
Abstract
Bile acids (BAs) are considered to be important diagnostic biomarkers to understand the pathophysiology of hepatobiliary and metabolic diseases. BAs regulate lipid and glucose metabolism by binding to farnesoid X receptor (FXR). To date, there were no reports on the effect of an exogenous FXR modulator, ivermectin (IVM), on the plasma BA profiles in rats. To explore the effect of IVM on plasma BA levels in rat, an ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC–MS/MS) method was developed and validated for simultaneous determination of seven major BAs in rat plasma. The developed method was selective, specific, accurate and precise for the quantification of plasma BAs. Sprague–Dawley rats were orally administered with IVM at a dose of 5 mg/kg once a day for 14 days and the plasma BAs were determined before and after IVM exposure using developed UHPLC–MS/MS method. Once-daily administration of IVM for 14 days resulted in significant reduction in cholic acid and deoxycholic acid levels while glycodeoxycholic acid and taurodeoxycholic acid levels were not affected. Interestingly, tauro-α-muricholic acid and tauro-β-muricholic acid levels were significantly increased. This study revealed that IVM has an important effect on plasma BA profiles in rats. This report provides an analytical methodology that can be applied to investigate the effect of drugs or pathophysiological factors on plasma BA levels.
Similar content being viewed by others

Introduction
Bile acids (BAs) are synthesized from cholesterol and then conjugated to glycine or taurine to form bile salts. These BAs are secreted into the bile duct and stored in gall bladder. Bile is released into the small intestine based on the stimuli received from food intake or other factors. The oral absorption of lipids, fat-soluble vitamins and other lipophilic nutrients present in the food is facilitated by the bile constituents, primarily by the bile salts. Most of the BAs are reabsorbed into the systemic circulation from the terminal ileum. Some BAs are converted to secondary BAs in the intestine by gut microbiota, some are excreted in the feces and some are metabolized to their respective sulfates and glucuronides and excreted in urine. All BAs that are reabsorbed into the systemic circulation enter into the liver and involve in the enterohepatic circulation (Chiang 2013; Ridlon et al. 2014; Dawson and Karpen 2015). BA homeostasis is maintained by various nuclear receptors and signaling pathways (Li and Chiang 2013). BA synthesis and their homeostasis is disrupted in diseases like primary biliary cholangitis, liver cirrhosis and non-alcoholic fatty liver disease. Altered total BA composition is observed in the inflammatory bowel syndrome, Crohn’s disease and ulcerative colitis. BAs, at higher concentrations, are reported to exhibit toxicity due to their surfactant like property and can contribute to hepatobiliary as well as intestinal disorders (Chiang 2017).
Farnesoid X receptor (FXR) is the crucial nuclear receptor expressed mainly in the liver and intestine which involves in the homeostasis and enterohepatic circulation of BAs. FXR also involves in the homeostasis of lipid and glucose and has become a promising target in the management of hyperlipidemia, diabetes, non-alcoholic fatty liver disease, cholestasis, primary biliary cholangitis and chronic inflammatory diseases of the liver and intestine (Massafra et al. 2018; Massafra and van Mil 2018; Wang et al. 2018a, b; Hou et al. 2019; Xi and Li 2020). Differential tissue expression and tissue specific functionalities of FXR is attracting more attention in exploring various ligands for its agonistic and/or antagonistic activities (Kim et al. 2007; Fiorucci et al. 2012; Kong et al. 2012).
Ivermectin (IVM) is a broad spectrum antiparasitic drug and had been identified as a highly selective FXR ligand. IVM was first categorized as a partial FXR agonist and later some studies reported its antagonistic activity to FXR. However, the FXR antagonistic activity of IVM was not well characterized in vivo (Jin et al. 2013; Jin et al. 2015; Qi et al. 2019, van de Wiel et al. 2019). IVM reduces serum cholesterol and glucose while increasing the insulin sensitivity by stimulating FXR in mice which causes reduction in body weight as well as hepatic lipid accumulation in diabetic mice. Since FXR is a BA-activated nuclear receptor and regulates BA homeostasis, it would be clinically important to study the in vivo effect of IVM in the regulation of BA homeostasis and quantitative measurement of important BAs including primary BAs cholic acid (CA), chenodeoxycholic acid (CDCA), secondary BAs deoxycholic acid (DCA) and its glyco/tauro- conjugates (G/T-DCA), murine specific tauro-α-muricholic acid (Tα-MCA) and tauro-β-muricholic acid (Tβ-MCA) in rats. Moreover, BAs such as DCA and GDCA were reported to be endogenous etiologic agents in gastrointestinal cancer (Bernstein et al. 2009). Further, GDCA and TDCA were significantly increased in nonalcoholic steatohepatitis patients with fibrosis and fibrosis mouse models (Xie et al. 2021). Therefore, specific and accurate estimation of these BAs can be highly advantageous in studying the effect of drugs like IVM on BA pool, in the diagnosis and detection of drug induced liver injury (DILI) as well as to predict BA related disorders (Schadt et al. 2016; Evangelakos et al. 2021, Saran et al. 2022). Though BAs are majorly present in bile, it is important to estimate the BAs in plasma as they are reabsorbed into the systemic circulation due to enterohepatic circulation (James et al. 2021).
