
Abstract
In recent malaria research, the complexity of the disease has been explored using machine learning models via blood smear images, environmental, and even RNA-Seq data. However, a machine learning model based on genetic variation data is still required to fully explore individual malaria risk. Furthermore, many Genome-Wide Associations Studies (GWAS) have associated specific genetic markers, i.e., single nucleotide polymorphisms (SNPs), with malaria. Thus, the present study improves the current state-of-the-art genetic risk score by incorporating SNPs mutation location on large-scale genetic variation data obtained from GWAS. Nevertheless, it becomes computationally expensive for hyperparameter optimization on large-scale datasets. Therefore, this study proposes a machine learning model that incorporates mutation location as well as a Genetic Algorithm (GA) to optimize hyperparameters. Besides that, a deep learning model is also proposed to predict individual malaria risk as an alternative approach. The analysis is performed on the Malaria Genomic Epidemiology Network (MalariaGEN) dataset comprising 20,817 individuals from 11 populations. The findings of this study demonstrated that the proposed GA could overcome the curse of dimensionality and improve resource efficiency compared to commonly used methods. In addition, incorporating the mutation location significantly improved the machine learning models in predicting the individual malaria risk; a Mean Absolute Error (MAE) score of 8.00E−06. Moreover, the deep learning model obtained almost similar MAE scores to the machine learning models, indicating an alternative approach. Thus, this study provides relevant knowledge of genetic and technical deliberations that can improve the state-of-the-art methods for predicting individual malaria risk.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Background
Over the past decades, researchers have been studying malaria risk factors, i.e., root causes of malaria, as this infectious disease has caused more than 400,000 human deaths annually [1]. Many Genome-Wide Associations Studies (GWAS) have been carried out since human genetics has been identified as one of the main malaria risk factors. Substantial studies have linked specific genetic variants, i.e., single nucleotide polymorphisms (SNPs), to malaria; and have shown that populations have different susceptibility or resistance levels towards malaria [2, 3]. More specifically, genetic differences are caused by genetic diversity that stems from various events shaping a peculiar population’s genetic structure and uniqueness. Thus, specific genetic markers in the human genome affect individuals’ susceptibility (possibly infected) and resistance (possibly protected) against malaria [3].
More and more GWAS has been applied to several malaria-endemic areas. It has successfully identified genetic markers that characterize the disease’s complexity. For example, genes such as HBB, ABO, ATP2B4, G6PD, CD40LG, FY, GYPA, GYPB, GYPC, HBA, HP, SCL4A1 have been associated with malaria susceptibility or resistance across different populations [4]. The genotype in each genetic marker in these genes is formed by two alleles, A and a, generally expressed as AA, Aa, and aa. In this regard, some observations in the literature have indicated that the sickle cell anemia traits of the hemoglobin gene can partially prevent malaria [3, 5,6,7]. To have sickle cell anemia, an individual needs to have two recessive alleles—one from the mother and one from the father. If the alleles are heterozygous (Aa), the individual tends to be resistant to the development of malaria. In contrast, if the alleles are homozygous (AA/aa), the individual is susceptible to the development of malaria. Their findings demonstrated that the genotype pattern (heterozygosity and homozygosity) of the genetic marker has significant implications to the association of malaria.
Besides that, an SNP mutation is one of the primary sources of genetic variation, where mutation changes the DNA sequences. Multiple prior studies [8, 9] have established that the effects of mutation may be neutral, harmful, or beneficial to individuals, depending on the characteristics of the disease or location. For example, the mutation location is crucial for the development of Parkinson’s disease [10]. In contrast, the mutation location does not affect the development of Juvenile-onset myopia [11]. Therefore, this study explores the correlation between mutation location and malaria genetic marker to incorporate this information for individual malaria risk prediction through a machine learning approach.
In this study, we chose a machine learning approach because the big data containing genetic variation collected from GWAS provides unprecedented opportunities for further exploration. As an alternative, we also provide a deep learning approach. More specifically, we are developing a risk prediction model based on regression algorithms. Existing prediction studies focusing on binary classification via one-class learning, deep learning, and ensemble learning have shown great promise in classifying biological data [12,13,14,15,16]. However, we are interested in finding out whether it is possible to quantify an individual's risk of malaria based on SNP genotype data for facilitating personalized prevention and treatment. Therefore, we are unable to use the existing classification approach that generates a “yes” or “no” outcome. Thus, this study aims to provide a score that reflects the level of risk in the presence of genetic markers based on an individual's genetic variation profile. The current state-of-the-art risk score is based on the weighted genetic risk score (wGRS) to estimate the cumulative contribution of genetic factors to a specific disease [17]. Inspired by the importance mentioned above, we will incorporate the risk score calculation information on genotype patterns and mutation locations into wGRS.
Another important aspect is determining the optimal number of SNPs that are required for malaria risk prediction. This is because the genetic basis of malaria's susceptibility or resistance is complex on several levels where many genes are involved [3]. For example, previous studies [3, 4, 18,19,20,21,22,23,24] have proved that the genetic marker, SNP rs334 from the HBB gene, is malaria's main genetic risk factor. In other words, SNP rs334 may give the most predictive ability, while other malaria-related SNPs may only improve the predictive power slightly. From a biological point of view, each genetic marker, i.e., SNP, has a certain degree of interaction and contribution to the development of the disease [25]. Statistical tests are commonly done in GWAS to determine the effect size of SNPs to see the implications for the association of malaria. However, there is still uncertainty on the most important SNPs for all populations due to population differentiation. Therefore, this study utilized 104 malaria-related SNPs identified in the literature review of 31 papers to determine the optimal number of SNPs required for malaria risk prediction.
One of the most extensive processes in training prediction models is hyperparameter optimization. This process determines the optimal hyperparameter values to achieve the best performance model. Hyperparameters have a significant impact on the performance of the model being trained because these values directly control the behaviour of the training algorithm [26]. Although studies have shown several approaches for optimizing hyperparameter values, grid search and random search have always been considered the most straightforward algorithms to implement. This is because grid search tries all possible value combinations [27], whereas random search randomly combines different values [28]. However, one of the disadvantages of grid search is that it is computationally expensive on large datasets [27]. On the other hand, the random search is not exhaustive, where the randomly combined values were chosen without any strategy or prior trial information [28]. Another common approach is Bayesian optimization. This approach does not sample every possible combination like the grid search but works more systematically than the random search. Nevertheless, it consumes more computing resources than both approaches [29]. Thus, to overcome the curse of dimensionality and improve resource efficiency, we propose a Genetic Algorithm (GA) with probability values, population, and generation sizes. Determining these values is still an open research question as they depend on the research domain [30].
