
Abstract
Background
Cymbidium goeringii, belonging to the Orchidaceae family, is an important ornamental plant with striking petals and lips. Extremely diversified floral patterns and morphologies make C. goeringii good research material to examine floral development of orchids. However, no floral organ-specific protein has been identified yet. To screen floral development associated proteins, four proteomes from petal (PE), lip (LI), gynostemium (GY), and sepal (SE) were analyzed using Tandem Mass Tag-based proteomic analysis.
Results
A total of 6626 unique peptides encoding 2331 proteins were identified in our study. Proteins in several primary metabolic pathways, including amino acid metabolism, energy metabolism, and lipid metabolism, were identified as differentially expressed proteins. Interestingly, most of the energy metabolism-related proteins highly expressed in SE, indicating that SE is an important photosynthetic organ of C. goeringii flower. Furthermore, a number of phytohormone-related proteins and transcription factors (TFs) were identified in C. goeringii flowers. Expression analysis showed that 1-aminocyclopropane-1-carboxylate oxidase highly expressed in GY, IAA-amino acid hydrolase ILR1-like 4 and gibberellin receptor 1 C greatly expressed in LI, and auxin-binding protein ABP20 significantly expressed in SE, suggesting a significant role of hormones in the regulation of flower morphogenesis and development. For TFs, GY-highly expressed bHLH13, PE-highly expressed WRKY33, and GY-highly expressed VIP1, were identified.
Conclusions
Mining of floral organ differential expressed enzymes and TFs helps us to excavate candidate proteins related to floral organ development and to accelerate the breeding of Cymbidium plants.
Similar content being viewed by others


Background
Orchidaceae family, the largest group of flowering plants on our planet, contains a number of horticulturally important and favored ornamental plants (de la Torre Llorente 2018). The genus Cymbidium, consisting of C. goeringii, C. sinense, C. faberi Rolfe, C. tortisepalum, and C. kanran, holds a large share of the flower market in China and other Southeast Asian countries (de la Torre Llorente 2018; Wang et al. 2020). C. goeringii, which is called the spring orchid, blooms from later January to early March; consequently, it is a widely required Spring Festival gift (Yang et al. 2017). Extremely diversified floral morphological patterns and colors make C. goeringii substantial commercial value (Yang et al. 2013).
With increased demand, many fascinating new orchid cultivars with altered floral characteristics have been developed by cross-breeding or mutation-breeding annually (Xiang et al. 2018). Many studies on C. goeringii floral trait-related genes, have been performed (Huang et al. 2012). For example, the FLOWERING LOCUS T (FT) orthologs from C. goeringii have been functional identified as a regulators of the vegetative to reproductive transition (Xiang et al. 2012). The low-temperature-induced transcriptomes of C. goeringii revealed that CgSVP gene plays an essential role in the regulation of flowering by interacting with two important flowering regulators, CgAP1 and CgSOC1 (Yang et al. 2019). Screening of a C. goeringii cDNA library identified a number of B and E class MADS-box genes, including AP-, AP2-, SEP-, DEF-, GLO- and AGL6-like genes, involving in the determination of perianth formation (Xiang et al. 2018). A recent RNA sequencing analysis of C. goeringii identified several floral scent biosynthesis-related genes (Ramya et al. 2019). However, no proteome of C. goeringii has been reported to date. Many flowering and floral development-related genes display floral organ-specific expression patterns (Suzuki et al. 2017). For example, a zinc finger protein EPF1, which is involved in the expression of the 5-enolpyruvylshikimate-3-phosphate synthase-encoding gene, is specifically expressed in petunia petals (Takatsuji et al. 1994). Several floral organ-specific promoters, such as Arabidopsis TCP3 and tobacco AP1-like, function in the regulation of floral transition, initiation and development (Katsutomo et al. 2016; Zhang et al. 2014). Furthermore, some floral organ-related hormones displayed organ-specific accumulation (Liu et al. 2013). In Arabidopsis, stamen development is controlled by organ-specific over-expression of ethylene synthesis gene CsACO2 (Duan et al. 2008). Thus, screening of floral organ-specific genes and proteins may help us to identify the regulation factors that are involved in floral development.
Recently, a gel-free tandem mass spectrometry (MS/MS)-based proteomics method with isobaric labeling reagents has been developed for accurate quantification of proteins (Smolikova et al. 2020). Liquid chromatograph (LC)-MS/MS based comparative proteomics of floral nectars revealed the differential expressed proteins involved in floral defense of various plant species, such as Nicotiana spp., Petunia hybrida and Datura stramonium (Silva et al. 2020). Using the gel-free MS/MS-based proteomics method, a large number of proteins can be obtained and more valuable genetic information can be mined (Hao et al. 2017). Compared with the traditional 2-D method, the quantitative analysis of protein accumulation level is also more accurate, which is helpful to screen differentially expressed proteins among floral organs. In our study, floral organ-specific proteome profiling of metabolic pathways and transcription factors may help to accelerate the breeding of Cymbidium plants.
