
Abstract
Interleukin-33 (IL-33) is an IL-1 family protein that induces a type-2 immune response. IL-33 is constitutively expressed in epithelial cells and released in response to the cell damage or stimulation by an allergen. The secreted protein is activated when the N-terminal domain is cleaved by a protease, and the active form signals downstream immune cells, such as eosinophils, by binding to the heterodimeric ST2:IL-1RAcP receptor complex on the cell surface. The binding stimulates an inflammatory response, and the abnormal inflammatory response can cause allergic diseases such as atopic dermatitis and asthma. Inhibition of the interaction between IL-33 and ST2 is an attractive target to control the inflammatory disease at the upstream of the signaling. However, discovering the chemical moieties that bind to the protein–protein interaction interface is a challenging task due to the relatively wide and shallow binding pocket compared to the enzyme’s active site. For the IL-33-specific binder discovery, a series of IL-33 mutants were designed, and an electrophile chemical library was screened. Herein, we described the backbone 1H, 15N, and 13C resonance assignments of three IL-33 (117–270) mutants. Based on the assignments, the binding site of a selected compound by this approach was determined by 2D NMR. These results provide valuable information for further studies in drug discovery targeting the IL-33 and ST2 interaction.
Similar content being viewed by others


Introduction
Over a hundred million patients suffer from atopic dermatitis worldwide. The symptoms can be initiated by a variety of factors. When allergens stimulate epithelial cells, TLSP and interleukin-33 (IL33) are released, which leads to the activation of type-2 immune cells (Imai 2019; Warren et al. 2021; Meephansan et al. 2013; Klonowska et al. 2018; Sugita and Akdis 2020). Conventional drugs, such as antihistamines or steroids, target symptom relief after immune response activation, but these are not the fundamental treatment. However, an allergic reaction can be efficiently regulated if IL-33 is controlled in the early stage of the immune response. IL-33 is activated by an N-terminal domain cleavage by a protease after it is secreted from epithelial cells (Imai 2019; Park et al. 2017). The mature form of IL-33 signals downstream immune cells through binding the heterodimeric ST2:IL-1RAcP receptor complex. IL-33 makes direct contact with its primary receptor ST2 (Liu et al. 2013; Lingel et al. 2009). We aim to find small molecules that interrupt the interaction so that the further allergic response can be inhibited. Protein–protein interaction (PPI) inhibitor discovery is very challenging because the binding surface is wide and shallow. In addition, structure changes may be induced by binding, and the binding affinity of small molecules is weak (Allen and Lumb 2020). Thus, we used a Cys-focused fragments library (Enamine) for compound screening. The compounds in the library have a unique binding moiety and a functional group that forms a covalent bond to the sulfhydryl group of cysteine at the ST2 receptor binding surface. For this purpose, intrinsic cysteine residues were mutated to alanine or serine, and a cysteine was introduced near the ST2 binding pocket of IL-33. Three mutants were constructed: IL-33 (117 to 270) C259A, IL-33 (117 to 270) C208S, C227A, C232A, and C259S, and IL-33 (117 to 270) C208S, C227A, C232A, C259S, and E269C. LC–MS was utilized to detect the complex formation. Ligand binding mode was described by a chemical shift perturbation (CSP) measurement of NMR. Resonance assignments of IL-33 mutants were carried out to map the compound binding site.
Mutant design and binding study
The target protein, human IL-33 S117-T270 constructs, has four intrinsic cysteine residues. To make compound binding occur at the hydrophobic binding surface specifically, the intrinsic cysteines were mutated to Ala or Ser, and a single cysteine was introduced near the binding interface by site-directed mutagenesis. We constructed, expressed, and purified the three mutants of IL33, named IL-33 (117 to 270) C259A, IL-33 (117 to 270) C208S, C227A, C232A, and C259S, and IL-33 (117 to 270) C208S, C227A, C232A, C259S, and E269C. We conducted the backbone-resonance assignment of the mutants based on the triple-resonance 3D experiments using 15N- and 13C- labeled mutant proteins or 3D NOESY and TOCSY experiments using 15N-labeled protein (Grzesiek, et al. 1992; Ovchinnikov et al. 1989).
Mutation assignment
IL-33 (117 to 270) C208S, C227A, C232A, and C259S, IL-33 4Cys-K.O.