In recent malaria research, the complexity of the disease was explored using machine learning algorithms, particularly malaria parasites and development stages, through blood smear images [31, 32]. In addition, environmental data were retrieved and trained by machine learning algorithms to link malaria transmission in climate change [33, 34]. Besides, RNA-Seq mosquito data has been extensively explored through different feature selection methods and classification algorithms to predict and detect novel genes for malaria infection [35,36,37,38,39]. The mentioned prediction strategies are considered successful. However, a machine learning prediction model based on genetic variation is still required to fully explore the genetic markers to facilitate personalized prevention and treatment. Therefore, this paper aims to provide a machine learning model that predicts individual malaria risk based on large-scale genetic variation data obtained from GWAS, particularly susceptibility or resistance genetic markers. As an alternative approach, this paper also aims to provide a deep learning model. The main contributions of this paper are summarized below:
-
Proposes optimal crossover, mutation, and parameter mutation probabilities, as well as population and generation sizes of GA for three machine learning models: Light Gradient Boosting Machine (LightGBM), Ridge Regression, and Support Vector Regression (SVR) in this domain;
-
Proposes a formula that incorporates mutation location information to improve the wGRS-based score;
-
Proposes a novel deep learning model as an alternative approach to predict individual malaria risk;
-
Provides a comprehensive analysis of the experimental results, and;
-
Proposes an optimal number of SNPs for malaria prediction.
The rest of this paper discusses materials and method; results and discussion; and conclusions.
Materials and method
Dataset
We used the Malaria Genomic Epidemiology Network (MalariaGEN) dataset, which consists of 20,854 individuals (10,791 malaria-affected individuals and 10,063 controls) from 11 global populations (Table 1). This dataset comes from the MalariaGEN Consortial Project 1 entitled: “Genome-wide study of resistance to severe malaria in eleven populations.” The consortial project structure has been expressed in [40], and the contributions of each partner’s studies and field sites to the project are acknowledged on the MalariaGEN website http://www.malariagen.net/.
Data preprocessing and data preparation
Our data preprocessing steps are as follows. (i) Data extraction: We extracted 122 malaria-related SNPs as variables from the MalariaGEN dataset. These SNPs were determined by reviewing and analysing 31 academic articles [2, 4, 18,19,20,21,22,23, 41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. (ii) SNPs that did not report effect size or availability in all populations were excluded from the analysis (18 SNPs). (iii) All unparseable values, such as data types and standard format errors, are converted to null representations. (iv) The Single Nucleotide Polymorphism database, in collaboration with EMBL-EBI European Variation Archive, assigns a unique ID to human genetic variation data, including SNPs [64]. These IDs are called rsIDs and appear in the format rs##. On the other hand, kgpIDs are identifiers created by Illumina during sequencing. The rsID of each SNP indicates variant information, including chromosome position. Any SNP assigned with kgpID is mapped to rsID (32 SNPs). (v) Samples without detailed information on the severe malaria subtype were excluded from the analysis (37 samples). This preprocessing procedure yielded 104 SNP variables from a total of 20,817 samples used in the model. The descriptions of all 104 SNPs are provided in Additional file 1.
This stage also imputes any missing genotypes based on the MalariaGEN dataset used in this study. Unfortunately, the existing genotype imputation software, such as IMPUTE2 [65] and Beagle [66] are based on publicly available reference datasets such as 1000 Genomes Project or HapMap 3. Therefore, we cannot estimate missing genotypes as we require the imputation to be more specific, i.e., based on population group and severe malaria subtypes. We developed a python program that imputes missing genotypes using the MalariaGEN dataset. The program first groups individuals based on their countries and then by their severe malaria subtypes. A comparison of a total of six SNPs for each missing genotype, i.e., three SNPs before and after the missing loci, is made before selecting the most common genotype data to impute the missing genotype.
On the other hand, for feature selection, we prepare the genotype data as a feature and class label data frame. Each column consists of an SNP with genotypes comprising major allele A and minor allele a, generally expressed as AA, Aa, and aa. The last column represents the class label and contains the binary classification of the individuals: 0—case (malaria-affected) and 1—control (healthy). However, for model development, we replace the binary classification in the last column with the wGRS-based risk scores instead.
Statistical analysis
Descriptive statistics
We performed descriptive statistics to understand the data characteristics and utilized kurtosis and skewness to identify the normality of the data. If the kurtosis and skewness fall within the range of [− 7, + 7] and [− 2, + 2], the data is considered normal [67, 68]. However, since we obtained the kurtosis and skewness within the range of [− 1.9, + 46.3] and [− 5.7, + 6.7], the data is non-normally distributed. These results further reaffirm the conclusion in [69, 70] that normally distributed medical data is an exception because real-world data is usually non-normally distributed and contains ordinal data. Therefore, we adopted a non-parametric test, the Spearman correlation coefficient, to evaluate the association between mutation location and malaria genetic marker.
Spearman correlation coefficient
We first implemented the Spearman correlation coefficient in Python using the scipy.stats.spearmanr() function. This function measures the degree of association between the mutation location and the malaria genetic marker in terms of strength and direction. It then returns the test result with correlation coefficient and p-value to indicate statistical significance.
Next, we generated 104 feature sets to observe the statistical significance of the association when the number of malaria genetic markers increases. These feature sets are based on the feature importance ranking obtained from feature selection. Thus, for example, the first feature set is composed of the top one feature, whereas the second feature set is composed of the top two features, and so on. More specifically, feature selection is performed on the preprocessed MalariaGEN dataset using the Logistic Regression and Recursive Feature Elimination (LR–RFE) method.