Results
Overview of the Tandem Mass Tag (TMT) LC-MS/MS data
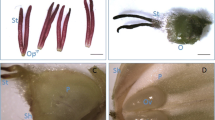
A picture showed the detail sites of the four floral organs, Sepal (SE), petal (PE), lip (LI), and gynostemium (GY) (Fig. 1a). The protein samples were extracted from the four floral organs (Fig. 1b). The basic chart of our experiment process is shown in Fig. 1c. In total, 45,774 peptide-spectral matches, 6626 unique peptides, 2331 protein groups, and 1,855 quantified proteins, were obtained (Fig. 1d). A boxplot of normalized densities are shown in Fig. 1e. The basic information for all the identified proteins, including protein IDs, protein annotations, peptide numbers, unique peptide numbers, sequence coverages, molecular weights, and sequence lengths, are listed in Additional file 1: Table S1.

Overview of the proteomic data. a A picture of typical Cymbidium goeringii flower. Samples from different floral organs, including gynostemium (GY): petal (PE), lip (LI), and sepal (SE) are showed in the picture. b The SDS-PAGE Gel electrophoretic graph for the samples from four different organ groups, including GY, PE, LI, and SE. M: protein marker. c Experimental strategy for the quantitative analysis of proteomes from four different floral organs. d Statistic results of protein identification. e Normalized MS intensity
Identification of the differential expressed proteins (DEPs) between different sample groups
Proteomic profiles of the floral organs, including GY, PE, LI and SE, is shown in Fig. 2a. All identified proteins were grouped into different clusters. In detail, the GY highly expressed proteins were grouped into Cluster VIII (462 proteins), the PE significantly expressed proteins were grouped into Clusters VI (225 proteins) and VII (149 proteins), the LI greatly expressed proteins were grouped into Cluster III (320 proteins), and the SE highly expressed proteins were classed into Cluster V (317 proteins) (Fig. 2b).

Analysis of differentially expressed proteins (DEPs) among the four floral organs. a A heatmap of the abundance of proteins in the four floral organs (N = 2). The heatmap scale ranges from − 2 to + 2 on a log2 scale. b–l MeV cluster analysis showed the numbers of floral organ-specific expressed proteins. The red oval indicated the number of organ specific expressed proteins. j The number of DEPs in different comparisons, including the GY vs. PE, GY vs. LI, GY vs. SE, PE vs. LI, PE vs. SE, LI vs. SE comparisons
The DEPs among different sample groups were analyzed and counted. In detail, 355 up- and 399 down-regulated proteins in the GY vs. PE comparison; 283 up- and 339 down-regulated proteins in the GY vs. LI comparison, 419 up- and 395 down-regulated proteins in the GY vs. SE comparison, 187 up- and 193 down-regulated proteins in the PE vs. LI comparison, 169 up- and 119 down-regulated proteins in the PE vs. SE comparison, and 337 up- and 252 down-regulated proteins in the LI vs. SE comparison, were identified (Fig. 2c). On the basis of the numbers of DEPs, there were great differences between GY and other three sample groups (PE, LI and SE) and limited differences between PE and two other sample groups (LI and SE).
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the DEPs
Most of the DEPs were assigned to 47 metabolic KEGG pathways belonging to 10 major categories. Interestingly, the DEPs in most comparisons were significantly enriched in the primary metabolism-related KEGG categories, including the amino acid-, energy-, and lipid-related categories. In the amino acid category, most of the DEPs were significantly enriched in the ‘biosynthesis of amino acids’ and ‘valine, leucine and isoleucine degradation’ KEGG terms; in the energy category, most of the DEPs were significantly enriched in the ‘carbon metabolism’, ‘pyruvate metabolism’ and ‘citrate cycle’ KEGG terms; and in the lipid category, most of the DEPs were significantly enriched in the ‘fatty acid degradation’, ‘α-linolenic acid metabolism’, ‘fatty acid metabolism’, and ‘biosynthesis of unsaturated fatty acids’ KEGG terms (Fig. 3). Interestingly, no significant KEGG term was identified in the FB vs. SE comparison.

KEGG enrichment analysis of all the DEPs in different comparisons. The significant P values of each KEGG term in the six comparisons were shown by heatmap. The heatmap scale ranges from 0 to 1. The red boxes indicated the significant differentially KEGG terms
Differential expression of amino acid metabolism-related proteins
In our study, 17 amino acid metabolism-related enzymes, including aspartate aminotransferase (AST), arogenate dehydratase 2 (ADT2), glycine cleavage system H protein (GCSH), cysteine synthase (CS), anthranilate synthase (ASA2), asparagine synthetase (ASNS), 3-isopropylmalate dehydrogenase 3 (IMD3), serine hydroxymethyltransferase 1 (SHMT1), isovaleryl-CoA dehydrogenase 1 (IVD1), S-adenosylmethionine synthase (SAM1), chorismate synthase (ARO2), glutamate–glyoxylate aminotransferase 2 (GGAT2), threonine dehydratase (ILV1), 3-isopropylmalate dehydratase 1 (LEU1), d-3-phosphoglycerate dehydrogenase (PHGDH), dihydrolipoyl dehydrogenase (DLD), and amino-methyltransferase (AMT) were identified in orchid (Additional file 2: Table S2).
Among the amino acid metabolism-related proteins, ASA2, ASNS, ADT2 and IMD3 highly expressed in the GY; SAM1, GGAT, ILV1, IVD, and ARO2 greatly expressed in the PE and LI; PHGDH highly expressed in the LI; and CSase, SHMT1, LEU1 and AMT lowly expressed in the GY (Fig. 4a).

