
Abstract
Background
Adenine (A), guanine (G), cytosine (C), thymine (T) and uracil (U) are building blocks and major constituents of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). The metabolic pathway of DNA and RNA is strongly related to A, G, C, T, and U.
Methods
Gas chromatography (GC) has been examined for the ease of separation of the nucleobases guanine (G), adenine (A), cytosine (C), thymine (T), and uracil (U) after precolumn derivatization with isobutyl chloroformate. GC was carried out from the column HP-5 (30 m × 0.32 mm id) with layer thickness 0.25 μm. The linear calibration curves were obtained with 0.5–50 μmol L−1 with limits of detection (LOD) 0.08–0.10 μmol L−1.
Results
The isolated DNA after acid hydrolysis from human blood and plants was analyzed, and amounts of T, A, C, and G were found in human blood within the ranges 0.521–2.625, 0.559–2.655, 0.490–2.915, and 0.450–2.739 μmol L−1, respectively, with relative standard deviation (RSD) 0.77–5.66 %. The ratios between T:A and G:C were 0.94–1.05 and 0.94–1.06, respectively. The values of (T + A)/(G + C) were within 0.90–1.07. Similarly from plant, T, A, C, and G were found within 0.48–2.915 nmolg−1 with RSD 0.88–5.00 %.
Conclusions
The method was applicable for the analysis of DNA nucleobases from biological samples.
Similar content being viewed by others


Background
Purine (guanine (G) and adenine (A)) and pyrimidine (thymine (T), cytosine (C), and uracil (U)) are the basic constituents of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), and their metabolic pathway is strongly associated with the development of DNA (Xia et al. 2009). The defects in metabolism of purines and pyrimidines result in the symptoms such as mental retardation, cardiovascular diseases, renal failure, gout, and toxemia (Sheng et al. 1991). Therefore, the determination of individual concentration of nucleobases or their ratio in DNA is important in biosciences and clinical diagnosis (Wang et al. 2002; Ba et al. 2012).
A number of analytical procedures have been proposed for the determination of nucleobases including electrochemical techniques (Ba et al. 2012; Wei et al. 2011; Huang et al. 2011; Li et al. 2013; Svorc and Kalchar 2014), high-performance liquid chromatography (Gill and Indyk 2007; Yeung et al. 2008; Graven et al. 2014), capillary electrophoresis (Ye and Jiang 2002; Rodriguez-Gomez and Carabias-Martinez 2014; Haunschmidt et al. 2008), spectroscopic methods (Amri et al. 2003; Heisler et al. 2002), chemiluminescence (Liu and Xue 2006), and mass spectrometry (Huang and Chang 2007). Capillary gas chromatography (GC) has high potential to separate closely related organic compounds in a short time. It is also free from the problem of disposing off the used solvents. However, the nucleobases are not sufficiently volatile and thermally stable to elute from the GC column. Thus, derivatization is required before their determination. The derivatization reagents used include bis(trimethylsilyl) trifluoroacetamide (Kuhara et al. 2011; Laking and Gehrke 1972); pentafluorobenzoyl chloride, pentafluorophenylsulfonyl chloride, or heptafluorobutyric anhydride (Nazareth et al 1984); N,N-tert-butyl-dimethylsilyl trifluoroacetamide (Glavin et al. 2006); and N-(tert-butyl-dimethylsilyl) N-methyl trifluoroacetamide (Horst et al. 2012). The silyl reagents are effective derivatizing reagents for nucleobases but require a nonaqueous medium for their derivatization. Isobutyl chloroformate (IBCF) has been used as a derivatization reagent for GC determination of primary and secondary low-molecular-mass aliphatic amines (Lundh and Akesson 1993); dicarboxylic acids and amino acids (Sobolevsk et al. 2004); and 4-(5-)methylimidazole (Fernando and Ferreira 1997). The derivatization is carried out in an aqueous medium. The present work examines IBCF as a derivatizing reagent for the GC determination of G, A, T, C, and U from aqueous-organic solvents. The work also optimizes the derivatization and GC elution conditions for the quantitation of nucleobases from acid-hydrolyzed DNA.