The statistical analysis results indicate a correlation between the mutation location and the malaria genetic marker for all the feature sets, as the coefficient values are between [− 1, + 1] [71]. The values within this range show a correlation; more specifically, values closer to − 1 or + 1 are considered to have a strong correlation, whereas values farther away are considered to have a weaker correlation. Moreover, the obtained p-values also show very significant differences (p < 0.001) for all feature sets, except for feature set 9, where p = 0.8873. The feature set 104 (containing all SNPs) indicated a correlation coefficient of − 0.0908 and p = 0.00E + 00. These results show that mutation location plays an important role in malaria development. To simply put it in layman’s terms, SNPs formed by the same genotype, e.g., AA, do not have the same importance in malaria development. Thus, we combine the mutation location information into the malaria risk prediction model.
To further elaborate on the relationship between the mutation location and the malaria genetic marker, we also observed the positive and negative correlations in the analysis. A positive correlation means that two variables move in the same direction, i.e., one increases and the other also increases. Conversely, a negative correlation means that two variables move in different directions, i.e., one increases and the other decreases. In our case, feature sets 2, 8, 10, and 12 obtained a positive correlation; feature set 2 has a very strong positive relationship with a correlation coefficient of 0.8754. In contrast, the remaining feature sets have a negative correlation. Figure 1 shows a graph containing the correlation coefficients against the number of features. The correlation coefficients and p-values for all the feature sets are summarized in Additional file 2. We consider p = 0.00E + 00 as very significant differences (p < 0.001).

Association between mutation location and malaria genetic marker with respect to correlation coefficients and feature sets based on Spearman correlation coefficient
Risk score
Inspired by the above results, we propose a formula that incorporates mutation location information to the wGRS + GF score. The proposed formula, namely, wGRS + GF + POS, combines the wGRS, genotype patterns, and mutation location information of genetic marker, i.e., SNP. These two formulas: wGRS + GF + POS and wGRS + GF, are utilized to calculate the target variable. The wGRS + GF is a baseline score.
wGRS + GF
The wGRS + GF is composed of wGRS and genotype frequency [72]. The wGRS is calculated by multiplying the number of risk alleles (0, 1, 2) with the estimated effect size reported for each variant. In contrast, the genotype frequency is calculated from the genotype data by using the Hardy–Weinberg equation. Thus, to generate wGRS + GF for each genotype, we multiple the genotype frequency with the wGRS obtained from each genotype. The calculation of wGRS + GF is summarized as: (risk allele * effect size) * genotype frequency = wGRS + GF.
wGRS + GF + POS
The wGRS + GF + POS is based on the wGRS + GF algorithm. The algorithm first encodes each genotype with a unique value in increments of 104 as there are 104 SNPs. (See Fig. 2a for details). Thus, for example, genotype AA is assigned with 104, whereas genotype AC is assigned with 208. Likewise, we encode each column name with a value in increments of 1. (See Fig. 2b for details). For example, rs334 is the first column in the prepared data and is assigned a value of 1. In contrast, rs2070724 is the second column in the prepared data and is assigned a value of 2. Next, for each genotype, we multiply the genotype encoding value with the column name encoding value to compute a value that we call as the mutation value. Continuing with the example, the mutation value of genotype AA in the first column is 1*104 = 104, while its mutation value in the second column is 2*104 = 208. Finally, we compute wGRS + GF + POS for each genotype by first multiplying wGRS with the genotype frequency and then dividing by the mutation value. The pseudocode of the proposed wGRS + GF + POS algorithm is presented in Fig. 3.

Overview of steps to encode genotypes and column names for the proposed wGRS + GF + POS algorithm

Pseudocode of the proposed wGRS + GF + POS algorithm
It is worth noting that there are 2,164,968 mutation values as there are 20,817 individuals with 104 SNPs each. Hence, we are not generating unique values for each genotype in each location as it is computationally expensive. Our initial exploration results indicated that using exponential functions create unique mutation values. However, our large dataset makes it computationally expensive to train the model as very large mutation values are computed. Furthermore, existing machine learning algorithms such as XGBoost returns an error if values larger than float32 are used.
Moreover, we expect the mutation values to overlap as; generally, each genotype in each location may have similar or little importance to malaria development. Thus, instead of randomly assigning weights to the mutation values, we leave them as they are. Another aspect worth noting is that we chose 104 as the incremental value as there are 104 SNPs. Thus, our approach can easily be updated to represent a new dataset, i.e., different number of genotypes, and has been experimentally tested on 20,817 samples.
Machine learning analysis
We performed feature selection using the LR–RFE method to generate 104 feature sets. These feature sets were earlier used for Spearman correlation coefficient analysis. Here, we explore the different feature sets to determine the optimal number of SNPs required for malaria risk prediction. Moreover, recent research has shown that the complex interaction between the SNPs increases the model's predictive ability [3]. Therefore, the target variable of each feature set is a risk score, which represents the cumulative effect of features calculated by using the wGRS + GF + POS and wGRS + GF.
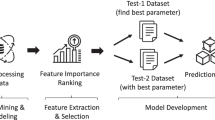
We split the preprocessed MalariaGEN dataset into two independent datasets: Test-1 (15%) and Test-2 (85%). Study [73] recommends the split percentage. Furthermore, we divided Test-1 and Test-2 into seven equally-sized random groups using sevenfold cross-validation to prevent overfitting. Hyperparameter optimization is done through a GA within the Test-1 dataset. We implemented three machine learning algorithms: LightGBM, Ridge Regression, and SVR to train the prediction model within the Test-2 dataset. Finally, we use the Mean Absolute Error (MAE) metric to measure the model’s performance based on wGRS + GF + POS and the wGRS + GF baseline model. MAE is a negatively-oriented score, where the lower the MAE value, the higher the prediction accuracy. Figure 4 shows the methodology flow chart in detail.

Methodology flow chart
Hyperparameter optimization
The most commonly used for hyperparameter optimization are the grid search and random search methods due to their simplicity and ease of use [27]. On the other hand, Bayesian optimization is known as a more efficient alternative [74]. However, the grid search uses an exhaustive search approach which becomes computationally expensive with large datasets. Moreover, the random search lacks strategies, and Bayesian optimization consumes more computational resources. Thus, we propose and implement a GA instead, where all the three optimization methods are used for benchmark comparisons.