Identification of amino acid metabolism- and lipid metabolism-related proteins. a Differential expression of amino acid metabolism-related proteins among four different floral organs. b Differential expression of lipid metabolism-related proteins among four different floral organs. The heatmap scale ranges from − 1.5 to 1.5
Differential expression of lipid metabolism-related proteins
Our study identified 19 fatty acid metabolism-related proteins, including 3-ketoacyl-CoA thiolase 2 (ACAA2), acyl-coenzyme A oxidase 3 (ACOX3), Acyl-[acyl-carrier-protein] desaturase 1 (BRENDA1), acetyl-CoA acetyltransferase (ACAT1), acyl-coenzyme A oxidase 3 (ACOX1), 3-oxoacyl-[acyl-carrier-protein] synthase (ACPS), Acyl-[acyl-carrier-protein] desaturase 2 (BRENDA2), multifunctional protein 1 (MFP1), 3-ketoacyl-CoA thiolase 1 (ACAA1), multifunctional protein 2 (MFP2), acyl-coenzyme A oxidase 4 (ACOX4), aldehyde dehydrogenase 3 (ADH 3), aldehyde dehydrogenase family 2 member B7 (ALDH2B7), alcohol dehydrogenase 3 (ADH3), 1uinone oxidoreductase-like protein 1 (CRYZL1), lipoxygenase 2.1 (LOX2.1), allene oxide synthase (CYP74A), lipoxygenase 2.2 (LOX2.2), Allene oxide cyclase 3 (AOC3), and 12-oxophytodienoate reductase 5 (OPR5), were identified in orchid (Additional file 3: Table S3).
Among the lipid metabolism-related proteins, ALDH2B7, LOX2.1 and OPR5 predominantly expressed in the GY sample; ALDH3 highly expressed in the PE sample; and most of these proteins, such as ACAA2, MEP2, ACAT1, ACOX1, ACPS, ADH3, ACOX4, CYP74A, MFP1, ACOX3, BRENDA2, ACAA1, BRENDA1, LOX2.2, and LOX2.2, greatly expressed in the LI and SE samples (Fig. 4b).
Differential expression of energy metabolism-related proteins
In the glycolysis pathway, a number of proteins, including two phosphoglucomutases (PGMs), one 6-phosphofructokinase (PFK), four fructose-bisphosphate aldolases (FBAs), one triosephosphate isomerase (TPI), three glyceraldehyde-3-phosphate dehydrogenases (GAPDHs), two phosphoglycerate kinases (PGKs), one phosphoglycerate mutase (PGAM), two enolases (ENOs), and six pyruvate kinases (PKs), were identified in orchid (Fig. 5a and Additional file 4: Table S4); for the TCA cycle, three citrate synthases (CSs), five isocitrate dehydrogenases (IDHs), one 2-oxoglutarate dehydrogenase (OGDH), two succinate dehydrogenases (SDHs), one fumarate hydratase 2 (FUM2), four malate dehydrogenases (MDHs), four aconitate hydratase 2 (ACO2) proteins, and one dihydrolipoyllysine-residue succinyltransferase (DLST), were identified in orchid (Fig. 5b and Additional file 4: Table S4); for the pyruvate metabolic pathway, three pyruvate dehydrogenase (PDH)-α subunits, two PDH-β subunits, one dihydrolipoamide acetyltransferase (DLAT), and one aldehyde dehydrogenase (ALDH), were identified in orchid (Fig. 5c and Additional file 4: Table S4).

Identification of energy metabolism-related proteins. a Identification of glycolysis-related proteins. b Differential expression of TCA cycle-related proteins among four different floral organs. c Differential expression of pyruvate metabolic pathway-related proteins among four different floral organs. d Differential expression of energy metabolism-related proteins among four different floral organs. The heatmap scale ranges from − 1.5 to 1.5
The expression patterns of the energy metabolism-related proteins are shown in Fig. 5d. Interestingly, most of these proteins displayed high expression levels in the SE sample.
Differential expression of hormone-related proteins
Phytohormones play important roles in floral organ development (Stewart et al. 2016). In our study, seven auxin-related proteins, including one auxin-binding protein (ABP20), four indole-3-acetic acid (IAA)-amino acid hydrolases (ILLs), one auxin transport protein (BIG), one auxin efflux carrier (PIN3), were identified (Table 1). In the GA signal pathway, one gibberellin-regulated protein 3 and one gibberellin receptor GID1C, were identified. Furthermore, one ethylene signaling pathway-related protein (1-aminocyclopropane-1-carboxylate oxidase 1, ACO1) and one ABA signaling pathway-related protein (abscisic acid receptor, PYL8) were also identified. Expression analysis showed that ACO1 highly expressed in the GY sample, ILL4 and GID1C greatly expressed in the LI sample, and ABP20 significantly expressed in the SE sample.