The development of an LC–MS/MS method of BAs is of great interest in the liver diseases and other metabolic diseases research area. Due to the structural similarities in some of the BAs, separation and quantification of isobaric BAs is challenging. Several LC–MS/MS based methods have been developed for detection, separation and quantification of different BAs in the recent years. Detailed analysis of the currently reported methods reveals either one or more of the issues such as complex sample preparation methods like solid phase extraction or liquid–liquid extraction (Tribe et al. 2010; Suzuki et al. 2013; Wegner et al. 2017), lack of important murine specific BAs such as Tα/β-MCA (Luo et al. 2018; Wang et al. 2018a, b; Danese et al. 2020; Fu et al. 2020; Hu et al. 2020; Shiffka et al. 2020), large injection volumes (> 2 µL) (Xiang et al. 2010; Yang et al. 2017; Zhang et al. 2022), long sample analysis run times (> 10 min) (Choucair et al. 2020; Gomez et al. 2020; Reiter et al. 2021; Shafaei et al. 2021; Zhang et al. 2022), lack of absolute quantification of BAs (Wang et al. 2018a, b; Prinville et al. 2020; Mireault et al. 2023), lack of baseline separation of isobaric BAs such as CDCA/DCA or Tα-MCA/Tβ-MCA (Huang et al. 2011; Bathena et al. 2013; Sarafian et al. 2015). There is a need to develop a method to draw crucial conclusions by incorporation of important human and rodent specific BAs in a simultaneous method for mechanistic understanding of BA homeostasis in rats and human. Therefore, a simple, rapid and reliable UHPLC–MS/MS method was developed and validated to simultaneously quantify seven BAs (CA, CDCA, GDCA, TDCA, Tα-MCA, Tβ-MCA and DCA) (Fig. 1). Further, the developed method was used to evaluate the effect of IVM on these BA profiles in rat plasma. To our knowledge this is the first paper to study the effect of IVM on BA levels in rats which may lead to further exploration of IVM as an FXR modulator.

Chemical structures of the seven bile acids analysed in the current study
Experimental materials
Cholic acid (CA) and chenodeoxycholic acid (CDCA) were purchased from TCI Chemicals (India) Private Limited, Hyderabad, India. Deoxycholic acid (DCA), glycodeoxycholic acid (GDCA), taurodeoxycholic acid (TDCA) and dimethyl sulfoxide (DMSO) were purchased from Sigma Aldrich (Merck) Chemicals Private Limited, Bangalore, India. Tauro-α-muricholic acid (Tα-MCA), tauro-β-muricholic acid (Tβ-MCA), cholic acid-d4, deoxycholic acid-d4, glycodeoxycholic acid-d4, taurodeoxycholic acid-d4 and taurocholic acid-d4 were purchased from Cayman Chemical Company, MI, USA. IVM and dipotassium EDTA were purchased from Sisco Research Laboratories Private Limited, Hyderabad, India. LC–MS grade methanol, acetonitrile, water and formic acid were purchased from Biosolve India Limited, Hyderabad, India. Male Sprague–Dawley rats and rat pooled plasma were procured from Hylasco Biotechnology Private Limited, Hyderabad, India.
Instrumentation
An Ultra-High Performance Liquid Chromatographic system (Nexera 40D-XS, Shimadzu Corporation, Kyoto, Japan) coupled with a SCIEX QTRAP® 4500 mass analyser attached with Turbo V™ and electron-spray ionization probe (Sciex, MA, USA) was used in the analysis. Acquisition and integration of the chromatograms was performed using Analyst software (Version 1.7). Aqueous mobile phase was filtered through a 0.22 μm Millipore® (MA, USA) filtration membrane using a vacuum pump. The samples were prepared using calibrated micropipettes (Eppendorf, Chennai, India). Vortex mixer (Tarsons India Private Limited) and refrigerated centrifuge (Eppendorf 5810R, Eppendorf, Hamburg, Germany) were used for sample processing while deep freezer (Panasonic, Osaka, Japan) was used for storing the samples during method development, validation and in vivo studies.
Preparation of charcoal stripped plasma
Dextran-coated charcoal (C6241, Sigma Aldrich, 0.025% w/w or 10:1) was vortex mixed at 4 °C for 16 h in 50 mL preparation buffer (0.25 M sucrose, 1.5 mM MgCl2, 10 mM HEPES, pH 7.4) at a concentration of 0.25% (w/v). 40 mL of this mixture was centrifuged at 2000 rpm for 10 min to pellet down the charcoal. The supernatant was decanted and 40 mL of blank plasma was added to the charcoal pellet and vortex mixed at 4 °C for 16 h. The mixture was centrifuged at 2000 rpm for 10 min and the supernatant was collected into a fresh centrifuge tube and repeated the centrifugation one more time. Following the centrifugation, the supernatant was separated and passed through 0.2 µm filter to get clear stripped plasma. The stripped plasma was aliquoted and stored at − 80 °C.
Stock solutions, calibration standards and quality control samples preparation
Primary stock solutions of each of the seven BAs and their respective deuterated internal standards (IS) were prepared, separately, at a concentration of 1 mg/mL using DMSO as the solvent. These primary stock solutions were divided into 0.25 mL aliquots and stored at − 80 °C to avoid repeated freezing and thawing. From each of the primary stock solutions of the seven BAs, 25 µL was drawn and added into a fresh centrifuge tube and diluted with 825 µL of diluent (methanol: water (1:1)) to yield a pooled stock solution containing each BA at concentration of 25,000 ng/mL. The pooled stock solution was used to prepare further working stock solutions to spike the charcoal stripped plasma to construct nine-point calibration curve for each of the seven BAs. The working stock solutions were prepared in the range of 25.03–25,000 ng/mL (25.03, 50.06, 250.31, 1251.56, 5006.25, 12515.63, 20025, 22500 and 25000 ng/mL) with the same diluent used in the preparation of pooled stock solution. The calibration curve range and calibration points values were set same for all the seven BAs. Each calibration curve standard, in the calibration curve range, was prepared separately by spiking 2 µL of appropriate working stock solution to 48 µL of charcoal stripped blank plasma. The calibration curve was developed in the range of 1–1000 ng/mL using nine calibration standards (1, 2, 10.01, 50.06, 200.25, 500.63, 801, 900 and 1000 ng/mL) for each of the BAs. The lower quality control (LQC), medium quality control (MQC) and higher quality control (HQC) standards were set at 3 ng/mL, 500 ng/mL and 840 ng/mL, respectively. The pooled internal standard working solution (2500 ng/mL) was prepared by aliquoting 2.5 µL of each of the internal standard primary stock solutions (2.5 µL × 5 = 12.5 µL) to a fresh centrifuge tube and diluting with 987.5 µL of diluent (1:1 mixture of methanol and water). All stock solutions were stored at − 20 °C until use.
