
Abstract
Background
Fusaric acid (FA) has been shown to stimulate the rapid development of disease symptoms, such as necrosis and foliar desiccation. In this study, we have evaluated the phytotoxicity of FA on tomato plants (Solanum lycopersicum L.). FA induced necrotic lesions in detached leaves, which are reminiscent of hypersensitive response (HR) lesions induced by plant-pathogen interactions and other abiotic stress factors.
Results
FA-treated tomato leaves exhibited visible necrotic lesion as a result of cell death which was evident by Evans blue staining, enhanced reactive oxygen species (ROS) levels and DNA degradation. Changes in the generation of O2.- and H2O2 as well as the activities of superoxide dismutase (SOD), catalase (CAT) and ascorbate peroxidase (APX) were examined in FA-treated tomato leaves. It was observed that FA exposure stimulated oxidative burst in the leaves, resulting in a lasting activation of O2.- and H2O2 production. After first day of FA application, the H2O2 scavenging enzymes CAT and APX showed a strong activity decrease followed by gradual recovery to the control level after 2 and 3 days.
Conclusion
A concomitant increase in ROS production, the down regulation of antioxidative enzymes activities and upregulation of lipid peroxidation were crucial for the onset of cell death. These results suggested that FA-induced damage might result from ROS pathways. Thus, our experiments provide a useful model plant system for research on FA-induced plant cell death.
Similar content being viewed by others


Background
Fusaric acid (FA), also known as 5-butylpicolinic acid, is a non-host specific phytotoxin of Fusarium species and suspected of being involved in pathogenecity (Gäumann [1957], Gäumann [1958]). FA has been shown to stimulate the rapid development of some disease symptoms, such as interveinal necrosis and foliar desiccation (Sutherland and Pegg [1992]). It has been detected in plants after Fusarium attack and was present in much higher concentration in plant tissues infected with a virulent strain than in those infected with an avirulent one (Harborne [1989]). At the subcellular level, FA has been found to induce many physico-chemical effects (Kuzniak et al. [1999]; Wu et al. [2008]; Sapko et al. [2011]). The effects of FA have also been described in terms of changes in the proton electrochemical gradient across the plasma membrane, increase in loss of electrolytes, decrease in cellular ATP level, and inhibition of some metalloenzymes (e.g. cytochrome oxidase) leading to respiratory impairment (Marre et al. [1993]; D’Alton and Etherton [1984]; Arias [1985]). Most studies have reported that FA shows toxic effects at concentration greater than 10-5 M. FA participates in fungal pathogenicity by decreasing plant cell viability. However, FA is also produced by non-pathogenic Fusaria, potential biocontrol agents for vascular wilt diseases in which FA at non-toxic concentrations (below 10-6 M) activates signal transduction components necessary for plant defense responses that contribute to biocontrol activity (Bouizgarne et al. [2006]).
Plants have developed complex protection systems to cope with pathogen attack. It has been reported that reactive oxygen species (ROS) and the enzymatic systems that govern their metabolism, for example superoxide dismutase (SOD), catalase (CAT) and peroxidases may play a role in pathogenesis (Baker and Orlandi [1995]). ROS, which are rapidly produced in plant cells after pathogen attack, are potentially involved in many defense processes including the hypersensitive response (HR), phytoalexin synthesis and oxidative cross-linking of plant cell wall proteins. Many non-host selective microbial phytotoxins are known to induce a generation of ROS and in many cases, disease symptom development can be explained by this mechanism (Heiser et al. [1998]). Therefore, the possibility that ROS might also be involved in the response to FA has been of interest.
Several lines of evidences suggest that, from among the generated ROS, H2O2 plays a central role in these plant defense responses (Mehdy et al. [1996]). On the other hand ROS exhibit deleterious impact on the cellular components and so plants possess effective mechanisms for detoxification of ROS in order to protect themselves from the toxic effects. SOD catalyze the dismutation of O2.- radicals to O2 and H2O2, and CAT and ascorbate peroxidase (APX) remove H2O2, thus playing a role of antioxidative key enzymes. Moreover, the Guaiacol type peroxidases may use H2O2 in the process of cell wall reinforcement by catalyzing the polymerization of cinnamyl groups into lignin, suberization and deposition of phenolic compounds. Hence the balance between ROS generation and breakdown may be critical for the outcome of plant-pathogen interaction. After failure of this balance, ROS accumulation occurs. Recently, the prominent role of ROS has been revealed in the induction, signalling and execution of plant cell death (Jabs [1999]; Apel and Hirt [2004]; Karuppanapandian et al. [2011]). The initial lower level of ROS may stimulate the production of higher amount of ROS through feedback regulation. The change of ROS content can trigger different signalling cascades leading to cell death (Breusegem and Dat [2006]). Moreover, there was a strong interplay between ROS and other signalling molecules (phytohormones) such as ethylene and salicylic acid (SA) during cell death (Overmyer et al. [2005]). Toxic level of ROS usually react with numerous cell components, causing a cascade of oxidative reactions and the consequent inactivation of enzymes, protein degradation and nucleic acid damage. In addition, excessive ROS can react with membrane lipids, generating lipid peroxides that can in turn lead to further damaging intermediates, such as malondialdehyde (MDA), to the point of inducing cellular dysfunction and/or cell death (Paciolla et al. [2004]; Maksymiec and Krupa [2006]; Schraudner et al. [1998]).