Methods
Reagents and solutions
The reagent-grade compounds G, A, C, T, and U (Sigma Chemical Co. St. Louis, USA); IBCF; chloroform and acetonitrile (Fluka, Buchs, Switzerland); methanol (Rdh Chemical Co. Spring Valley, CA); and pyridine (E-Merck, Darmstadt, Germany) were used.
Guaranteed reagent-grade hydrochloric acid (37 %), sodium bicarbonate, acetic acid, potassium chloride, sodium acetate, boric acid, sodium tetraborate, ammonium chloride, sodium carbonate, and ammonia solution (E-Merck, Darmstadt, Germany) were used.
Buffer solutions (0.2 M) between pH 1 and 12 at unit interval were prepared from the following: potassium chloride adjusted pH with hydrochloric acid (pH 1–2), acetic acid-sodium acetate (pH 3–6), ammonium acetate (pH 7), boric acid-sodium tetraborate (pH 7.5–8.5), sodium bicarbonate-sodium carbonate (pH 9), ammonia solution-ammonium chloride (pH 10), and potassium hydroxide-potassium chloride (pH 11–12).
The stock solutions (1 mmol L−1) of G, A, C, T, and U were prepared in water-methanol (1:1 V/V). Further solutions were prepared by suitable dilution.
Equipment
GC studies were carried out on Agilent model 6890 network GC system, connected with flame ionization detection (FID) and split injector (Agilent Technologies, Santa Clara, CA, USA), hydrogen generator (Parker Balston, Analytical Gas Systems, H2-90, Parker Hannifin, Haverhill, USA), and pure nitrogen (British Oxygen Company (BOC), Karachi, Pakistan); computer with ChemStation software controlled the gas chromatograph.
Capillary column HP-5 (30 m × 0.32 mm id) with film thickness 0.25 μm (J&W Scientific GC column, Wilmington, NC, USA) was used throughout the study.
GC-FID analysis and sample preparation
An aliquot of solution 0.5–1.0 mL containing 0.5–50 μmol L−1 A, T, C, U, and G was added to 0.5 mL of solvent containing acetonitrile-water-pyridine-methanol (40:40:10:10 V/V/V/V), 0.5 mL carbonate buffer (pH 9), and 0.25 mL of IBCF (10 % in methanol). The mixture was mixed well and placed for 15 min in ultrasonic water bath at room temperature (30 °C). Chloroform (0.5 mL) was then added and the contents were shacked well. The two layers were then allowed to separate, and an aliquot of organic layer was pipetted out in a screw-capped sample vial. The 1-μL solution was then injected on the column HP-5 at initial column temperature 100 °C for 1 min, followed by heating rate of 30 °C min−1 up to 280 °C and hold time at maximum temperature 2 min. The temperatures for injection and detection systems were 290 °C. The split ratio was 10:1, and carrier gas (nitrogen) flow rate was 1.5 mL min−1. The FID flow rate for nitrogen as make-up gas was fixed at 45 mL min−1; air and hydrogen flow rates were 450 and 40 mL min−1, respectively.
Sample collection and sample preparation
Five blood samples were collected by vein puncture in tubes of EDTA by verbal consent at Molecular Biology Laboratory, Liaquat University of Medical and Health Sciences (LUMHS), Jamshoro. The isolation of the DNA from blood was carried out at LUMHS by the reported procedure by Ali et al. (2013), following inorganic method (Grimberg et al. 1989). An aliquot of isolated DNA was obtained and was used in the next step as received.
A total of three samples of plant leaves, one from each (cotton, rice, and jasmine), were collected, and isolation of DNA was carried out at the laboratories of Nuclear Institute of Agriculture (NIA), Tando Jam. The isolation of DNA was carried out by a slightly modified procedure reported by Choudhary et al. (Choudhary et al. 2003). Leaves (5 g) were cleaned, washed, and ground with 5 mL of cetyl trimethyl ammonium bromide (C.TAB) buffer. After grinding, the mixture was kept in an ice machine for 20–30 min and was then allowed to achieve room temperature. Five milliliters of isoamyl alcohol-chloroform mixture (1:24) was added, and contents were shacked well. The mixture was centrifuged for 30 min at 4000g. The upper layer was collected, and 5 mL of isoamyl alcohol-chloroform (1:24) was added again into it, and the process was repeated. Then, 2-propanol (5 mL) was added to the aqueous layer, and off-white threads were collected at the bottom of the tube and were centrifuged for 5 min at 1000g. The residue was dissolved in 1 mL of tris buffer.