The Test-1 dataset is used to choose the optimized hyperparameters for LightGBM, Ridge Regression, and SVR algorithms. More specifically, we optimized the following hyperparameters: for LightGBM, the maximum tree leaves [8, 16, 32], the maximum tree depth [4, 6, 8], the subsample ratio of the training instance [0.6, 0.8, 1.0], and the subsample ratio of columns when constructing each tree [0.6, 0.8, 1.0]; for Ridge Regression, regularization strength [1, 10, 100, 1000]; and finally for SVR, the kernel type [linear, rbf] and regularization parameter [1, 2, 3, 4, 5].
To date, researchers have proposed several variants of GA for different research domains [75]. Generally, GA comprises initial population development, fitness evaluation, parent selection, crossover, and mutation. Figure 5 summarizes the pseudocode of GA, which we refer to as Algorithm GA. For each feature set, Algorithm GA first initializes the population with random parameters according to the defined population size and then calculates the fitness values of all populations. This is followed by the crossover and mutation steps for breeding. The main difference of the existing GA lies in the chosen crossover, mutation, and parameter mutation probabilities indicated in Steps 7, 11, and 12 of Algorithm GA. We chose a crossover probability of 0.5, a mutation probability of 0.2, and a parameter mutation probability of 0.15, as it worked sufficiently well by achieving good MAE scores.

Pseudocode of the proposed GA
It is worth noting that determining the population and generation sizes is still an open research question as it depends on the research domain and selected features [30]. An optimal solution for one dataset or domain is not necessarily optimal for another dataset or domain. Therefore, in our research, we explore the optimal combination value of these sizes by implementing the population size [population size = 5, 10, 15, 20] and the generation size [generation size = 1, 2, 3, 4, 5, 10, 15, 20], and evaluate their performance based on MAE. Furthermore, studies have shown that the selection method employed by GA to choose the parents for breeding affects the MAE of the final model. Thus, choosing a suitable selection method is also crucial. Therefore, we implemented rank-based selection and tournament selection to observe the difference in generating the generations for breeding.
As noted earlier, we compared the performance of the proposed GA with three methods: grid search, random search, and Bayesian optimization. We evaluated these methods’ MAE, execution time, and peak random access memory (RAM) usage in optimizing the hyperparameter values for the three machine learning models. More specifically, we compared feature sets 20, 40, 60, 80, and 100 to provide an overview performance of running small and large feature sets. Thus, the purpose of this experiment is to gauge the performance of different hyperparameter optimization methods on genetic variation data ranging from 416,340 SNPs to 2,081,700 SNPs.
Alternative approach: deep learning model for individual malaria risk prediction
The deep learning model for individual malaria risk prediction of this paper is shown in Fig. 6. The input layer contains eight neurons, a bias initializer of 0.1, and an exponential linear unit (ELU) activation. The features are transformed via MinMaxScaler and fed using the input_dim parameter. Moreover, there are eight neurons with ELU activation on both the hidden layers. Finally, the output layer consists of one neuron representing the single risk score. All layers contain the HeUniform kernel initializer.

Deep learning model for malaria risk prediction
We chose the ELU activation as it is known to reduce computational complexity by pushing the mean activation toward zero during training and thereby increasing the learning speed [76]. In contrast, we chose the number of neurons via the trial-and-error approach since unnecessary increments of neurons can lead to overfitting, and insufficient neurons can lead to underfitting [77]. Lastly, we use the Adam optimizer as it is an adaptive optimization algorithm that is effective in solving practical deep learning problems even on large datasets [78].
For hyperparameter optimization and training of the deep model, the Test-1 dataset is used to select the optimized hyperparameters, while the Test-2 dataset is used to train the prediction model with the optimal hyperparameter values. The performance of the model is also measured using the MAE metric. We search hyperparameters by grid search and report the results using the best values for the batch size [4, 8, 16, 20], epoch [10, 20, 30, 40], and learning rate [0.0001, 0.001, 0.01].
Implementation details
We implemented the proposed approach using the python programming language while utilizing the sklearn library for the machine learning models and the Keras library for the deep learning model. The experiments were run on a Mac machine equipped with a 2.7 GHz Quad-Core Intel Core i7 processor and 16 GB of memory.
Results and discussion
This study established machine learning models to predict the risk score of individuals developing malaria, using the MalariaGEN dataset, with 20,817 samples and 104 SNPs as features. Moreover, a deep learning model is also proposed as an alternative approach. Based on the feature importance ranking obtained by the LR–RFE method, 104 feature sets are generated to explore different feature sets for predicting malaria risk. Of note, a recent study [79] used LR–RFE to rank feature importance and selection to find the optimal feature set in breast cancer prediction. This study developed several models to predict breast cancer using the cytological characteristics obtained from the breast fine needle aspiration test. It was concluded that LR–RFE contributed to better classification performance and improved model accuracy. Hence, we chose to use LR–RFE on genetic variation to observe the effectiveness of improving the accuracy of risk score prediction.
We implemented the wGRS + GF + POS algorithm to calculate the risk score as the target variable for each feature set and the wGRS + GF algorithm as the baseline. Each feature set is split into two independent datasets (Test-1 and Test-2). For each feature set in the Test-1 dataset, GA is used to optimize the hyperparameter values of the three machine learning models: LightGBM, Ridge Regression, and SVR. Meanwhile, grid search is used to optimize the hyperparameter values of the deep learning model. Then, we use the Test-2 dataset to train the model with the optimal hyperparameter values. The performance evaluation of the experiment is shown below.
Optimizing LightGBM, Ridge regression, and SVR hyperparameters
Based on our experiments, for all three machine learning models, the optimal population size is 5, and the generation size is 4. Our results indicated that the performance degrades when population size increases to 10, 15, and 20. On the other hand, there is little difference in performance when the generation size is 1 or 2.
We implemented two selection methods for the GA to observe the difference in selecting parents for breeding. Our results indicated that a machine learning model is affected by the selection method used in the GA. Some models perform better in tournament selection, while others perform better with the rank-based selection instead. Generally, LightGBM and SVR performed better in tournament selection, while Ridge Regression performed better in rank-based selection. These results will be used for further analysis: hyperparameter optimization benchmark comparisons and model training. The optimal hyperparameter values obtained for LightGBM, Ridge Regression, and SVR are summarized in Additional file 3.