Collection of rat plasma
Under light isoflurane anesthesia, 200 µL of blood was collected from retro-orbital plexus using rat bleeding capillary tubes. The blood was collected into fresh microcentrifuge tubes containing anticoagulant solution (200 mM K2EDTA in normal saline) at a concentration of 2% v/v (in the blood collected). The blood samples were kept in the wet ice until centrifugation. The blood samples were centrifuged in a cooling centrifuge at 10000 rpm for 10 min at 4 °C and the supernatant plasma was collected carefully, labelled and stored at -80 °C until UHPLC–MS/MS analysis.
Sample preparation
A simple and convenient protein precipitation method was utilized for plasma sample preparation. Ice-cold acetonitrile containing 100 ng/mL of each of the internal standards was used to precipitate the proteins. The protein precipitating solvent was prepared just before to use by adding 5 µL of each of the internal standard primary stock solutions (1 mg/mL) (5 µL × 5 = 25 µL) into 49.975 mL of ice-cold acetonitrile. The plasma sample preparation was done as follows. 50 µL of plasma was added to a fresh 0.5 mL centrifuge tube and 200 µL of ice-cold acetonitrile containing internal standards was added to precipitate the proteins. The tube was vortexed for 2 min and then centrifuged at 10000 rpm for 10 min at 4 °C and the supernatant was transferred into 96-well plate for UHPLC–MS/MS analysis. In the case of samples whose concentrations were higher than the upper limit of quantification (ULOQ), the samples were diluted with acetonitrile as required and centrifuged for 5 min at 10000 rpm at 4 °C. The supernatant obtained was subjected to UHPLC–MS/MS analysis.
Method development
Individual BAs at 100 ng/mL concentration were introduced into the mass spectrometer by infusion syringe pump to identify and optimize compound specific mass spectrometric parameters. The mass analyzer conditions were optimized by studying the effect of ion-source temperature, ion-spray voltage, flow rates of curtain gas, nebulizer gas and drying gas, declustering potential, collision energy, entry and exit potential etc. Pseudo-Multiple reaction monitoring (Pseudo-MRM) method was employed for quantification of the BAs. In the preliminary chromatographic method development trials, simultaneous separation of seven BAs and the internal standards was achieved by optimizing various LC conditions like mobile phase composition, flow rate, column chemistry and oven temperature as well as peak shape/properties, separation and intensity. Different ammonium salts including acetate, formate, carbonate and bicarbonate along with pH modifiers were tried as the aqueous phase component. The effect of methanol and acetonitrile as the non-aqueous phase component of the mobile phase was also studied. Further, the mobile phase gradient, flow rate and column temperature were also optimized.
Chromatographic and mass spectrometric conditions
The chromatographic separation of the seven bile salts was achieved on ACQUITY UPLC® HSS T3 column (1.8 µm, 2.1 × 100 mm) (Waters India Private Limited, Bangalore, India). The mobile phase consisted of eluent A (water containing 0.1% v/v formic acid) and eluent B (100% acetonitrile). The mobile phase was pumped in a gradient elution mode as per the following program: at 0 min—5% B, 0.5 min—5% B, 1 min—35% B, 2 min—30% B, 3 min—30% B, 4 min—40% B, 5 min—45% B, 6 min—70% B, 7 min—100% B, 8 min—100% B, 9 min—5% B and at 10 min—5% B. The mobile phase flow rate was fixed at 0.45 mL/min. 2 µL of sample was injected for analysis. The column temperature and autosampler temperature were set at 40 °C and 15 °C, respectively. The column was pre-saturated with the same mobile phase gradient program and the chromatography system was stabilized for 60 min by observing the baseline prior to actual analysis. The mass spectrometer was operated with the electron spray ionization probe in negative polarity. The ion source temperature (TEM) was set to 500 °C. The curtain gas (CUR) was set at 35 L/h, collision gas (CAD) was set to medium, ion spray voltage (IS) at 4500 V, nebulizing gas (GS1) at 50 psi and drying gas (GS2) at 45 psi. Nitrogen was used as both nebulizing as well as drying gas. Pseudo-MRM was used in the method development as well as the analysis of the samples obtained from the in vivo studies. Peak integration was done for each BA and their respective internal standard at their retention times. The ratio of peak area of a specific analyte to its corresponding internal standard was calculated.
Method validation
Since BAs are endogenous molecules, a surrogate matrix (charcoal stripped plasma) was employed in the current method for method validation. The method was validated for selectivity, specificity, carryover, sensitivity, linearity, matrix effect, recovery, accuracy and precision, dilution integrity, reinjection reproducibility and stability according to FDA Guidance for Industry-Bioanalytical Method Validation and Study Sample Analysis (Guideline 2022).