Acid hydrolysis of extracted DNA
Perchloric acid (1 mL) was added to a 100-μL isolated DNA sample in a screw-capped tube and was heated at water bath for 1 h at 95–100 °C. The pH of the solution was adjusted to 7 with 2 M sodium hydroxide and was filtered. The solution 0.2–0.5 mL was treated as a GC-FID analytical procedure. The quantitation of the nucleobases G, C, A, T, and U was carried out from a freshly prepared external calibration curve using linear regression equation y = ax + b.
G, A, C, and T contents from acid-hydrolyzed spiked DNA samples using linear calibration
The acid-hydrolyzed DNA (0.5 mL) was taken in duplicate. A part was processed as discussed in the GC-FID procedure, and the other was added to 0.5 mL of the solution containing G, C, A, and T (1 μmol L−1) each, and again, the procedure of GC-FID was repeated. The quantitation was carried out from the external calibration curve and from the increase in response with added standard.
Results and discussion
GC separation
The nucleobases G, C, A, T, and U were eluted from the GC column HP-5 after derivatization with IBCF and were separated from the derivatizing reagent easily. Each of the compounds indicated a single peak. Attempt was made to separate all the compounds simultaneously with short retention times. Different temperature programs for column oven and nitrogen flow rates were examined. Initial column oven temperature was varied from 90 to 120 °C with an interval of 10 °C; column heating rate was varied from 15 to 35 °C min−1 up to 280 °C with an interval of 5 °C min−1; and nitrogen as a carrier was varied from 1 to 2 mL min−1 at an interval of 0.5 mL min−1. Better separation with symmetrical peaks was obtained with run time of 9 min when eluted with initial column temperature of 100 °C for 1 min, followed by heating rate 30 °C min−1 up to 280 °C with a nitrogen flow rate of 1.5 mL min−1 (Fig. 1). The repeatability of the separation was examined in terms of retention times (n = 6), and relative standard deviation (RSD) was observed within 2 %.

Chromatogram of separation of standards of nucleobases (1) A, (2) U, (3) T, (4) G, and (5) C as IBCF derivatives. GC conditions as in Fig. 2
Optimization of derivatization
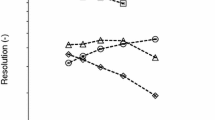
The separation was used to optimize the derivatization conditions simultaneously for all the nucleobases in terms of pH, solvent system for reaction, amount of derivatizing reagent added per analysis, reaction time, and solvent for the extraction of the compounds. The conditions which gave maximum response were considered optimum. The pH was varied between 1 and 12 at a unit interval, and better response was obtained at pH 9 and 10, and pH 9 was selected (Fig. 2). Solvent system water, methanol, acetonitrile, and solvent system proposed by Husek (1998) for amino acids acetonitrile-water-methanol-pyridine (40:40:10:10) were examined, and solvent system acetonitrile-water-methanol-pyridine gave better results and was selected. The addition of IBCF (10 % in methanol) as a derivatizing reagent was varied from 0.1 to 0.4 mL at an interval of 0.05 mL, and the addition of the derivatizing reagent was not critical as long as excess of the reagent was available. However, the addition of 0.25 mL was selected for a broader calibration range. After mixing of the reagent, the contents were sonicated for 5–20 min at an interval of 5 min. Maximum response was observed with reaction time of 15 min and above, and 15 min was selected. As extraction solvents of the derivatives, 1,2-dichloroethane, chloroform, and ethyl acetate were examined, but chloroform was used as proposed for related compounds (Husek 1991). After derivatization, GC response (average peak area/peak height (n = 4)) was examined for the compounds investigated, but no change in response was indicated up to 18 h.