Following this, Table 2 shows a performance comparison between four methods: proposed GA, grid search, random search, and Bayesian optimization, in optimizing hyperparameter values. The proposed GA only showed marginal differences compared to other methods in terms of MAE scores. All methods obtained almost similar MAE scores on all feature sets. However, the LightGBM and SVR models performed better with the proposed GA in execution times. In contrast, the grid search and random search methods optimize the Ridge Regression in slightly less time than the proposed GA. This may probably be because the LightGBM and SVR models have several hyperparameters compared to the Ridge Regression model, which only has a single parameter. Nevertheless, the proposed GA always performed better than the Bayesian optimization method. Besides that, the proposed GA also consumed the least amount of peak RAM usage. However, as expected, the Bayesian optimization method consumed the most memory for all the feature sets. On the other hand, the peak memory usage of the grid search and random search methods is between the proposed GA and Bayesian optimization methods. Generally, the proposed GA performed significantly better in execution times and peak memory usage. Thus, we can conclude that the proposed GA can be considered an alternative approach for optimizing genetic variation data or models with multiple hyperparameter values as it can overcome the curse of dimensionality and improve resource efficiency.
Malaria risk prediction via machine learning model
Figure 7 shows a graph containing the MAE scores against the number of features based on wGRS + GF + POS as the target variable. The baseline models based on wGRS + GF are presented in Fig. 8.

Performance analysis of the wGRS + GF + POS-based model with respect to MAE scores and feature sets

Performance analysis of the baseline model with respect to MAE scores and feature sets
Interestingly, the results in Fig. 7 show that compared to Fig. 8, the MAE scores obtained with different feature sets are much lower by incorporating mutation location information. The MAE scores in Fig. 7 are between 8.00E−06 and 2.18E−04, indicating that the wGRS + GF + POS-based models outperform the baseline models shown in Fig. 8, with MAE scores only ranging between 3.38E−03 and 6.71E-01. Recall that a lower MAE score indicates higher prediction accuracy, as MAE is a negatively-oriented score. The best performing model is LightGBM, which achieves an MAE score of 8.00E−06 when training on a single feature, i.e., rs334, and an MAE score of 5.50E−05 when training on all 104 features. Thus, compared with the baseline models, there is a significant improvement in prediction accuracy. As for the baseline model, the best performing model is also LightGBM, which achieves an MAE score of 3.38E-03 on a single feature, i.e., rs334, and an MAE score of 6.39E-01 on all the 104 features. The difference in MAE scores further highlights the contribution of mutation location in predicting malaria risk. A summary of the MAE scores obtained for each feature set based on the wGRS + GF + POS-based models and the baseline models are given in Additional file 4.
However, it is insufficient to train with a single feature as recent research has shown that the complex interaction between the features increases the model's predictive ability [3]. Therefore, it is important to determine the optimal number of features for malaria prediction. Further analysing the wGRS + GF + POS-based models indicated that when the model is trained with LightGBM and Ridge Regression, the model's performance decreases when the number of features used for training increases. However, when trained with SVR, the model's performance increases when the number of features is increased.
Moreover, we also noticed that for LightGBM, 52 features are typically optimal sufficient to train the model with an MAE score of 5.10E−05. This is because no significant changes were observed in the MAE scores when the number of features increased. Likewise, for Ridge Regression, the optimal number of features is 53 with an MAE score of 6.00E−05. In contrast, for SVR, the optimal number of features is 103 with an MAE score of 6.70E−05. Thus, we can conclude that generally, the LightGBM performs best in predicting malaria risk based on genetic variation data in terms of the number of features required and the MAE score obtained, i.e., LightGBM achieves better performance with fewer features. This is a conclusion drawn from a computer science perspective based on the obtained MAE scores using 20,817 samples.
Malaria risk prediction via deep learning model
The MAE scores obtained for each feature set of the wGRS + GF + POS-based models and the baseline models are summarized in Additional file 5. Moreover, the optimal hyperparameters used are presented in Additional file 6.
The MAE scores of the models based on wGRS + GF + POS range from 6.00E-05 to 3.94E-03; the best performing model used 13 features in contrast to the worst-performing model, which used 22 features. However, generally, regardless of the number of features employed, all wGRS + GF + POS-based models obtained much lower MAE scores than baseline models. The MAE scores of the baseline models range from 3.51E−03 to 6.30E−01, with the best performing model trained on a single feature while the worst performing model trained on 98 features. Moreover, when trained with 104 features, the wGRS + GF + POS-based model outperformed the baseline model with an MAE score of 5.71E−04 instead of 6.16E−01. This further confirms that prediction accuracy can be significantly improved when mutation location information is provided.
However, regarding finding the optimal number of features, we observed that the prediction accuracy did not improve significantly when the number of features increased or decreased. In other words, lower MAE scores were obtained with fewer or more features. Thus, we hardly determined the optimal number of SNPs required for malaria prediction. This observation is different than the observation of the previous machine learning models, where prediction accuracy was affected by the number of SNPs used as features. Another noticeable observation is that there is only a slight difference in MAE scores obtained between the machine learning and deep learning models. Thus, we can conclude that the deep learning model can serve as an alternative approach for predicting individual malaria risk based on genetic variation data.
Conclusions
This study developed a novel GA that optimizes the hyperparameters of three machine learning models: LightGBM, Ridge Regression, and SVR in this domain. The leading idea takes advantage of GA in parallel searching from a population of points, providing higher efficiency, significant running time, and more robustness. Based on the statistical analysis results that evaluate the association between mutation location and malaria genetic marker, generally, we obtained significant correlation coefficients and p-values for all feature sets. Therefore, as expected, the machine learning and deep learning models showed substantial performance improvement when mutation location was incorporated to predict the risk of malaria.
The potential application of this study is to provide relevant knowledge of genetic and technical deliberations, which can enhance state-of-the-art methods or as an alternative to quantify individual malaria risk, conduct risk score analysis, and facilitate personalized prevention and treatment. Moreover, this study utilizes large-scale genetic variation data obtained from the MalariaGEN dataset. This dataset provides a good generalization for malaria risk prediction in different populations. This is because a large amount of genotyping data from diverse populations is usually required to obtain reasonable prediction generalization. In terms of future work, it would be interesting to see the ability of our proposed method with populations from different continents. Besides that, we did not include phenotype data as the focus of our study was on genetic variation data. We plan to investigate the model with different types of biological data, such as protein interaction, phenotype, and gene expression data.