Selectivity was established with lower limit of quantification (LLOQ) samples and blank matrix, while specificity was evaluated by comparing zero, LLOQ and only BA samples for any interference at the retention time of each of the BAs. Carryover effect in the blank samples following the injection of ULOQ sample was also evaluated. The limit of any interference in the above three method validation parameters was set to < 20% and < 5% for analytes and internal standards, respectively. A nine-point calibration curve ranging from 1 to 1000 ng/mL was constructed in stripped plasma in triplicate and least-square linear regression with 1/x2 weighting factor was used to determine calibration curve equations with higher R2PRESS values. The mean accuracy of the back calculated concentration should be within ± 20% and ± 15% for LLOQ and all the other calibration standards, respectively. Intra-day (n = 12) and inter-day (n = 18) accuracy (%bias) and precision (percent coefficient of variation, %CV) were evaluated using LLOQ, LQC, MQC and HQC samples with the limits of ± 15% and ≤ 15% for accuracy and precision, respectively. Extraction recovery studies were conducted using LQC, MQC, HQC and IS (100 ng/mL) samples in stripped plasma in replicates (n = 6). The pre-extracted samples were compared with the post-extracted samples, where > 50% recovery was expected. Matrix effect was evaluated for LQC, HQC and IS samples in stripped plasma in replicates (n = 6) and compared with neat standard samples of the same concentrations. The accuracy and precision should be ± 15% and ≤ 15%, respectively. Stability of the samples was assessed by subjecting LQC and HQC samples (n = 6) to different conditions viz., auto-sampler/injector stability (up to 24 h at 15 °C), bench-top stability, freeze–thaw stability (5 cycles) and long-term stability at − 20 °C and − 80 °C (18 days) and quantified using a freshly prepared calibration curve. Stock solution and working stock solution stabilities of BAs at LQC and HQC levels as well as working stock solution stability of IS at room temperature were assessed in replicates (n = 6) for 24 h and compared with the corresponding freshly prepared samples. The percentage deviation and precision compared to time zero samples should be within ± 15% and ≤ 15%, respectively. LQC, MQC and HQC samples in replicate (n = 6) were subjected to reinjection reproducibility assessment under a calibration curve. A sample containing 2000 ng/mL concentration of each of the BAs was diluted with stripped plasma by 5-folds and 20-folds to evaluate the dilution integrity against a calibration curve. The accuracy and precision should be within ± 15% and < 15%, respectively, for both reinjection reproducibility and dilution integrity tests.
Effect of IVM on plasma concentrations of BAs
The protocol for animal experiments was reviewed and approved by our institutional animal ethics committee (IAEC) prior to the study. Male rats of 6–8 weeks’ age having 220–250 gm body weight were procured and quarantined for 5 days in our institute’s animal house. The rats were acclimatized to the conditions in the animal house where the temperature and relative humidity were maintained at 22 ± 1 °C and 50 ± 10%, respectively, with 12 h dark–light cycle. The rats were provided free access to regular rat chow diet and water. IVM formulation was prepared by dissolving the drug in a solvent containing the mixture of 2% DMSO, 2% tween 80 and 96% purified water. Following the quarantine period, IVM formulation was administered to rats (n = 6) through oral route using gavage, at a dose of 5 mg/kg and dosing volume of 5 mL/kg body weight. Rats were fasted for at least 4 h prior to the dosing of IVM formulation on each day of dosing. The animals were administered with IVM formulation once a day at a fixed time (11:00 AM on every day) for 14 consecutive days. Blood samples were collected by retro orbital plexus method at predose (1 h before administering the first dose) and at 2 h after dosing on the day-14. The blood samples were processed immediately after collection and the plasma samples were labelled properly and stored at − 80 °C until UHPLC–MS/MS analysis.
Statistical analysis
Least-square simple linear regression analysis was employed to determine the calibration equation. The difference in the BA concentrations in the samples collected at predose and day-14 was analyzed by using a paired t-test (two-sided) at 5% significance level (α = 0.05).
Results
Method development
Mass spectrometric conditions were optimized for each of the BAs and all the internal standards in negative ESI mode. Nebulizer gas, heat block temperature and desolvation gases were optimized simultaneously. For each of the BAs and internal standards, Q1 masses were detected, and the DPs were optimized. Pseudo-MRM was applied in the method for quantification of individual BAs. The optimized MRM details of BAs and internal standards are presented in Table 1. The summary of the trials conducted on two different instruments viz., Shimadzu Prominence UFLC coupled with Shimadzu 8040 triple quadrupole mass analyser (Additional file 1: Table S1 and Fig. S1) and Agilent 1290 Infinity II Quaternary System coupled to 6470 Triple Quad LC/MS System (Additional file 1: Table S2 and Fig. S2) prior to achieving the optimized method conditions is presented in the Additional file 1. The optimized method was developed using UHPLC–MS/MS (Nexera 40D-XS-SCIEX QTRAP® 4500) system. Chromatographic separation was performed on HSS-T3 column (1.8 µm, 2.1 × 100 mm) using mobile phase composition containing water with 0.1% v/v formic acid as the aqueous phase (eluent A) and acetonitrile as the organic phase (eluent B) in a gradient program. The optimized method conditions resulted in good peak properties, separation and peak intensity for the analytes. Considering detector sensitivity and reproducibility, the injection volume was set as 2 µL. The specific identification of isobaric BAs such as CDCA/DCA and Tα-MCA/Tβ-MCA were confirmed by comparing their retention times in mixture and individual BA injections with the same method conditions.
Method validation
The developed method was validated as per the FDA guidelines for its intended purpose to quantitatively determine BAs in rat plasma. Charcoal stripped blank rat plasma was used as a surrogate matrix in the current UHPLC–MS/MS method to avoid the possible effect of endogenous BAs in the analysis. The complete stripping of the from the plasma was confirmed by the absence of selected BAs at their specific retention times in the chromatogram of stripped blank plasma.
Selectivity, specificity and carryover
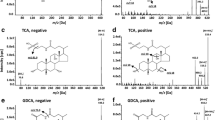
Pseudo-MRM transitions and efficient chromatographic separation enabled the highly selective and specific analysis of BAs in plasma. Typical chromatograms obtained from four scenarios as mentioned in methods were analyzed and no significant interferences were observed either for analytes or for internal standards at the specified concentration levels at their respective retention times. Differentiation of isobaric compounds like CDCA-DCA and TαMCA-TβMCA was achieved by separating them at different retention times. However, some other isobaric compounds containing identical molecular weights to that of selected BAs were observed in the unstripped blank plasma at different retention times. No interference of these compounds was observed during the analysis. Accordingly, no analyte response was observed in the subsequent blank samples after the ULOQ injections in the carryover studies. Chromatograms of stripped blank plasma and a calibration curve standard are presented in Figs. 2 and 3, respectively. The unstripped plasma sample chromatogram was shown in Additional file 1: Figure S3.