Differential expression of cell division and pigment production
In total, seven cell division-related proteins, including five Cell Division Cycle family proteins, and two Filamenting Temperature-Sensitive mutant Z (FTS-Z) proteins, were identified (Additional file 5: Table S5). Among these cell division-related proteins, no organ-specific expressed proteins were identified. Furthermore, five pigment production-related proteins, including two Isoflavone reductase like (IRL) proteins, and three CYP family proteins, were detected (Additional file 5: Table S5). Expression analysis showed that two IRL proteins highly expressed in the GY.
Differential expression of transcription factors (TFs)
In total, 15 TFs, including two bHLHs, two CAMTAs, two HBPs, two LWDs, two WRKYs, one AGL9, one BIM2, one MADS2, one PUR1, and one VIP1, were identified (Table 2). Most of these TFs constitutively expressed in the four floral organs. Expression analysis showed that bHLH13 and VIP1 highly expressed in the GY sample and WRKY33 greatly expressed in the PE sample.
Discussion
Owing to its multiple flowering patterns, C. goeringii has a high economic value and is widely favored in East Asia (Chung et al. 2011; Hyun et al. 2012). However, limited genetic information on C. goeringii is available and the molecular mechanism responsible for floral patterning is also largely unknown. In the present study, we have analyzed the spatial protein expression pattern in flowers by comparing the protein profiles of four floral organs.
Comparative proteomic analyses have been previously applied to identify the DEPs in Cymbidium plants (Chen et al. 2018a; Li et al. 2014). Using traditional 2-D technology, a large number of protein spots were detected in C. ensifolium, but only 30 differentially expressed spots were excised and analyzed using MALDI-TOF/TOF (Li et al. 2014). Another 2-D analysis identified 103 DEPs and 104 DEPs responsive to drought in C. sinense and C. tracyanum, respectively (Li et al. 2018). In the present study, we have identified 2331 proteins, which is more than the previously published works. As expected, most of the quantified proteins showed floral organ-specific expression pattern (Fig. 2a). The comprehensive information will help us to investigate novel proteins that are potentially associated with the floral development of C. goeringii.
Although the physiological and molecular characteristics of flowers are well-recognized in the model plants, the primary metabolism during the floral development process of orchids is largely unknown (Muller et al. 2010). Most DEPs were enriched in several primary metabolic pathways, such as amino acid metabolism, energy metabolism, and lipid metabolism pathways (Fig. 3), indicating that significant differences in primary metabolism among four different floral organs of C. goeringii. In flowering plants, carbohydrates provide energy and precursors for the secondary metabolism in floral organs(Borghi and Fernie 2017) and serve as nutritional rewards for pollinators (Roy et al. 2017). In medicinal Chrysanthemum, soluble sugar and amino acid contents were significantly induced during the floral development process (Ma et al. 2016). The cycles of carbohydrate hydrolysis are involved in pollen development, pollen tube growth, and pollination (Pacini et al. 2006). In our study, a number of carbohydrate hydrolysis-related enzymes and their floral organ-specific expression patterns were revealed in C. goeringii (Fig. 4). In flowers, photosynthesis mostly occurs in sepals and young petals, and carbon resources are then transferred to the other floral organs (Muller et al. 2010). C. goeringii flowers have three large and green sepals (Fig. 1a). Interestingly, most of the energy metabolism-related proteins highly expressed in the SE, indicating that it is an important photosynthetic organ of C. goeringii flowers.
In flowering plants, various phytohormones, such as gibberellins (GAs), jasmonates (JAs), auxins, brassinosteroids (BRs), and cytokinins (CKs), play significant roles in the regulation of flower morphogenesis and development (Song et al. 2013). Auxin’s capacity to regulate aspects of growth and development has been deeply characterized in orchids (Novak et al. 2014). For example, auxin plays an essential role in pollination-induced ovary growth and inflorescence initiation in Dendrobium orchids (Ketsa et al. 2006). ABP20, an important auxin receptor protein, controls the first event in the auxin action process (Lazarus et al. 1991). In our study, ABP20 highly expressed in the SE sample, suggesting active auxin signal transduction during SE growth. To regulate the indole-3-acetic acid (IAA) levels, IAA-amino acid hydrolase and IAA-amido synthetase function in the permanent inactivation and temporary storage of auxin (LeClere et al. 2002). In C. goeringii flowers, three out of four ILLs are ubiquitous in all the floral organs, suggesting an essential role of auxin homeostasis in floral patterning and development (Yamaguchi et al. 2017). GID1 is a soluble GA receptor widely identified in various plant species (Nakajima et al. 2006; Ueguchi-Tanaka et al. 2005). In our study, differential expression analysis showed that the gibberellin receptor GID1C of C. goeringii was a LI-specifically expressed protein, suggesting a potential role of GID1C in the growth and development of flower lip. ACO catalyzes the conversion of 1-aminocyclopropane-1-carboxylate (ACC) to ethylene (Nadeau et al. 1993). In petunia flowers, the ACO gene showed a pistil-specific and ethylene-regulated expression pattern (Sanchez and Mariani 2002). The ACO1 of C. goeringii predominantly expressed in the GY organ, indicating a greater level of ethylene in GY than other floral organs. Ethylene plays an important role in the regulation of floral and organ abscission (Kucko et al. 2019). High ethylene accumulation in GY might promote the senescence of floral organs of C. goeringii.