To summarize, the novelties of this study are as follows (i) proposed a novel GA with optimal crossover, mutation, and parameter mutation probabilities, as well as population and generation sizes, for three machine learning models: LightGBM, Ridge Regression, and SVR, in this domain (ii) proposed a formula that incorporates mutation location information that improved the wGRS-based score, (iii) proposed a deep learning model as an alternative approach to predict individual malaria risk, (iv) evaluated the experimental results, and (v) proposed an optimal number of SNPs for malaria prediction.
Availability of data and materials
The datasets analysed during the current study are available in the MalariaGEN Consortial Project 1 (https://www.malariagen.net/projects/consortial-project-1).
Abbreviations
- ELU:
-
Exponential linear unit
- GA:
-
Genetic algorithm
- GWAS:
-
Genome-Wide Associations Studies
- LightGBM:
-
Light Gradient Boosting Machine
- MAE:
-
Mean Absolute Error
- MalariaGEN:
-
Malaria Genomic Epidemiology Network
- RAM:
-
Random access memory
- SNP:
-
Single nucleotide polymorphisms
- SVR:
-
Support Vector Regression
- wGRS:
-
Weighted genetic risk score
References
World Health Organization. World malaria report 2020: 20 years of global progress and challenges. World Health Organization. 2020. https://www.who.int/docs/default-source/malaria/world-malaria-reports/9789240015791-double-page-view.pdf?sfvrsn=2c24349d_5. Accessed 17 Nov 2021.
De Mendonça VR, Goncalves MS, Barral-Netto M. The host genetic diversity in malaria infection. J Trop Med. 2012;2012: 940616.
Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77(2):171–92.
Rockett KA, Clarke GM, Fitzpatrick K, Hubbart C, Jeffreys AE, Malaria Genomic Epidemiology Network, et al. Reappraisal of known malaria resistance loci in a large multicenter study. Nat Genet. 2014;46(11):1197–204.
Archer NM, Petersen N, Clark MA, Buckee CO, Childs LM, Duraisingh MT. Resistance to Plasmodium falciparum in sickle cell trait erythrocytes is driven by oxygen-dependent growth inhibition. Proc Natl Acad Sci. 2018;115(28):7350–5.
Williams TN, Mwangi TW, Roberts DJ, Alexander ND, Weatherall DJ, Wambua S, et al. An immune basis for malaria protection by the sickle cell trait. PLoS Med. 2005;2(5): e128.
Luzzatto L. Sickle cell anaemia and malaria. Mediterr J Hematol Infect Dis. 2012;4(1): e2012065.
Loewe L. Genetic mutation. Nat Educ. 2008;1(1):113.
Loewe L, Hill WG. The population genetics of mutations: good, bad and indifferent. Philos Trans R Soc Lond B Biol Sci. 2010;365(1544):1153–67.
Selvaraj S, Piramanayagam S. Impact of gene mutation in the development of Parkinson’s disease. Genes Dis. 2019;6(2):120–8.
Mutti DO, Semina E, Marazita M, Cooper M, Murray JC, Zadnik K. Genetic loci for pathological myopia are not associated with juvenile myopia. Am J Med Genet. 2002;112(4):355–60.
Kaufmann J, Asalone K, Corizzo R, Saldanha C, Bracht J, Japkowicz N. One-class ensembles for rare genomic sequences identification. In: Appice A, Tsoumakas G, Manolopoulos Y, Matwin S, editors. International conference on discovery science. Cham: Springer International Publishing; 2020.
Basavegowda HS, Dagnew G. Deep learning approach for microarray cancer data classification. CAAI Trans Intell Technol. 2020;5(1):22–33.
Tonkovic P, Kalajdziski S, Zdravevski E, Lameski P, Corizzo R, Pires IM, et al. Literature on applied machine learning in metagenomic classification: a scoping review. Biology. 2020;9(12):453.
Fuhad KMF, Tuba JF, Sarker MRA, Momen S, Mohammed N, Rahman T. Deep learning based automatic malaria parasite detection from blood smear and its smartphone based application. Diagnostics. 2020;10(5):329.
Pattanaik PA, Mittal M, Khan MZ. Unsupervised deep learning CAD scheme for the detection of malaria in blood smear microscopic images. IEEE Access. 2020;8:94936–46.
Hüls A, Krämer U, Carlsten C, Schikowski T, Ickstadt K, Schwender H. Comparison of weighting approaches for genetic risk scores in gene-environment interaction studies. BMC Genet. 2017;18(1):115.
Ndila CM, Uyoga S, Macharia AW, Nyutu G, Peshu N, Ojal J, et al. Human candidate gene polymorphisms and risk of severe malaria in children in Kilifi, Kenya: a case-control association study. Lancet Haematol. 2018;5(8):e333–45.
Shelton JM, Corran P, Risley P, Silva N, Hubbart C, Jeffreys A, et al. Genetic determinants of anti-malarial acquired immunity in a large multi-centre study. Malar J. 2015;14(1):333.
Manjurano A, Sepúlveda N, Nadjm B, Mtove G, Wangai H, Maxwell C, et al. USP38, FREM3, SDC1, DDC, and LOC727982 gene polymorphisms and differential susceptibility to severe malaria in Tanzania. J Infect Dis. 2015;212(7):1129–39.
Toure O, Konate S, Sissoko S, Niangaly A, Barry A, Sall AH, et al. Candidate polymorphisms and severe malaria in a Malian population. PLoS ONE. 2012;7(9): e43987.
Jallow M, Teo YY, Small KS, Rockett KA, Deloukas P, Clark TG, et al. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat Genet. 2009;41(6):657–65.
Band G, Le QS, Jostins L, Pirinen M, Kivinen K, Jallow M, et al. Imputation-based meta-analysis of severe malaria in three African populations. PLoS Genet. 2013;9(5): e1003509.
Gouagna LC, Bancone G, Yao F, Yameogo B, Dabiré KR, Costantini C, et al. Genetic variation in human HBB is associated with Plasmodium falciparum transmission. Nat Genet. 2010;42(4):328–31.
Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322(5903):881–8.