Effect of pH on IBCF derivatization (x-axis = pH, y-axis = peak height) and GC elution of T, U, C, A, and G from the column HP-5 (30 m × 0.32 mm id) at column temperature 100 °C for 1 min with a programmed heating rate at 30 °C min−1 up to 280 °C and held at maximum temperature for 2 min. The nitrogen flow rate was 1.5 mL min−1 with split ratio 10:1. The detector and injector temperatures were 290 °C. The flow rate for FID was set fixed for nitrogen as make-up gas 45 mL min−1, air 450 mL min−1, and hydrogen 40 mL min−1
Analytical method validation
The linearity of calibration curves for all the five nucleobases was evaluated by recording average peak height/peak area (n = 4) against concentration and was observed in the range of 0.5–50 μmol L−1 with coefficient of determination (r 2) within the range 0.990–0.994. The limits of detection (LOD) and limits of quantitations (LOQ) were measured as signal-to-noise ratios of 3:1 and 10:1 and were observed within 0.08–0.10 and 0.24–0.30 μmol L−1, respectively (Table 1). The repeatability of the separation, derivatization, and quantitation was examined in intra (n = 5)- and inter (n = 5)-day variation by the same operator with the concentrations of 2, 10, and 20 μmol L−1 in each nucleobase in terms of peak area/peak height and retention time, and RSDs were observed within 4.5 %. Four test solutions of nucleobases G, C, A, T, and U were analyzed within the calibration range, and relative error was observed within 0.58–1.04 %.
The presence of ascorbic acid, glucose, methylparaben, gum acacia, Na, K, Ca, Mg, Cl, CO3, SO4, and NO3 did not affect the determination of the nucleobases, when present at least twice the concentration of nucleobases.
Sample analysis
The five human blood DNA samples and three plant leaf DNA samples were collected and hydrolyzed by an acid, and were examined for GC quantitation of the nucloebases, after precolumn derivatization with IBCF. Fresh calibration curves were prepared, and quantitations were made by external calibration using linear regression equation. The results of the analysis are summarized in Table 2, and a typical chromatogram is shown in Fig. 3. Nucleobases G, A, C, and T were detected from both blood and plant DNA samples, but U was observed below LOD. The peaks of the nucleobases were identified by comparing the peaks with retention times of the standard nucleobases. The amounts of G, A, C, and T from the blood samples were observed (μmol L−1): 0.988–2.739, 1.012–2.655, 1.040–2.915, and 0.994–2.625, respectively, with RSD within 0.76–5.66 % (Table 2). The ratios between T:A and G:C for each blood sample were calculated and were within 0.94–1.05 and 0.94–1.06, respectively. The values of (T + A)/(G + C) for DNA from human blood were calculated within 0.90–1.07. Similarly, three samples of plant leaf DNA were analyzed, and amounts of G, A, C, and T found were (nmol g−1) 0.480–2.739, 0.559–0.899, 0.490–2.915, and 0.521–0.952, respectively, with RSD within 0.88–5.00 %. Similarly, the ratios between T:A and G:C for each plant sample were calculated and were observed within 0.94–1.05 and 0.94–1.04, and also, the ratios between (T + A)/(G + C) for each sample were observed within 1.00–1.009, respectively. The samples from acid-hydrolyzed DNA from blood and plants were spiked with 1 μmol L−1 for each G, A, C, and T, and analysis was carried out using the procedure discussed in the GC-FID portion. The results agreed with external calibration, and recoveries were calculated within 99–105 % with RSD 0.50–4.55 %.

Chromatogram of the acid-hydrolyzed human blood DNA samples (1) A, (2) T, (3) G, and (4) C as IBCF derivatives. GC conditions as in Fig. 1
Comparative study
Now, comparing the present work with the reported GC procedures (Table 3) indicates that most of the reported procedures are based on precolumn derivatization with different silylation reagents, which requires nonaqueous media for their derivatization (Laking and Gehrke 1972; Nazareth et al. 1984; Gelijkens et al. 1981; Tekel et al. 1998), whereas the present work is based on the use of aqueous-organic phase. The present method indicates comparable or better sensitivity with shorter retention times than the reported procedure. The use of an inexpensive reagent with a shorter reaction time for derivatization is an added advantage of the present method. The results of the analysis agreed with the reported values for C by Xia et al. (2011) and C, T, and A with Xia et al. (2009) for human plasma samples. The reported values for plant DNA samples also agreed with Zhao et al. (2003).
Conclusions
A GC procedure has been developed for a complete separation and determination of nucleobases after precolumn derivatization with IBCF. The derivatization is carried out from an aqueous-organic phase. The method is sufficiently sensitive to be used for the analysis of DNA after acid hydrolysis. The method is reproducible with RSD within 6 %.