Recently, a number of flowering-related TFs, including the MYB, bHLH and C2H2 families, have been identified in model plants (Chen et al. 2018b; Zhou et al. 2019). In Arabidopsis, CIB1, a typical bHLH TF, is involved in the regulation of floral induction (Wang et al. 2018) and WRKY75 is a positive regulator of flowering initiation (Zhang et al. 2018). CpWRKY71, a WRKY TF of wintersweet, promotes flowering (Huang et al. 2019). Moreover, basic region/leucine zipper motif (bZIP) TFs regulate various biological processes, including signal transduction, defence responses, maturation and flower development (Jakoby et al. 2002). VIP1 is a classic bZIP protein regulating the mannitol responses (Tsugama et al. 2014). In the present study, a GY-highly expressed bHLH13, a PE-highly expressed WRKY33, and a GY-highly expressed VIP1, were identified in C. goeringii. Mining of floral organ differentially expressed TFs may help identify candidate regulators related to floral organ development.
Conclusions
A comprehensive proteomic profile of C. goeringii, aimed at discovering proteins participating in floral organ patterning and development, has been developed. A total of 2,331 protein groups, of which 1,855 proteins were quantified, were identified in four floral organs of C. goeringii. A differential expression analysis showed that most DEPs were enriched in amino acid, lipid, and energy metabolism. Furthermore, hormone-related proteins were identified, suggesting a significant role of hormones in the regulation of flower morphogenesis and development. Three floral organ differentially expressed TFs, bHLH13, WRKY33 and VIP1, were identified, which will aid in the identification of candidate regulators related to floral organ development.
Methods
Plant material and sampling
Five-year-old Cymbidium goeringii seedlings were planted in a greenhouse at Zhejiang Academy of Agriculture Science at a temperature of 26 ± 1 °C with a light/dark cycle of 8/16 h and 65–75% relative humidity. In March 2019, samples from various floral organs, including the SE, PE, LI, and GY, were harvested for organ-specific proteomic analysis. Each sample was collected from 20 independent flowers and a pack of about 6–7 flowers was treated as one biological replicate. There were three biological replicates for each floral organ group.
Protein isolation and extraction
An appropriate 500 mg of samples from each floral organ were ground into power in mortar with liquid N2. After adding of 500 µL lysis buffer, each sample was boiled for 5 min, sonicated for 5 min, and precipitated with trichloroacetic acid solution for 12 h. The lysis buffer contains 4% of sodium dodecyl sulfate, 100 mM Tris, 100 mM dithiothreitol and adjusted to pH 7.8. After 12,000×g centrifugation at 4 °C for 10 min, the supernatants were collected and the remaining debris was discarded. The protein samples were quantified by bicinchoninic acid assay (Walker 1994). Ten µg of each protein sample was added with loading buffer to 5:1 (v/v) and kept in boiling water bath for 5 min. Then, the resulting samples were checked by 8–16% SDS-polyacrylamide gel electrophoresis.
Protein digestion and pretreatment
For protein digestion, 300 µg of each sample was added with dithiothreitol resulting in a final concentration of 100 mM. The samples were kept in boiling water bath for 5 min and cooled until room temperature. The samples were added with 200 µL of urea buffer containing 8 M urea and 150 mM Tris-HCl and centrifuged at 12,000×g for 10 min. After centrifugation, supernatant was discarded. Subsequently, the sample were shaken and alkylated with 100 µL of 50 mM iodoacetamide buffer for 1 min. After 30 min incubation in darkness, the precipitants were extracted by centrifugation at 12,000×g for 15 min. Then, samples were washed by 100 µL of urea buffer twice and followed by 100 µL of NH4HCO3 buffer twice. Finally, the resulting samples were added with 40 µL of trypsin buffer, containing 6 µg trypsin in 40 µL of NH4HCO3 buffer, shaken at 600 rpm at 37 ℃ for 16 h.
For desalination, the resulting samples were put into a tube by centrifugation at 12,000×g for 15 min, and added with 0.1% trifluoroacetic acid solution. The remaining samples were desalinated in a C18 Cartridge (Sigma-Aldrich).
Tandem Mass Tag (TMT) labeling and fractionation of peptides
After peptide quantification, about 100 µg of each sample was harvested and labeled with one unit of TMT reagents as the provider’s instructions (Thermo Fisher Scientific). Briefly, the peptide samples were dissolved in 100 µL of 0.05 M tetraethylammonium bromide solution and the TMT reagents were dissolved in 41 µL of anhydrous acetonitrile solution, respectively. Then, the above two solutions were mixed and incubated for 1 h. The reaction was stopped by adding 8 µL of 5% hydroxylamine for 15 min.
The TMT-labeled peptide solution was separated by a Pierce™ High pH Reversed Phase Peptide Fractionation column on an Agilent high-performance liquid chromatographer (HPLC) system. Thirty fractions were harvested for each sample and concatenated to 15 fractions for further analysis (Yu et al. 2020).