Assignment of the IL-33 (117 to 270) C208S, C227A, C232A, and C259S mutant was done by acquiring standard triple resonance experiments (HNCACB, HNCOCACB, HNCACO, and HNCO) on cryo-probe equipped 700 MHz NMR spectrometer (Bruker, Korea Basic Science Institute) at 208 K. The data were processed by NMRpipe and analyzed by NMRview (Delaglio et al. 1995; Johnson 2004). The sequential backbone assignment was performed with the assistance of the CYANA auto assignment program (Schmidt and Guntert 2012). The 1H-15N HSQC spectrum was well-dispersed, and 137 residues were assigned among 147 non-proline residues. Two residues, Q167 and H168, located at the dynamic loop between \(\beta 4{-}\beta 5\), and four residues, N222, M223, H224, S225, at the β-turn between \(\beta 8{-}\beta 9\), were not identified by the triple resonance experiments. Strip plots of the HNCACB spectrum show sequential connectivity from E172 to D175 (Fig. 1).

HNCACB strip plot of the IL-33 (117 to 270) C208S, C227A, C232A, and C259S mutant showing sequence connectivity from E172 to D175
IL-33 (117 to 270) C208S, C227A, C232A, C259S, and E269C
For the IL-33 (117 to 270) C208S, C227A, C232A, C259S, and E269C mutant resonance assignment, cross-peaks of the 1H-15N HSQC spectrum were peak-picked by NMRview (Johnson 2004). Then, the assignment was accomplished by comparing its spectrum with the 1H-15N HSQC spectrum of IL-33 4Cys-K.O. The two spectra show similar dispersion patterns, as shown in Fig. 2. There was a little change due to a single mutation close to the c-terminal residue. S268 is located next to the mutated residue. The chemical sift of S268, therefore, is moved to higher ppm at the 1H-15N HSQC spectrum of IL-33 (117 to 270) C208S, C227A, C232A, C259S, and E269C. Other peaks, as shown in Fig. 2, presented small changes; therefore, the assignments were easily traced down. A total of 115 residues were assigned among 147 non-proline residues. The number of assigned residues was less than IL-33 4Cys-K.O. but it was enough to describe CSP at the PPI surface.

Overlay of the 2D 1H-15N HSQC spectra with one letter code and residue number of IL-33 (117 to 270) C208S, C227A, C232A, and C259S (Black) and IL-33 (117 to 270) C208S, C227A, C232A, C259S, and E269C (Gray)
IL-33 (117 to 270) C259A
The purpose of the screening was to identify a chemical moiety with a non-covalent binding affinity to the target site of the IL-33 protein. Although wild-type IL-33 (117 to 270) can be used for the binding study, the wild-type protein is known to be very unstable due to oxidation of the cysteine residues in the protein (Cohen et al. 2015). We tested the stabilities of the mutants and found that IL-33 (117 to 270) C259A was stable against the oxidation. In addition, the C259 residue is in the long loop between \(\beta 11{-}\beta 12\), which is not involved in the binding to the ST2 receptor (Lingel et al. 2009). Thus, we planned to use this mutant for further binding studies with non-covalent inhibitors. We expressed and purified the 15N-labeled IL-33 (117 to 270) C259A mutant, and the protein remained stable during NMR experiments. The 1H-15N HSQC spectrum of C259A was well-dispersed with uniform intensities. Sequence-specific assignments were confirmed by 3D 15N-NOESY and TOCSY on an 800 MHz NMR spectrometer (Bruker, Korea Basic Science Institute) (Ovchinnikov et al. 1989). The TOCSY data was acquired using 60-ms DIPSI2 spin-lock mixing, and NOESY data were acquired using a 100-ms mixing time. In total, 138 residues were assigned among the 147 non-proline residues.
CSP binding study with a selected inhibitor
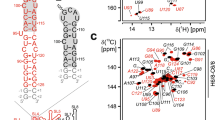
We obtained target site-specific binders from screening and characterized the binding by 2D NMR experiments (Kim et al. 2021). To confirm that the selected compound had a non-covalent binding affinity, the reactive warhead was linked through a long and flexible linker containing more than ten hydrocarbons (Fig. 3A). The binding reaction was performed with the IL-33 (117 to 270) C208S, C227A, C232A, C259S, and E269C mutants. The 2D 1H-15N HSQC spectra of the labeled proteins were measured in the presence and absence of the compound (Fig. 3B). The site-specific interactions between the IL-33 protein and the compound were visualized by a CSP measurements based on the 1H-15N HSQC spectra (Williamson 2013). The residues with significant CSP were mapped on the wild-type IL-33 X-ray crystal structure (Fig. 3C, PDB ID: 4K3C) (Liu et al. 2013).