Chromatogram of charcoal stripped rat blank plasma representing respective pseudo-MRM transitions of: A Cholic acid (407.20/407.20), B Chenodeoxycholic acid (391.20/391.20), C Deoxycholic acid (391.3/391.3), D Glycodeoxycholic acid (448.30/448.30), E Taurodeoxycholic acid (498.20/498.20), F Tauro-α-muricholic acid (514.10/514.10), G Tauro-β-muricholic acid (514.20/514.20)

Chromatogram of seven bile acids (each at a concentration of 500 ng/mL) and their respective internal standards (each at a concentration of 100 ng/mL) in stripped plasma
Linearity
The least-square linear regression equations constructed using ‘1/x2’ as the weighting factor yielded higher adjusted R2 (> 0.9965) and R2PRESS (> 0.9936) values compared to unweighted or any other weighting factors (1/x or 1/y or 1/y2). The slope and intercept values of the calibration equations, obtained from the least-square linear regression analysis with ‘1/x2’ as the weighting factor, of all the seven BAs are presented in Additional file 1: Table S3. All the seven BAs showed a linear response in the calibration range of 1–1000 ng/mL. Higher adjusted R2 (> 0.9965) and R2PRESS (> 0.9936) and low standard error of estimates (< 23.57) for the calibration equations of all the seven BAs indicate the ability of the regression equations to predict the concentrations of unknown samples as close as possible to the actual concentrations. The samples which exceed the calibration range were reanalyzed by diluting them to five folds with diluent.
Accuracy and precision
The results obtained from accuracy and precision studies of the developed method are presented in Table 2. The accuracy values, expressed in terms of %bias, for all the BAs across all the samples (LLOQ, LQC, MQC and HQC) were between − 8.77 and 13.5%, suggesting that the method is accurate for quantification of the seven BAs in plasma. The intra-day precision, expressed in terms of %CV, for all the BAs across all the samples (LLOQ, LQC, MQC and HQC) was between 3.04 and 14.23%, while the intra-day precision was between 3.14 and 12.47%. The %CV values for the intra-day and inter-day precision studies were well within the acceptable limit of < 15%, indicating that the developed method is precise for quantification of the seven BAs.
Extraction recovery and matrix effect
The extraction recoveries of BAs following the protein precipitation from the striped plasma samples at three different QC levels (LQC, MQC and HQC levels) and internal standards at 100 ng/mL are presented in Table 3. The mean recovery values of the seven BAs varied between 74.14% (for DCA at LQC level) to 87.86% (for Tα-MCA at HQC level) with %CV of not more than 13.25% for any of the BAs at any QC level. The mean recovery values of internal standards varied between 81.22% (for d4-DCA) to 92.98% (for d4-TCA) with %CV of not more than 4.72% for any of the internal standards.
The overall accuracy and precision of the matrix effect on the analysis of the seven BAs at two QC levels is presented in Table 4. The highest %bias (accuracy) was 13.86% (for TDCA) and the precision (%CV) was not more than 11.39% for the matrix effect on any of the BAs, at any of the two QC levels. For each of the seven BAs, at two different QC levels, no significant suppression or enhancement was observed in the response. Therefore, it can be inferred that there is no matrix effect on the analysis of the seven BAs. Similarly, no significant matrix effect was also observed in the analysis of internal standards. The highest %bias for the matrix effect on any of the internal standards was − 13.55% (for d4-DCA) and the precision (%CV) was not more than 13.81% for any of the internal standards. These results suggest that the method exhibited no significant matrix effect.
Stability, reinjection reproducibility and dilution integrity
Results obtained from the stability studies of the seven BAs evaluated using three QC standards (LQC, MQC and HQC) when subjected to different stress conditions were shown in Additional file 1: Table S4. All the seven BAs were stable in the stripped plasma when subjected to five cycles of freeze–thaw (− 20 °C to room temperature) on five consecutive days and long-term stability for a period of 18 days when stored at − 20 °C as well as − 80 °C. No significant degradation was observed in the processed samples when placed on bench top at room temperature (25 ± 2 °C) or in the autosampler racks (15 ± 0.5 °C) for a period of 24 h. The primary stock solutions and the working standard solution of the BAs and the working standard solutions of internal standards were stable for 24 h when stored at room temperature (25 ± 2 °C). The %deviation was within the acceptable limits of ± 15% for the stock solutions of BAs and internal standards used in the study. Stabilities of BAs at different conditions and stock solution stabilities are reported in Additional file 1: Tables S4 and S5, respectively.
The mean accuracy and precision values of the reinjected QC standards (LQC, MQC and HQC) are given in Additional file 1: Table S6. The accuracy was ranging from − 4.70% (for DCA at MQC) to 5.97% (for Tβ-MCA at LQC) and precision was less than 8.5% across all QC levels for all BAs. The accuracy and precision values of the dilution integrity samples (5-folds and 20-folds dilution) were within the specifications of ± 15% and < 15% of the nominal concentration. The results obtained from dilution integrity studies are presented are given in Additional file 1: Table S7.