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis
After re-dissolution, protein samples were used for LC-MS/MS analysis. Samples were chromatographically separated by a nano flow rate EasynLC1200 chromatographic system (Thermo-Scientific). The solvent system consisting of Solution A (0.1% aqueous solution of formic acid) and solution B (0.1% formic acid and 85% acetonitrile). Chromatographic column was balanced with 95% of solution A at a flow rate of 0.75. The sample was uploaded to Trap C18 column (100 μm × 20 mm, 5 μm) and was separated by C18 chromatographic column (75 μm × 150 mm, 3 μm). The liquid phase separation gradient is set as follows: 0–5 min, B solution from 5 to 8%; 5–50 min, B solution from 8 to 23%; 50–60 min, B solution from 23 to 40%; 60–65 min, B solution from 40 to 100%; 65–75 min, B solution maintain at 100%. The following ion source parameters, including spray voltage 1.8 kV, capillary temperature 275 °C and declustering potential 100 V, were set. The mass spectrometer was run using a data-dependent Top-20 acquisition mode, switching automatically between MS and MS/MS.
After separation, data dependent acquisition MS was carried out by Q Exactive Plus mass spectrometer (Thermo Scientific). The essential parameters were shown as follows: analysis time is set at 75 min; detection mode is set at positive ion; parent ion scanning range is set from 300 to 1800 m/z; first stage mass spectrometry resolution (70,000) is set at m/z 200; AGC target is set at 3E6, and Level 2 maximum IT is set at 30 ms. Peptide secondary mass spectrometry analysis was performed according to the following sets: 20 highest intensity parent ions were collected after each full scan, second stage mass spectrometry resolution (17,500) is set at m/z 200, AGC target is set at 1e5, Level 2 MaTimum is set at 60 ms, and MS2 Activation Type is set at HCD.
Database search
The raw LC-MS/MS data is uploaded into MaxQuant software (ver. 1.6.0.16) for database searching. The database used in the library is UniProt-Apostasia shenzhenica-21681-20181217, from the protein database on the web site https://www.uniprot.org/uniprot/?query=taTonomy:10090. The number of proteins is 21,681. A precursor mass 6 ppm was set as initial search. The main parameters are set as follows: isobaric label is TMT 6pleT, enzyme is Trypsin, reporter mass tolerance is set at 0.005 Da, MaT missed cleavage is 2, peptide search tolerance is set at 4.5 ppm, peptide tolerance of first round search is 20 ppm. MS/MS tolerance is 20 ppm, FiTed modification is carbamidomethyl, and variable modifications are OTidation and Acetyl. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD019636.
Protein quantification and differentially expressed protein (DEPs)
The intensity ratios of the TMT reporter ions in MS/MS from raw data were applied to analyze the fold changes of each protein between two different groups. The expression level of each protein was mean-normalized to center the distribution of quantitative values. Protein quantitation was calculated as the median ratio of corresponding unique peptides. DEPs were screened in six different comparisons with cut-off of ratio fold as > 1.2 or < 0.83 and P values < 0.05. Expression levels of proteins were analyzed by hierarchical clustering method. To reach the criterion of the hierarchical clustering, the significant P values were transformed into Z-score by log transformation.
Enrichment analysis of the DEPs
A two-tailed Fisher’s exact text was performed to analyze Gene Ontology (GO) and Encyclopedia of Genes and Genomes (KEGG) enrichment of the DEPs against all identified proteins. The categories that were at least enriched in one of the clusters with P value < 0.05 were filtered out, and the filtered P values were transformed by the formula: adjusted P = −log10 (P value).
Availability of data and materials
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD019636.
References
Borghi M, Fernie AR (2017) Floral metabolism of sugars and amino acids: implications for pollinators’ preferences and seed and fruit set. Plant Physiol 175:1510–1524
Chen C, Zeng L, Ye Q (2018a) Proteomic and biochemical changes during senescence of Phalaenopsis ‘Red Dragon’ petals. Int J Mol Sci 19:1317
Chen D, Yan W, Fu LY, Kaufmann K (2018b) Architecture of gene regulatory networks controlling flower development in Arabidopsis thaliana. Nat Commun 9:4534
Chung MY, Nason JD, Chung MG (2011) Significant demographic and fine-scale genetic structure in expanding and senescing populations of the terrestrial orchid Cymbidium goeringii (Orchidaceae). Am J Bot 98:2027–2039
de la Torre Llorente D (2018) Conservation status of the family Orchidaceae in spain based on European, National, and Regional Catalogues of Protected Species. Sci World J. 2018:7958689
Duan QH, Wang DH, Xu ZH, Bai SN (2008) Stamen development in Arabidopsis is arrested by organ-specific overexpression of a cucumber ethylene synthesis gene CsACO2. Planta 228:537–543