Wu J, Chen XY, Zhang H, Xiong LD, Lei H, Deng SH. Hyperparameter optimization for machine learning models based on Bayesian optimization. J Electron Sci Technol. 2019;17(1):26–40.
Bergstra J, Bengio Y. Random search for hyper-parameter optimization. J Mach Learn Res. 2012;13(10):281–305.
Elgeldawi E, Sayed A, Galal AR, Zaki AM. Hyperparameter tuning for machine learning algorithms used for arabic sentiment analysis. Inform. 2021;8(4):79.
Yu T, Zhu H. Hyper-Parameter Optimization: A Review of Algorithms and Applications. 2020; arXiv preprint. https://arxiv.org/abs/2003.05689.
Vrajitoru D. Large population or many generations for genetic algorithms? Implications in information retrieval. In: Crestani F, Pasi G, editors. Soft computing in information retrieval. Heidelberg: Physica; 2000.
Poostchi M, Silamut K, Maude RJ, Jaeger S, Thoma G. Image analysis and machine learning for detecting malaria. Transl Res. 2018;194:36–55.
Das DK, Ghosh M, Pal M, Maiti AK, Chakraborty C. Machine learning approach for automated screening of malaria parasite using light microscopic images. Micron. 2013;45:97–106.
Sharma V, Kumar A, Panat L, Karajkhede G, Lele A. Malaria outbreak prediction model using machine learning. Int J Adv Res Comput Eng Technol. 2015;4(12):4415–9.
Thakur S, Dharavath R. Artificial neural network based prediction of malaria abundances using big data: a knowledge capturing approach. Clin Epidemiol Glob Health. 2019;7(1):121–6.
Arowolo MO, Adebiyi MO, Adebiyi AA. A genetic algorithm approach for predicting ribonucleic acid sequencing data classification using KNN and decision tree. Telkomnika. 2020;19(1):310–6.
Arowolo MO, Adebiyi MO, Adebiyi AA. An efficient PCA Ensemble learning approach for prediction of RNA-Seq malaria vector gene expression data classification. Int J Eng Res Technol. 2020;13(1):163–9.
Arowolo MO, Adebiyi MO, Aremu C, Adebiyi AA. A survey of dimension reduction and classification methods for RNA-Seq data on malaria vector. J Big Data. 2021;8(1):1–17.
Adebiyi MO, Arowolo MO, Olugbara O. A genetic algorithm for prediction of RNA-seq malaria vector gene expression data classification using SVM kernels. Bull Electr Eng Inform. 2021;10(2):1071–9.
Arowolo MO, Adebiyi MO, Adebiyi AA. Enhanced dimensionality reduction methods for classifying malaria vector dataset using decision tree. Sains Malays. 2021;50(9):2579–89.
Network MGE. New insights into malaria susceptibility from the genomes of 17,000 individuals from Africa, Asia, and Oceania. Nat Commun. 2019;10:1–19.
Busby GB, Band G, Le QS, Jallow M, Bougama E, Mangano VD, et al. Admixture into and within sub-Saharan Africa. Elife. 2016;5: e15266.
Network MGE. A novel locus of resistance to severe malaria in a region of ancient balancing selection. Nature. 2015;526(7572):253–7.
Clark TG, Fry AE, Auburn S, Campino S, Diakite M, Green A, et al. Allelic heterogeneity of G6PD deficiency in West Africa and severe malaria susceptibility. Eur J Hum Genet. 2009;17(8):1080–5.
Wilson JN, Rockett K, Jallow M, Pinder M, Sisay-Joof F, Newport M, et al. Analysis of IL10 haplotypic associations with severe malaria. Genes Immun. 2005;6(6):462–6.
Apinjoh TO, Anchang-Kimbi JK, Njua-Yafi C, Ngwai AN, Mugri RN, Clark TG, et al. Association of candidate gene polymorphisms and TGF-beta/IL-10 levels with malaria in three regions of Cameroon: a case–control study. Malar J. 2014;13(1):236–47.
Manjurano A, Clark TG, Nadjm B, Mtove G, Wangai H, Sepulveda N, et al. Candidate human genetic polymorphisms and severe malaria in a Tanzanian population. PLoS ONE. 2012;7(10): e47463.
Eid NA, Hussein AA, Elzein AM, Mohamed HS, Rockett KA, Kwiatkowski DP, Ibrahim ME. Candidate malaria susceptibility/protective SNPs in hospital and population-based studies: the effect of sub-structuring. Malar J. 2010;9(1):119.
Ayodo G, Price AL, Keinan A, Ajwang A, Otieno MF, Orago AS, et al. Combining evidence of natural selection with association analysis increases power to detect malaria-resistance variants. Am J Hum Genet. 2007;81(2):234–42.
Atkinson A, Barbier M, Afridi S, Fumoux F, Rihet P. Evidence for epistasis between hemoglobin C and immune genes in human P. falciparum malaria: a family study in Burkina Faso. Genes Immun. 2011;12(6):481–9.
Dewasurendra RL, Suriyaphol P, Fernando SD, Carter R, Rockett K, Corran P, et al. Genetic polymorphisms associated with anti-malarial antibody levels in a low and unstable malaria transmission area in southern Sri Lanka. Malar J. 2012;11(1):281.
Mombo LE, Ntoumi F, Bisseye C, Ossari S, Lu CY, Nagel RL, et al. Human genetic polymorphisms and asymptomatic Plasmodium falciparum malaria in Gabonese schoolchildren. Am J Trop Med Hyg. 2003;68(2):186–90.
Walley AJ, Aucan C, Kwiatkowski D, Hill AV. Interleukin-1 gene cluster polymorphisms and susceptibility to clinical malaria in a Gambian case–control study. Eur J Hum Genet. 2004;12(2):132–8.
Santos SD, Clark TG, Campino S, Suarez-Mutis MC, Rockett KA, Kwiatkowski DP, et al. Investigation of host candidate malaria-associated risk/protective SNPs in a Brazilian Amazonian population. PLoS ONE. 2012;7(5): e36692.
Gelabert P, Olalde I, de Dios T, Civit S, Lalueza-Fox C. Malaria was a weak selective force in ancient Europeans. Sci Rep. 2017;7(1):1377.