Effect of IVM on plasma concentrations of BAs
In the in vivo study, effect of oral administration of IVM (at 5 mg/kg body weight, once-daily for 14 days) on the plasma concentrations of seven BAs was studied. A comparison of plasma concentrations of the seven BAs before and after IVM treatment is shown in Fig. 4. The plasma concentrations of CA were significantly (P = 0.003) lesser in the day-14 samples compared to the predose samples. There was no significant (P = 0.352) difference in the CDCA concentrations before and after treatment of IVM. However, a statistically (P = 0.0324) significant reduction was observed in the DCA concentrations from predose to day-14. Statistically no significant difference was observed in the concentrations of GDCA and TDCA at predose and day-14 of IVM treatment. Interestingly, in contrast to the CA or DCA results where the concentrations decreased at day-14, there was a significant increase in the concentrations of Tα-MCA (P = 0.02) and Tβ-MCA (P = 0.045) at day-14 compared to predose. Overall, oral administration of IVM (once-daily for 14 days) resulted in significant reduction in CA and DCA levels; significant increase in the Tα-MCA and Tβ-MCA levels and no significant change in the CDCA levels.

Plasma concentrations of seven bile acids before and after oral dosing of IVM in rats (5 mg/kg once-daily for 14-days, n = 6). ns = not significant, * = significant (0.01 < P < 0.05), ** = very significant (0.001 < P < 0.01)
Discussion
The LC–MS/MS based methods, reported in the literature, for detection, separation and quantification of BAs in the recent years lack validated method for this specific set of human and murine BAs. This provoked us to develop and validate a simultaneous UHPLC–MS/MS method which can be advantageous to the research community for studying metabolic disease mechanisms in relation to BA accumulation or in regulation of BAs homeostasis. Development and validation of LC–MS/MS method for simultaneous estimation of BAs is challenging due to structural similarities and presence of isobaric BAs. The cross talk between the isobaric BAs is also a major challenge in their absolute quantification.
In the current method, charcoal stripped plasma was used instead of normal plasma to avoid the interference of endogenous BAs. Further, the use of stable isotope labelled internal standards enhances the reliability of the method for accurate and precise quantification of analytes. The developed method offers the advantage of uniform calibration curve range (1–1000 ng/mL) having same calibration points for all the seven BAs. In addition, the developed method employs a simple, cost and time saving approach of protein precipitation for the sample preparation instead of solid phase extraction or liquid–liquid extraction methods. The method offers higher sensitivity with the LLOQ values of 1 ng/mL with baseline separation of isobaric BAs with short run time of 10 min without any requirement of equilibration of column between the runs. Due to the high sensitivity of the developed method even with a small injection volume of 2 µL, the current method would avoid unnecessary deposition of the sample in the mass analyzer which improves the life of the instrument. The developed method was validated for all the parameters as per the regulatory guidelines which was found missing in many of the reported methods.
The synthesis of BAs in humans and murine happen via two pathways, the classical pathway and alternative pathway. Classical pathway produces two major BAs, CA and CDCA, while alternative pathway produces only CDCA. In murine, CDCA is further converted into α-MCA and β-MCA, which upon conjugation with taurine produces Tα-MCA and Tβ-MCA as the major BAs. Meanwhile, CDCA, DCA and CA are the natural FXR agonists with order of potency of CDCA > DCA > CA, whereas Tα-MCA and Tβ-MCA were identified as natural FXR antagonists (Hofmann 1999; Xue et al. 2021). Once-daily dosing of IVM for 14 days reduced the plasma concentration of CA and DCA while it increased the plasma concentrations of Tα-MCA and Tβ-MCA. Based on the FXR negative feedback mechanism (Chiang 2015; Jia et al. 2021), we assume that IVM, could be acting as an FXR agonist, inhibiting CYP7A1 enzyme (rate limiting enzyme in classical BAs synthesis) and thereby reducing CA synthesis in the classical pathway. As DCA is the secondary metabolite derived from CA, the levels of DCA were also reduced in line with the CA levels. IVM did not cause a significant change in the concentration of CDCA. This could be possibly due to the balancing out effect of IVM on two different pathways. On one hand IVM may be acting as FXR agonist which causes the inhibition of CYP7A1/CYP8B1 enzyme (and thereby the classical pathway) and reduction in the synthesis of CDCA while on the other hand IVM may be activating the CYP27A1/CYP7B1 enzymes involved in the synthesis of CDCA by alternative pathway. The increase in concentrations of Tα-MCA and Tβ-MCA could be due to the possible activation of CYP2C70 enzyme which is responsible for the conversion of CDCA (produced from either classical or alternative pathway) into α/β-MCA and the eventual formation of their taurine conjugates. The above results indicate that IVM has an opposite effect on classical and alternative BA synthesis pathways by inhibition and activation, respectively, and result in significant changes in the BA haemostasis. However, further studies are required to elucidate the exact mechanism involved in inhibition of the classical pathway and activation of the alternative pathway of BAs synthesis by IVM.
Conclusions
A simple, sensitive, reliable and reproducible UHPLC–MS/MS method was developed and validated for the quantitative analysis of seven important BAs in charcoal stripped rat plasma. The developed method had a uniform calibration curve range of 1–1000 ng/mL for all the BAs. The isobaric BAs pairs of Tα-MCA/Tβ-MCA and CDCA/DCA were well resolved for their accurate and precise quantification. The method was successfully applied to study the effect of IVM on the plasma concentration of the seven BAs. The results obtained suggest that IVM significantly affect the plasma concentration of the seven major BAs by acting both on the classical pathway and alternate pathway of BA synthesis. Further, this study provides useful insights regarding the FXR modulation in relation to BA homeostasis in rats upon treatment with IVM. The described UHPLC–MS/MS method can be further employed in the future preclinical studies to discover the detailed mechanisms of IVM or any other molecule of interest on BA homeostasis in rats.