Hao J, Guo H, Shi X, Wang Y, Wan Q, Song YB, Zhang L, Dong M, Shen C (2017) Comparative proteomic analyses of two Taxus species (Taxus × media and Taxus mairei) reveals variations in the metabolisms associated with paclitaxel and other metabolites. Plant Cell Physiol 58:1878–1890
Huang W, Fang Z, Zeng S, Zhang J, Wu K, Chen Z, Teixeira da Silva JA, Duan J (2012) Molecular cloning and functional analysis of three FLOWERING LOCUS T (FT) homologous genes from Chinese Cymbidium. Int J Mol Sci 13:11385–11398
Huang R, Liu D, Huang M, Ma J, Li Z, Li M, Sui S (2019) CpWRKY71, a WRKY transcription factor gene of Wintersweet (Chimonanthus praecox), promotes flowering and leaf senescence in Arabidopsis. Int J Mol Sci 20:5325
Hyun YS, Kim J, Chung KW (2012) Development of polymorphic microsatellite markers for Cymbidium goeringii (Orchidaceae). Am J Bot 99:e193–198
Jakoby M, Weisshaar B, Droge-Laser W, Vicente-Carbajosa J, Tiedemann J, Kroj TF, Parcy F (2002) bZIP transcription factors in Arabidopsis. Trends Plant Sci 7:106–111
Katsutomo S, Yamaguchi H, Kasajima I, Narumi T, Ohtsubo N (2016) Generation of novel floral traits using a combination of floral organ-specific promoters and a chimeric repressor in Torenia fournieri Lind. Plant Cell Physiol. 57(6):1319–31
Ketsa S, Wisutiamonkul A, van Doorn WG (2006) Auxin is required for pollination-induced ovary growth in Dendrobium orchids. Funct Plant Biol 33:887–892
Kucko A, Wilmowicz E, Ostrowski M (2019) Spatio-temporal IAA gradient is determined by interactions with ET and governs flower abscission. J Plant Physiol 236:51–60
Lazarus CM, Napier RM, Yu LX, Lynas C, Venis MA (1991) Auxin-binding protein--antibodies and genes. Symp Soc Exp Biol 45:129–148
LeClere S, Tellez R, Rampey RA, Matsuda SP, Bartel B (2002) Characterization of a family of IAA-amino acid conjugate hydrolases from Arabidopsis. J Biol Chem 277:20446–20452
Li X, Xu W, Chowdhury MR, Jin F (2014) Comparative proteomic analysis of labellum and inner lateral petals in Cymbidium ensifolium flowers. Int J Mol Sci 15:19877–19897
Li JW, Chen XD, Hu XY, Ma L, Zhang SB (2018) Comparative physiological and proteomic analyses reveal different adaptive strategies by Cymbidium sinense and C. tracyanum to drought. Planta 247:69–97
Liu D, Liu X, Meng Y, Sun C, Tang H, Jiang Y, Khan MA, Xue J, Ma N, Gao J (2013) An organ-specific role for ethylene in rose petal expansion during dehydration and rehydration. J Exp Bot 64:2333–2344
Ma CH, Chu JZ, Shi XF, Liu CQ, Yao XQ (2016) Effects of enhanced UV-B radiation on the nutritional and active ingredient contents during the floral development of medicinal chrysanthemum. J Photochem Photobiol B 158:228–234
Muller GL, Drincovich MF, Andreo CS, Lara MV (2010) Role of photosynthesis and analysis of key enzymes involved in primary metabolism throughout the lifespan of the tobacco flower. J Exp Bot 61:3675–3688
Nadeau JA, Zhang XS, Nair H, O’Neill SD (1993) Temporal and spatial regulation of 1-aminocyclopropane-1-carboxylate oxidase in the pollination-induced senescence of orchid flowers. Plant Physiol 103:31–39
Nakajima M, Shimada A, Takashi Y, Kim YC, Park SH, Ueguchi-Tanaka M, Suzuki H, Katoh E, Iuchi S, Kobayashi M, Maeda T, Matsuoka M, Yamaguchi I (2006) Identification and characterization of Arabidopsis gibberellin receptors. Plant J Cell Mol Biol. 46:880–889
Novak SD, Luna LJ, Gamage RN (2014) Role of auxin in orchid development. Plant Signal Behav 9:e972277
Pacini E, Guarnieri M, Nepi M (2006) Pollen carbohydrates and water content during development, presentation, and dispersal: a short review. Protoplasma 228:73–77
Ramya M, Park PH, Chuang YC, Kwon OK, An HR, Park PM, Baek YS, Kang BC, Tsai WC, Chen HH (2019) RNA sequencing analysis of Cymbidium goeringii identifies floral scent biosynthesis related genes. BMC Plant Biol 19:337
Roy R, Schmitt AJ, Thomas JB, Carter CJ (2017) Review: Nectar biology: From molecules to ecosystems. Plant Sci 262:148–164
Sanchez AM, Mariani C (2002) Expression of the ACC synthase and ACC oxidase coding genes after self-pollination and incongruous pollination of tobacco pistils. Plant Mol Biol 48:351–359
Silva F, Guirgis A, von Aderkas P, Borchers CH, Thornburg R (2020) LC-MS/MS based comparative proteomics of floral nectars reveal different mechanisms involved in floral defense of Nicotiana spp., Petunia hybrida and Datura stramonium. J Proteomics 213:103618
Smolikova G, Gorbach D, Lukasheva E, Mavropolo-Stolyarenko G, Bilova T, Soboleva A, Tsarev A, Romanovskaya E, Podolskaya E, Zhukov V, Tikhonovich I, Medvedev S, Hoehenwarter W, Frolov A (2020) Bringing new methods to the seed proteomics platform: challenges and perspectives. Int J Mol Sci 21:9162
Song S, Qi T, Huang H, Xie D (2013) Regulation of stamen development by coordinated actions of jasmonate, auxin, and gibberellin in Arabidopsis. Mol Plant 6:1065–1073