Caetano CP, Kraaijenbrink T, Tuladhar NM, Driem GLV, Knijff P, Tyler-Smith C, et al. Nepalese populations show no association between the distribution of malaria and protective alleles. J Mol Genet Med. 2006;2(1):101–6.
Ravenhall M, Campino S, Sepúlveda N, Manjurano A, Nadjm B, Mtove G, et al. Novel genetic polymorphisms associated with severe malaria and under selective pressure in North-eastern Tanzania. PLoS Genet. 2018;14(1): e1007172.
Kariuki SM, Rockett K, Clark TG, Reyburn H, Agbenyega T, Taylor TE, et al. The genetic risk of acute seizures in African children with falciparum malaria. Epilepsia. 2013;54(6):990–1001.
Flori L, Delahaye NF, Iraqi FA, Hernandez-Valladares M, Fumoux F, Rihet P. TNF as a malaria candidate gene: polymorphism-screening and family-based association analysis of mild malaria attack and parasitemia in Burkina Faso. Genes Immun. 2005;6(6):472–80.
Clark TG, Diakite M, Auburn S, Campino S, Fry AE, Green A, et al. Tumor necrosis factor and lymphotoxin-α polymorphisms and severe malaria in African populations. J Infect Dis. 2009;199(4):569–75.
Dunstan SJ, Rockett KA, Quyen NT, Teo YY, Thai CQ, Hang NT, et al. Variation in human genes encoding adhesion and proinflammatory molecules are associated with severe malaria in the Vietnamese. Genes Immun. 2012;13(6):503–8.
Maiga B, Dolo A, Touré O, Dara V, Tapily A, Campino S, et al. Human candidate polymorphisms in sympatric ethnic groups differing in malaria susceptibility in Mali. PLoS ONE. 2013;8(10): e75675.
Diakite M, Achidi EA, Achonduh O, Craik R, Djimde AA, Evehe MS, et al. Host candidate gene polymorphisms and clearance of drug-resistant Plasmodium falciparum parasites. Malar J. 2011;10(1):250.
Sepúlveda N, Manjurano A, Campino SG, Lemnge M, Lusingu J, Olomi R, et al. Malaria host candidate genes validated by association with current, recent, and historical measures of transmission intensity. J Infect Dis. 2017;216(1):45–54.
National Center for Biotechnology Information. About dbSNP Reference (rs) number. 2021. https://www.ncbi.nlm.nih.gov/snp/docs/RefSNP_about/. Accessed 17 Nov 2021.
Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6): e1000529.
Browning BL, Zhou Y, Browning SR. A one-penny imputed genome from next-generation reference panels. Am J Hum Genet. 2018;103(3):338–48.
Byrne BM. Structural equation modeling with AMOS: basic concepts, applications, and programming. 2nd ed. New York: Taylor and Francis Group; 2010.
Hair JF, Black WC, Babin BJ, Anderson RE. Multivariate data analysis: a global perspective. 7th ed. New Jersey: Pearson Education Inc; 2010.
Micceri T. The unicorn, the normal curve, and other improbable creatures. Psychol Bull. 1989;105(1):156–66.
Rochon J, Gondan M, Kieser M. To test or not to test: Preliminary assessment of normality when comparing two independent samples. BMC Med Res Methodol. 2012;12(1):81.
Schober P, Boer C, Schwarte LA. Correlation coefficients: appropriate use and interpretation. Anesth Analg. 2018;126(5):1763–8.
Tai KY, Dhaliwal J, Balasubramaniam V. Leveraging Mann-Whitney U test on large-scale genetic variation data for analysing malaria genetic markers. Malar J. 2022;21(1):79.
Dobbin KK, Simon RM. Optimally splitting cases for training and testing high dimensional classifiers. BMC Med Genomics. 2011;4(1):31.
Nguyen V. Bayesian Optimization for Accelerating Hyper-Parameter Tuning. In: 2019 IEEE Second International Conference on Artificial Intelligence and Knowledge Engineering (AIKE); June 3–5, 2019.
Katoch S, Chauhan SS, Kumar V. A review on genetic algorithm: past, present, and future. Multimed Tools Appl. 2021;80(5):8091–126.
Nwankpa CE, Ijomah W, Gachagan A, Marshall S. Activation functions: Comparison of trends in practice and research for deep learning. 2018; arXiv preprint. https://arxiv.org/abs/1811.03378.
Uzair M, Jamil N. Effects of hidden layers on the efficiency of neural networks. In: 2020 IEEE 23rd International Multitopic Conference (INMIC); Nov 5–7, 2020.
Kingma DP, Ba JL. Adam: A method for stochastic optimization. 2017; arXiv preprint. https://arxiv.org/abs/1412.6980.
Mathew TE. A logistic regression with recursive feature elimination model for breast cancer diagnosis. Int J Emerg Technol. 2019;10(3):55–63.
Acknowledgements
This study makes use of data generated by MalariaGEN. A full list of the investigators who contributed to the generation of the data is available from www.MalariaGEN.net. Funding for this project was provided by Wellcome Trust (WT077383/Z/05/Z) and the Bill & Melinda Gates Foundation through the Foundation of the National Institutes of Health (566) as part of the Grand Challenges in Global Health Initiative.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
TKY performed analysis, interpreted the results, designed the algorithms, designed the models, and drafted the manuscript. JD supervised the project, interpreted the results, designed the algorithms, designed the models, and revised the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Description of the 104 SNPs used in this study.
Additional file 2:
Correlation coefficients and p-values between mutation location and malaria genetic marker using Spearman correlation coefficient.
Additional file 3:
Summary of the optimal hyperparameter values obtained for LightGBM, Ridge Regression, and SVR via GA.
Additional file 4:
Comparison of MAE scores of different feature sets results with wGRS + GF + POS-based and baseline models when trained using machine learning approach.
Additional file 5:
Comparison of MAE scores of different feature sets results with wGRS + GF + POS-based and baseline models when trained using deep learning approach.
Additional file 6:
Summary of the optimal hyperparameter values obtained for deep learning model via grid search.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tai, K.Y., Dhaliwal, J. Machine learning model for malaria risk prediction based on mutation location of large-scale genetic variation data. J Big Data 9, 85 (2022). https://doi.org/10.1186/s40537-022-00635-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40537-022-00635-x