Availability of data and materials
All data generated or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BA:
-
Bile acid
- FXR:
-
Farnesoid X receptor
- IVM:
-
Ivermectin
- CA:
-
Cholic acid
- CDCA:
-
Chenodeoxycholic acid
- DCA:
-
Deoxycholic acid
- GDCA:
-
Glycodeoxycholic acid
- TDCA:
-
Taurodeoxycholic acid
- Tα-MCA:
-
Tauro alpha muricholic acid
- Tβ-MCA:
-
Tauro beta muricholic acid
- SD:
-
Sprague–Dawley
- UHPLC–MS/MS:
-
Ultra-high performance liquid chromatography–tandem mass spectrometer
- IS:
-
Internal standard
- FDA:
-
Food and drugs administration
- QC:
-
Quality control
- IAEC:
-
Institutional animal ethics committee
- ESI:
-
Electrospray ionization
- MRM:
-
Multiple reaction monitoring
- PRESS:
-
Predicted residual error sum of squares
References
Bathena SP, Mukherjee S, Olivera M, Alnouti Y. The profile of bile acids and their sulfate metabolites in human urine and serum. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;942–943:53–62.
Bernstein H, Bernstein C, Payne CM, Dvorak K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol. 2009;15(27):3329–40.
Chiang JY. Bile acid metabolism and signaling. Compr Physiol. 2013;3(3):1191–212.
Chiang JY. Negative feedback regulation of bile acid metabolism: impact on liver metabolism and diseases. Hepatology. 2015;62(4):1315–7.
Chiang JY. Recent advances in understanding bile acid homeostasis. F1000Res. 2017;6:2029.
Choucair I, Nemet I, Li L, Cole MA, Skye SM, Kirsop JD, Fischbach MA, Gogonea V, Brown JM, Tang WHW, Hazen SL. Quantification of bile acids: a mass spectrometry platform for studying gut microbe connection to metabolic diseases. J Lipid Res. 2020;61(2):159–77.
Danese E, Negrini D, Pucci M, De Nitto S, Ambrogi D, Donzelli S, Lievens PM, Salvagno GL, Lippi G. Bile acids quantification by liquid chromatography-tandem mass spectrometry: method validation, reference range, and interference study. Diagnostics. 2020;10(7):462.
Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56(6):1085–99.
Evangelakos I, Heeren J, Verkade E, Kuipers F. Role of bile acids in inflammatory liver diseases. Semin Immunopathol. 2021;43(4):577–90.
Fiorucci S, Zampella A, Distrutti E. Development of FXR, PXR and CAR agonists and antagonists for treatment of liver disorders. Curr Top Med Chem. 2012;12(6):605–24.
Fu X, Xiao Y, Golden J, Niu S, Gayer CP. Serum bile acids profiling by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and its application on pediatric liver and intestinal diseases. Clin Chem Lab Med. 2020;58(5):787–97.
Gomez C, Stucheli S, Kratschmar DV, Bouitbir J, Odermatt A. Development and validation of a highly sensitive LC-MS/MS method for the analysis of bile acids in serum, plasma, and liver tissue samples. Metabolites. 2020;10(7):282.
Guideline, I. H. (2022) Bioanalytical method validation and study sample analysis M10. ICH Harmonised Guideline: Geneva, Switzerland.
Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159(22):2647–58.
Hou Y, Fan W, Yang W, Samdani AQ, Jackson AO, Qu S. Farnesoid X receptor: an important factor in blood glucose regulation. Clin Chim Acta. 2019;495:29–34.
Hu T, An Z, Shi C, Li P, Liu L. A sensitive and efficient method for simultaneous profiling of bile acids and fatty acids by UPLC-MS/MS. J Pharm Biomed Anal. 2020;178:112815.
Huang J, Bathena SP, Csanaky IL, Alnouti Y. Simultaneous characterization of bile acids and their sulfate metabolites in mouse liver, plasma, bile, and urine using LC-MS/MS. J Pharm Biomed Anal. 2011;55(5):1111–9.
James SC, Fraser K, Young W, Heenan PE, Gearry RB, Keenan JI, Talley NJ, Joyce SA, McNabb WC, Roy NC. Concentrations of fecal bile acids in participants with functional gut disorders and healthy controls. Metabolites. 2021;11(9):612.
Jia W, Wei M, Rajani C, Zheng X. Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein Cell. 2021;12(5):411–25.
Jin L, Feng X, Rong H, Pan Z, Inaba Y, Qiu L, Zheng W, Lin S, Wang R, Wang Z, Wang S, Liu H, Li S, Xie W, Li Y. The antiparasitic drug ivermectin is a novel FXR ligand that regulates metabolism. Nat Commun. 2013;4:1937.
Jin L, Wang R, Zhu Y, Zheng W, Han Y, Guo F, Ye FB, Li Y. Selective targeting of nuclear receptor FXR by avermectin analogues with therapeutic effects on nonalcoholic fatty liver disease. Sci Rep. 2015;5:17288.
Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48(12):2664–72.
Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56(3):1034–43.
Li T, Chiang JY. Nuclear receptors in bile acid metabolism. Drug Metab Rev. 2013;45(1):145–55.
Luo L, Aubrecht J, Li D, Warner RL, Johnson KJ, Kenny J, Colangelo JL. Assessment of serum bile acid profiles as biomarkers of liver injury and liver disease in humans. PLoS ONE. 2018;13(3):e0193824.
Massafra V, van Mil SWC. Farnesoid X receptor: a “homeostat” for hepatic nutrient metabolism. Biochim Biophys Acta Mol Basis Dis. 2018;1864(1):45–59.
Massafra V, Pellicciari R, Gioiello A, van Mil SWC. Progress and challenges of selective farnesoid X receptor modulation. Pharmacol Ther. 2018;191:162–77.
Mireault M, Prinville V, Ohlund L, Sleno L. Semi-targeted profiling of bile acids by high-resolution mass spectrometry in a rat model of drug-induced liver injury. Int J Mol Sci. 2023;24(3):2489.
Prinville V, Ohlund L, Sleno L. Targeted analysis of 46 Bile acids to study the effect of acetaminophen in rat by LC-MS/MS. Metabolites. 2020;10(1):26.
Qi W, Clark JM, Suvorov A, Park Y. Ivermectin decreases triglyceride accumulation by inhibiting adipogenesis of 3T3-L1 preadipocytes. Food Chem Toxicol. 2019;131:110576.