Stewart D, Graciet E, Wellmer F (2016) Molecular and regulatory mechanisms controlling floral organ development. FEBS J 283:1823–1830
Suzuki JY, Amore TD, Calla B, Palmer NA, Scully ED, Sattler SE, Sarath G, Lichty JS, Myers RY, Keith LM, Matsumoto TK, Geib SM (2017) Organ-specific transcriptome profiling of metabolic and pigment biosynthesis pathways in the floral ornamental progenitor species Anthurium amnicola Dressler. Sci Rep 7:1596
Takatsuji H, Nakamura N, Katsumoto Y (1994) A new family of zinc finger proteins in petunia: structure, DNA sequence recognition, and floral organ-specific expression. Plant Cell 6:947–958
Tsugama D, Liu S, Takano T (2014) Analysis of functions of VIP1 and its close homologs in osmosensory responses of Arabidopsis thaliana. PLoS ONE 9:e103930
Ueguchi-Tanaka M, Ashikari M, Nakajima M, Itoh H, Katoh E, Kobayashi M, Chow TY, Hsing YI, Kitano H, Yamaguchi I, Matsuoka M (2005) GIBBERELLIN INSENSITIVE DWARF1 encodes a soluble receptor for gibberellin. Nature 437:693–698
Walker JM (1994) The bicinchoninic acid (BCA) assay for protein quantitation. Methods Mol Biol 32:5–8
Wang S, Li L, Xu P, Lian H, Wang W, Xu F, Mao Z, Zhang T, Yang H (2018) CRY1 interacts directly with HBI1 to regulate its transcriptional activity and photomorphogenesis in Arabidopsis. J Exp Bot 69:3867–3881
Wang H, Park SY, Song SH, San ML, Kim YC, Ham TH, Kim DY, Kim TS, Lee J, Kwon SW (2020) Analysis of complete chloroplast genome sequence of Korean landrace Cymbidium goeringii. 3 Biotech 10:29
Xiang L, Li X, Qin D, Guo F, Wu C, Miao L, Sun C (2012) Functional analysis of FLOWERING LOCUS T orthologs from spring orchid (Cymbidium goeringii Rchb. f.) that regulates the vegetative to reproductive transition. Plant physiol Biochem. 58:98–105
Xiang L, Chen Y, Chen L, Fu X, Zhao K, Zhang J, Sun C (2018) B and E MADS-box genes determine the perianth formation in Cymbidium goeringii Rchb.f. Physiol Plant 162:353–369
Yamaguchi N, Huang J, Xu Y, Tanoi K, Ito T (2017) Fine-tuning of auxin homeostasis governs the transition from floral stem cell maintenance to gynoecium formation. Nat Commun 8:1125
Yang JB, Tang M, Li HT, Zhang ZR, Li DZ (2013) Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol Biol 13:84
Yang F, Zhu G, Wei Y, Gao J, Liang G, Peng L, Lu C, Jin J (2019) Low-temperature-induced changes in the transcriptome reveal a major role of CgSVP genes in regulating flowering of Cymbidium goeringii. BMC Genomics 20:53
Yu C, Luo X, Zhang C, Xu X, Huang J, Chen Y, Feng S, Zhan X, Zhang L, Yuan H, Zheng B, Wang H, Shen C (2020) Tissue-specific study across the stem of Taxus media identifies a phloem-specific TmMYB3 involved in the transcriptional regulation of paclitaxel biosynthesis. Plant J Cell Mol Biol 103:95–110
Zhang J, Yan G, Wen Z, An YQ, Singer SD, Liu Z (2014) Two tobacco AP1-like gene promoters drive highly specific, tightly regulated and unique expression patterns during floral transition, initiation and development. Planta 239:469–478
Zhang L, Chen L, Yu D (2018) Transcription factor WRKY75 interacts with DELLA proteins to affect flowering. Plant Physiol 176:790–803
Zhou A, Sun H, Dai S, Feng S, Zhang J, Gong S, Wang J (2019) Identification of transcription factors involved in the regulation of flowering in Adonis amurensis through combined RNA-seq transcriptomics and iTRAQ proteomics. Genes 10:305
Acknowledgements
We thanks Dehui Qin (Zhejiang Academy of Agriculture Science) for reading this manuscript and Xiaozhong Chen (Zhejiang Orchid Association) and Dechu Ye (Xuekou Town Government Resident) for providing plant materials.
Funding
This study was supported by the National Natural Science Foundation of China (31801891, 41907213), the Tree Breeding Project of Zhejiang Province, China (2016C02065). There is no role of the funding body in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
CS conceived and designed the study. QS collected the samples. YC, ZX, and QS performed the experiments. YC and QS analyzed the data. CS wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The basic information of all identified proteins.
Additional file 2: Table S2.
The basic information of amino acid metabolism-related proteins.
Additional file 3: Table S3.
The basic information of fatty acid metablism-related proteins.
Additional file 4: Table S4.
The basic information of energy metabolism-related proteins.
Additional file 5: Table S5.
The basic information of cell division and pigment related proteins.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Xu, Z., Shen, Q. et al. Floral organ-specific proteome profiling of the floral ornamental orchid (Cymbidium goeringii) reveals candidate proteins related to floral organ development. Bot Stud 62, 23 (2021). https://doi.org/10.1186/s40529-021-00330-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40529-021-00330-9