References
Ali S, Ahmed B, Bashir S, Mahmmod N. Linkage analysis for Dfnb 23/Usher1f locus. J Life Sciences. 2013;10:310–5.
Amri CE, Baron MH, Maurel MC. Adenine and RNA in mineral samples. Surface-enhanced Raman spectroscopy (SERS) for picomole detections. Spectro Chim Acta A. 2003;59:2645–54.
Ba X, Luo L, Ding Y, Zhang Z, Chu Y, Wang B. Poly(alizarin red)/graphene modified glassy carbon electrode for simultaneous determination of purine and pyrimidine. Anal Chim Acta. 2012;752:94–100.
Choudhary K, Mathar N, Choudhary OP, Pillai U. Protocol for isolation of genomic DNA form dry and fresh leaves of Vigna species suitable for rapid and restriction digestion. Adv Biol Res. 2003;2:83–9.
Fernando JO, Ferreira MA. Gas chromatographic-mass spectrometric determination of 4-(5) methylimidazole in ammonia caramel colour using ion-pair extraction and derivatization with isobutylchloroformate. J Chromatogr A. 1997;786:299–308.
Gelijkens CF, Smith DL, McCloskey JA. Capillary gas chromatography of pyrimidines and purines: N,O-peralkyl and trifluoroacetyl-N,O-alkyl derivatives. J Chromatogr. 1981;225:291–9.
Gill BD, Indyk HE. Development and application of a liquid chromatographic method for analysis of nucleotides and nucleosides in milk and infant formulas. Int Diary J. 2007;17:596–605.
Glavin DP, Cleaves HJ, Buch A, Schubert M, Aubrey A, Bada JL, Mahaffy PR. Sublimation extraction coupled with GC-MS: a new technique for future in situ analysis of purines and pyrimidines on Mars. Planet Space Sci. 2006;54:1584–91.
Graven P, Tambalo M, Scapozza L, Perozzo R. Purine metabolite and energy charge analysis of Trypanosoma brucei cells in different growth phases using an optimized ion-pair RP-HPLC/UV for the quantification of adenine and guanine pools. Exp Parasitol. 2014;141:28–38.
Grimberg J, Nawosehik S, Bellusis L, Mckee R, Truck A, Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989;17:8390.
Haunschmidt M, Buchberger W, Klampfl CW. Investigations on the migration behaviour of purines and pyrimidines in capillary electromigration techniques with UV detection and mass spectrometric detection. J Chromatogr A. 2008;1213:88–92.
Heisler I, Keller J, Tauber R, Sutherland M, Funchs H. A colorimetric assay for the quantitation of free adenine applied to determine enzymatic activity of ribosome-inactivating proteins. Anal Biochem. 2002;302:114–22.
Horst SM, Yelle RV, Buch A, Carrasco N, Cerno-gora G, Dutuit O, Quirico E, Sciamma-O’Brien E, Smith MA, Somogyl A, Szopa C, Thissen R, Vuitton V. Formation of amino acids and nucleotide bases in a Titan atmosphere simulation experiment. Astrobiology. 2012;12:1–9.
Huang KJ, Niu DJ, Sun JY, Han CH, Wu ZW, Li YL, Xiong XQ. Novel electrochemical sensor based on functionalized graphene for simultaneous determination of adenine and guanine in DNA. Colloids Surf B. 2011;81:543–9.
Huang YF, Chang HT. Analysis of adenosine triphosphate and glutathione through gold nanoparticles assisted laser desorption/ionization mass spectrometry. Anal Chem. 2007;79:4852–9.
Husek P. Chloroformates in gas chromatography as general purpose derivatizing agents. J Chromatogr B. 1998;717:57–91.
Husek P. Rapid derivatization and gas chromatographic determination of amino acids. J Chromatogr. 1991;552:289–98.
Kuhara T, Ohdol C, Ohse M. Simple gas chromatography-mass spectrometric procedure for diagnosing pyrimidine degradation defects for prevention of severe anti cancer side effects. J Chromatogr B. 2011;758:61–75.
Laking BD, Gehrke CW. Gas-liquid chromatographic analysis of purine and pyrimidine bases in hydrolysates of nucleic acid. Clin Chem. 1972;18:810–3.