Reiter S, Dunkel A, Metwaly A, Panes J, Salas A, Haller D, Hofmann T. Development of a highly sensitive ultra-high-performance liquid chromatography coupled to electrospray ionization tandem mass spectrometry quantitation method for Fecal bile acids and application on Crohn’s disease studies. J Agric Food Chem. 2021;69(17):5238–51.
Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30(3):332–8.
Sarafian MH, Lewis MR, Pechlivanis A, Ralphs S, McPhail MJ, Patel VC, Dumas ME, Holmes E, Nicholson JK. Bile acid profiling and quantification in biofluids using ultra-performance liquid chromatography tandem mass spectrometry. Anal Chem. 2015;87(19):9662–70.
Saran C, Sundqvist L, Ho H, Niskanen J, Honkakoski P. Novel bile acid-dependent mechanisms of hepatotoxicity associated with tyrosine kinase inhibitors. J Pharmacol Exp Therap. 2022;380(2):114–25.
Schadt HS, Wolf A, Pognan F, Chibout SD, Merz M, Kullak-Ublick GA. Bile acids in drug induced liver injury: Key players and surrogate markers. Clin Res Hepatol Gastroenterol. 2016;40(3):257–66.
Shafaei A, Rees J, Christophersen CT, Devine A, Broadhurst D, Boyce MC. Extraction and quantitative determination of bile acids in feces. Anal Chim Acta. 2021;1150:338224.
Shiffka SJ, Jones JW, Li L, Farese AM, MacVittie TJ, Wang H, Swaan PW, Kane MA. Quantification of common and planar bile acids in tissues and cultured cells. J Lipid Res. 2020;61(11):1524–35.
Suzuki Y, Kaneko R, Nomura M, Naito H, Kitamori K, Nakajima T, Ogawa T, Hattori H, Seno H, Ishii A. Simple and rapid quantitation of 21 bile acids in rat serum and liver by UPLC-MS-MS: effect of high fat diet on glycine conjugates of rat bile acids. Nagoya J Med Sci. 2013;75(1–2):57–71.
Tribe RM, Dann AT, Kenyon AP, Seed P, Shennan AH, Mallet A. Longitudinal profiles of 15 serum bile acids in patients with intrahepatic cholestasis of pregnancy. Am J Gastroenterol. 2010;105(3):585–95.
van de Wiel SMW, Bijsmans I, van Mil SWC, van de Graaf SFJ. Identification of FDA-approved drugs targeting the farnesoid x receptor. Sci Rep. 2019;9(1):2193.
Wang H, He Q, Wang G, Xu X, Hao H. FXR modulators for enterohepatic and metabolic diseases. Expert Opin Ther Pat. 2018a;28(11):765–82.
Wang X, Wang F, Lu Z, Jin X, Zhang Y. Semi-quantitative profiling of bile acids in serum and liver reveals the dosage-related effects of dexamethasone on bile acid metabolism in mice. J Chromatogr B Analyt Technol Biomed Life Sci. 2018b;1095:65–74.
Wegner K, Just S, Gau L, Mueller H, Gerard P, Lepage P, Clavel T, Rohn S. Rapid analysis of bile acids in different biological matrices using LC-ESI-MS/MS for the investigation of bile acid transformation by mammalian gut bacteria. Anal Bioanal Chem. 2017;409(5):1231–45.
Xi Y, Li H. "Role of farnesoid X receptor in hepatic steatosis in nonalcoholic fatty liver disease. Biomed Pharmacother. 2020;121:109609.
Xiang X, Han Y, Neuvonen M, Laitila J, Neuvonen PJ, Niemi M. High performance liquid chromatography-tandem mass spectrometry for the determination of bile acid concentrations in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878(1):51–60.
Xie G, Jiang R, Wang X, Liu P, Zhao A, Wu Y, Huang F, Liu Z, Rajani C, Zheng X, Qiu J, Zhang X, Zhao S, Bian H, Gao X, Sun B, Jia W. Conjugated secondary 12alpha-hydroxylated bile acids promote liver fibrogenesis. EBioMedicine. 2021;66:103290.
Xue R, Su L, Lai S, Wang Y, Zhao D, Fan J, Chen W, Hylemon PB, Zhou H. Bile acid receptors and the gut-liver axis in nonalcoholic fatty liver disease. Cells. 2021;10(11):2806.
Yang T, Shu T, Liu G, Mei H, Zhu X, Huang X, Zhang L, Jiang Z. Quantitative profiling of 19 bile acids in rat plasma, liver, bile and different intestinal section contents to investigate bile acid homeostasis and the application of temporal variation of endogenous bile acids. J Steroid Biochem Mol Biol. 2017;172:69–78.
Zhang X, Liu X, Yang J, Ren F, Li Y. Quantitative profiling of bile acids in feces of humans and rodents by ultra-high-performance liquid chromatography–quadrupole time-of-flight mass spectrometry. Metabolites. 2022;12(7):633.
Acknowledgements
The authors are thankful to Central Animal Facility, Birla Institute of Technology and Science-Pilani, Hyderabad Campus, Hyderabad, Telangana, India.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
TR conceived and designed the experiments; TR, GT performed the experiments, analysed the data; TR contributed reagents/materials/analysis tools; TR, GT wrote the paper. PR supervised, reviewed and edited the paper. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal experiments described in our study were approved by Institutional animal ethics committee (IAEC), BITS-Pilani, Hyderabad Campus, India.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supporting Information and Supplementary Tables & Figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mullapudi, T.V.R., Ravi, P.R. & Thipparapu, G. Simultaneous determination of seven bile acids to study the effect of ivermectin on their plasma levels in rat by UHPLC–MS/MS. J Anal Sci Technol 14, 44 (2023). https://doi.org/10.1186/s40543-023-00408-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-023-00408-y