assay method also has potential practical applications and can be transferred to other sensing platforms for the detection of other types of biological substances.
Availability of data and materials
Almost all details of experimental data are presented in the article or additional file.
References
Alarcon B, Vicedo B, Aznar R. PCR-based procedures for detection and quantification of Staphylococcus aureus and their application in food. J Appl Microbiol. 2006;100(2):352–64.
Becker RE, Bubeck Wardenburg J. Staphylococcus aureus and the skin: a longstanding and complex interaction. Skinmed. 2015;13(2):111–9.
Brakstad OG, Aasbakk K, Maeland JA. Detection of Staphylococcus aureus by polymerase chain reaction amplification of the nuc gene. J Clin Microbiol. 1992;30(7):1654–60.
Chen H, Zhang J, He Y, Lv Z, Liang Z, Chen J, Li P, Liu J, Yang H, Tao A, Liu X. Exploring the role of Staphylococcus aureus in inflammatory diseases. Toxins (basel). 2022;14:7.
Deng L, Schilcher K, Burcham LR, Kwiecinski JM, Johnson PM, Head SR, Heinrichs DE, Horswill AR, Doran KS. Identification of key determinants of Staphylococcus aureus vaginal colonization. Mbio. 2019;10:6.
Gao R, Zhong Z, Gao X, Jia L. Graphene oxide quantum dots assisted construction of fluorescent aptasensor for rapid detection of pseudomonas aeruginosa in food samples. J Agric Food Chem. 2018;66(41):10898–905.
Gherardi G. Staphylococcus aureus infection: pathogenesis and antimicrobial resistance. Int J Mol Sci. 2023;24:9.
Gill AAS, Singh S, Thapliyal N, Karpoormath R. Nanomaterial-based optical and electrochemical techniques for detection of methicillin-resistant Staphylococcus aureus: a review. Mikrochim Acta. 2019;186(2):114.
Hassan M, Vittal R, Raj JM, Chakraborty G. Loop-mediated isothermal amplification (LAMP): a sensitive molecular tool for detection of Staphylococcus aureus in meat and dairy product. Braz J Microbiol. 2022;53(1):341–7.
Huang Z, Yu X, Yang Q, Zhao Y, Wu W. Aptasensors for Staphylococcus aureus risk assessment in food. Front Microbiol. 2021;12: 714265.
Kadariya J, Smith TC, Thapaliya D. Staphylococcus aureus and staphylococcal food-borne disease: an ongoing challenge in public health. Biomed Res Int. 2014;2014: 827965.
Li Y, Wang J, Wang S, Wang J. Rolling circle amplification based colorimetric determination of Staphylococcus aureus. Mikrochim Acta. 2020;187(2):119.
Li D, Liu L, Huang Q, Tong T, Zhou Y, Li Z, Bai Q, Liang H, Chen L. Recent advances on aptamer-based biosensors for detection of pathogenic bacteria. World J Microbiol Biotechnol. 2021;37(3):45.
Linzner N, Loi VV, Fritsch VN, Antelmann H. Thiol-based redox switches in the major pathogen Staphylococcus aureus. Biol Chem. 2021;402(3):333–61.
Locke A, Fitzgerald S, Mahadevan-Jansen A. Advances in optical detection of human-associated pathogenic bacteria. Molecules. 2020;25:22.
Lyon LM, Doran KS, Horswill AR. Staphylococcus aureus fibronectin-binding proteins contribute to colonization of the female reproductive tract. Infect Immun. 2023;91(1): e0046022.
Rajapaksha P, Elbourne A, Gangadoo S, Brown R, Cozzolino D, Chapman J. A review of methods for the detection of pathogenic microorganisms. Analyst. 2019;144(2):396–411.
Song B, Wang J, Yan Z, Liu Z, Pan X, Zhang Y, Zhang X. Microfluidics for the rapid detection of Staphylococcus aureus using antibody-coated microspheres. Bioengineered. 2020;11(1):1137–45.
Suaifan G, Al Nobani SWA, Shehadeh MB, Darwish RM. Engineered colorimetric detection of Staphylococcus aureus extracellular proteases. Talanta. 2019;198:30–8.
Wang H, Hecht S, Kline D, Leber AL. Staphylococcus aureus and methicillin resistance detection directly from pediatric samples using PCR assays with differential cycle threshold values for corroboration of methicillin resistance. J Microbiol Methods. 2019;159:167–73.
Wang Y, Wang Z, Zhan Z, Liu J, Deng T, Xu H. Fluorescence detection of Staphylococcus aureus using vancomycin functionalized magnetic beads combined with rolling circle amplification in fruit juice. Anal Chim Acta. 2022;1189: 339213.
Xu X, Wang J, He Y, Wu X. Low-speed centrifugation based isolation and personal glucose meter assisted synchronous quantification of Pseudomonas aeruginosa in nursing home-acquired pneumonia. Anal Biochem. 2023;665: 115051.
Acknowledgements
The authors thank financial support from Hebei Province traditional Chinese medicine scientific research project (No. 2021286).
Funding
Funding is provided by Hebei Province traditional Chinese medicine scientific research project (Grant No. 2021286).
Author information
Authors and Affiliations
Contributions
YG performed related experiments, analyzed data, wrote original manuscript, supported the researched, designed the approach, assisted writing of the manuscript. CL, YW, and XY assisted data analysis, data collection and processing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The manuscript does not contain clinical or trial studies on patients, humans, or animals.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1
. Oligonucleotide Sequences used in this research. Table S2. A briefcomparison of the approach with former ones. Fig. S1. The SYBR Green I signals during the RCA process.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, Y., Li, C., Wang, Y. et al. Functional rolling circle amplification-based sensitive determination and low-speed centrifugation-based isolation of Staphylococcus aureus. J Anal Sci Technol 14, 45 (2023). https://doi.org/10.1186/s40543-023-00409-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-023-00409-x