A Chemical structure of the IL-33 binder. A reactive maleimide functional group is linked through a flexible linker. B Selected 1H-15N HSQC of IL-33 (117 to 270) C208S, C227A, C232A, C259S, and E269C in the presence (red) and absence (black) of binder compound. C The residues presenting modest and large CSP are depicted in yellow and red, respectively
Conclusion
In this study, three IL-33 mutants were designed for the screening of the selective binding compounds by NMR spectroscopy. Their 13C/15N- or 15N- labeled samples were prepared and backbone resonance assignments were accomplished. The assignment data can be used for drug screening and provided the valuable information for the 3D structure determination of protein–drug complexes. The ligand binding mode was assessed based on the measured NMR signal changes of IL-33 caused by a new ligand binding at the ST2 receptor binding interface. This approach can be widely applied to the PPI inhibitor screening and new drug designing based on complex structure determination.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its Additional file 1]. Additional File 1 including the list of the chemical shift of 1HN, 15N, 13C \(\alpha\) and 13C \(\beta\) of the three IL-33 mutants is available at www.ejast.org.
Abbreviations
- TLSP:
-
Thymic stromal lymphopoietin
- ST2:
-
Suppression of tumorigenicity 2
- PPI:
-
Protein–protein interaction
- NMR:
-
Nuclear magnetic resonance
- CSP:
-
Chemical shift perturbation
- LC–MS:
-
Liquid chromatography–mass spectrometry
- NOESY:
-
Nuclear Overhauser effect
- TOCSY:
-
Total correlation spectroscopy
- HSQC:
-
Hetero-single quantum correlation
- CYANA:
-
Combined assignment and dynamics algorithm for NMR approach
References
Allen SJ, Lumb KJ. Protein-protein interactions: a structural view of inhibition strategies and the IL-23/IL-17 axis. Adv Protein Chem Struct Biol. 2020;121:253–303.
Cohen ES, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. 2015;6(1):8327.
Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93.
Grzesiek S, et al. Proton, carbon-13, and nitrogen-15 NMR backbone assignments and secondary structure of human interferon-. gamma. Biochemistry. 1992;31(35):8180–90.
Imai Y. Interleukin-33 in atopic dermatitis. J Dermatol Sci. 2019;96(1):2–7.
Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Protein NMR Tech. 2004;278:313–52.
Kim Y, et al. Rational design, synthesis and evaluation of oxazolo [4, 5-c]-quinolinone analogs as novel interleukin-33 inhibitors. Chem Asian J. 2021;16(22):3702–12.
Klonowska J, et al. New cytokines in the pathogenesis of atopic dermatitis—new therapeutic targets. Int J Mol Sci. 2018;19(10):3086.
Lingel A, et al. Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors—insight into heterotrimeric IL-1 signaling complexes. Structure. 2009;17(10):1398–410.
Liu X, et al. Structural insights into the interaction of IL-33 with its receptors. Proc Natl Acad Sci. 2013;110(37):14918–23.
Meephansan J, et al. Expression of IL-33 in the epidermis: the mechanism of induction by IL-17. J Dermatol Sci. 2013;71(2):107–14.
Ovchinnikov YA, et al. Overcoming the overlap problem in the assignment of ’H NMR spectra of larger proteins by use of three-dimensional heteronuclear lH-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: application to interleukin. Biochemistry. 1989;28(6):150–6.
Park S-H, et al. IL-33-matured dendritic cells promote Th17 cell responses via IL-1β and IL-6. Cytokine. 2017;99:106–13.
Schmidt E, Guntert P. A new algorithm for reliable and general NMR resonance assignment. J Am Chem Soc. 2012;134(30):12817–29.
Sugita K, Akdis CA. Recent developments and advances in atopic dermatitis and food allergy. Allergol Int. 2020;69(2):204–14.
Warren KJ, et al. Neutralization of IL-33 modifies the type 2 and type 3 inflammatory signature of viral induced asthma exacerbation. Respir Res. 2021;22(1):1–14.
Williamson MP. Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16.
Acknowledgements
We thank Eunhee Kim and Hae-Kap Cheong in Korea Basic Science Institute (KBSI) for their help for acquiring NMR spectrometer.
Funding
This work was supported by the National Research Foundation of Korea Grant (2019R1A6A1A03031807) and by KBSI R&D Program (C23403).
Author information
Authors and Affiliations
Contributions
MK carried out the experiments and wrote the manuscript. SK and CH helped analyzing NMR data. SS synthesized chemicals. YB and YJ were in charge of overall direction and planning of research. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table 1.
Resonance assignment of IL-33 (117 to 270) C208S, C227A, C232A, and C259S mutant. Table 2. Resonance assignment of IL-33 (117 to 270) C208S, C227A, C232A, C259S, and E269C mutant. Table 3. Resonance assignment of IL-33 (117 to 270) C259A mutant.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, M., Kim, S., Heo, C.H. et al. Backbone assignment and inhibitor binding studies of IL-33 mutants by NMR spectroscopy. J Anal Sci Technol 14, 32 (2023). https://doi.org/10.1186/s40543-023-00392-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-023-00392-3