Li C, Qin Y, Ling Y. Electrocatalytic oxidation and simultaneous determination of guanine and adenine on (2,6-pyridinedicarboxylic acid)/graphene composite film modified electrode. J Electroanal Chem. 2013;704:44–9.
Liu E, Xue BC. Flow injection determination of adenine at trace level based on luminol K2Cr2O7 chemiluminescence in a micellar medium. J Pharm Biomed Anal. 2006;41:649–53.
Lundh T, Akesson B. GC determination of primary and secondary low-molecular-mass aliphatic amines in urine using derivatization with IBCF. J Chromatogr. 1993;617:191–6.
Nazareth A, Joppich M, Adbul Baky S, Connell KO, Setissh A, Giese RW. Electrophore labeling and alkylation of standards of nucleic acid pyrimidine bases for alkylation by gas chromatography with electron-capture detection. J Chromatogr. 1984;314:201–10.
Rodriguez-Gomez E, Carabias-Martinez R. Development of a procedure for the isolation and enrichment of modified nucleosides and nucleobases from urine prior to their determination by capillary electrophoresis–mass spectrometry. J Pharm Biomed Anal. 2014;88:489–96.
Sheng RS, Ni F, Cotton TM. Determination of purine bases by reversed-phase high performance liquid chromatography using real-time surface-enhanced Raman spectroscopy. J Anal Chem. 1991;63:437–42.
Sobolevsk TG, Revelsky AI, Revelsky IA, Miller B, Oriedo V. Simultaneous determination of fatty, dicarboxylic and amino acids based on derivatization with isobutyl chloroformate followed by gas chromatography—positive ion chemical ionization mass spectrometry. J Chromatogr B. 2004;800:101–7.
Svorc L, Kalchar K. Modification-free electrochemical approach for sensitive monitoring of purine DNA bases: simultaneous determination of guanine and adenine in biological samples using boron-doped diamond electrode. Sens Actuators B. 2014;104:332–42.
Tekel J, Tahotna S, Vaverkova S. Gas chromatographic method for determination of uracil herbicides in roots of Echinacea angustifolia Moench (Asteraceae). J Pharm Biomed Anal. 1998;16:753–6.
Wang HS, Ju HX, Chen HY. Simultaneous determination of guanine and adenine in DNA using an electrochemically pretreated glassy carbon electrode. Anal Chimica Acta. 2002;461:243–50.
Wei Y, Hiang QA, Li MG, Huang XJ, Fang B, Wang L. Electrochemical and spectroscopic Studies of ssDNA damage induced by hydrogen peroxide using graphene based nanomaterials. Electrochim Acta. 2011;56:8571–3.
Xia JF, Liang QL, Liang XP, Wang YM, Hu P, Li P, Luo GA. Ultraviolet and tandem mass spectrometry for simultaneous quantification of 21 pivotal metabolites in plasma from patients with diabetic nephropathy. J Chromatogr B. 2009;877:1930–6.
Xia JF, Wang ZH, Liang QL, Wang YM, Li P, Luo GA. Correlations of six related purine metabolites and diabetic nephropathy in Chinese type 2 diabetic patients. Clin Chim Acta. 2011;412:940–5.
Ye CF, Jiang S. Determination of monophosphate nucleotides by capillary electrophoresis inductively coupled plasma mass spectrometry. J Analyst. 2002;127:1324–7.
Yeung P, Ding L, Casley WL. HPLC assay with UV detection for determination of RBC purine nucleotide concentrations and application for biomarker study in vivo. J Pharm Biomed Anal. 2008;47:377–882.
Zhao HQ, Wang X, Li HM, Yang B, Yang HJ, Huang L. Characterization of nucleosides and nucleobases in natural cordyceps by HILIC-ESI/TOF and HILIC-ESI/MS. Molecules. 2003;18:9755–69.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
The author ROZZB has done all the experiment work. Co authors MYK and TMJK supervised ROZZB during his experimant work. All the authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Brohi, R.O.Z.Z., Khuhawar, M.Y. & Khuhawar, T.M.J. GC-FID determination of nucleobases guanine, adenine, cytosine, and thymine from DNA by precolumn derivatization with isobutyl chloroformate. J Anal Sci Technol 7, 10 (2016). https://doi.org/10.1186/s40543-016-0090-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-016-0090-9