The present investigation portrays the phytotoxic action of FA towards tomato (Solanum lycopersicum L.). HR-like lesions were observed in excised leaves after FA infiltration. As the first step in elucidating the possible mechanisms of toxicity of FA against tomato, we analyzed the role of ROS in response to FA and the changes in the levels of several antioxidant enzymes triggered by FA stress. Microscopy and staining techniques were used to verify the toxicity and to identify the visualization of H2O2 and O2.-in situ. Our experiments provide a useful model system for the research on plant cell death resulting from FA.
Methods
Plant material and growth conditions
Tomato (Solanum lycopersicum L.) belongs to Solanaceae family and is an important model plant. The seeds of tomato (cv. Sel-7, susceptible to Fusarium oxysporum f. sp. Lycopersici) were obtained from Indian Institute of Vegetable Research, Varanasi, India and plants were grown in soil in green house with 14 h light and 10 h dark regime, at 27 ± 0.5°C. The plants were grown in thermocol pots of 6 cm diameter and 15 cm height containing autoclaved soil. The plants were allowed to grow and when they attained 6-8 leaves (3-4 weeks old), they were treated with FA.
FA treatment
Fusaric acid was extracted and purified from the culture filtrate of Fusarium oxysporum f. sp. lycopersici (causal agent of Fusarium wilt of tomato) (Venter and Steyn [1998]) grown at 25°C in 500 ml flasks containing 100 ml potato dextrose broth (PDB) and then analysed by TLC (Stefan [2005]) and HPLC (Venter and Steyn [1998]). The purified and characterized FA was stored at 4°C until further use. Crystalline FA standard (5-butylpicolinic acid) was supplied by MP Biomedicals, CA, USA.
Fusaric acid treatment of tomato leaves was performed by infiltration of leaf tissue with the help of a 1 ml syringe fitted with 25 guage needle in the midrib just above the petiole on the lower side of the leaf. One ml FA solution was infiltrated to each leaflet of fully expanded tomato leaves (Kuzniak et al. [1999]). Control plants were infiltrated with sterile distilled water. The second and third fully expanded leaves from the plant base were used for the experiments. Oxidative burst, antioxidant enzyme activities, lipid peroxidation and ethylene evolution in the leaves of tomato plants were measured at 0, 4, 8, 12, 16, 20, 24, 36, 48 and 72 h after FA treatment.
Measurement of cell death
Evans blue staining was used as a marker of cell death and cell death was measured in terms of Evans blue uptake 72 h after infiltration with various concentrations of FA (50 μg/ml, 100 μg/ml, 150 μg/ml, 200 μg/ml, 250 μg/ml and 300 μg/ml). Control leaves were infiltrated with sterile distilled water.
Evans blue staining to assess cell death was carried out by gentle heating the FA-treated and control leaves for 1 min in a freshly prepared solution of phenol, lactic acid, glycerol and distilled water (1:1:1:1) containing 20 mg/ml Evans blue. After boiling, leaf tissues were then clarified overnight in a solution of 2.5 gm/l chloral hydrate on a platform shaker at 27°C and 160 rpm. The leaf tissues were then transferred on glass slides and observed under the light microscope to see the amount of cell death in FA-treated as well as control leaves.
To estimate the amount of cell death in tomato leaves induced by FA treatment, the methodology of Baker and Mock ([1994]) was followed. The FA-treated and control leaf tissues were submerged in 10 ml plastic beakers containing 1 ml of 0.25% Evans blue and incubated on a platform shaker at 27°C and 80 rpm for 20 min. The beaker contents were poured into a small Buchner funnel and the tissues were rinsed well with deionized water until no more blue stain was eluted. The tissues were then transferred to 1.5 ml microfuge tubes. One half ml of 1% aqueous sodium dodecyl sulphate (SDS) was added in each tube to release the trapped Evans blue from the cells. The tissues were then ground using a pestle-mortar and the homogenates were diluted with 0.5 ml deionized water. The tubes were vortexed and centrifuged at 9000 g for 3 min. 0.8 ml aliquot of the supernatant was removed and optical density was determined spectrophotometrically (UV-VIS Spectrophotometer 2202, Systronics, India) at 600 nm.
Nitro blue tetrazolium reducing activity
Detached leaves from the plants were subjected to the treatments and their respective controls were immersed in 50 mM potassium phosphate buffer (pH 7.8) containing 0.1% nitroblue tetrazolium (NBT) and 10 mM sodium azide (NaN3). The leaves were vacuum infiltrated during 2 min and then incubated for 2 h in the dark (without vacuum) and further immersed in 96% (v/v) ethanol to completely eliminate the chlorophyll. Superoxide production was visualized as a purple formazan deposit within the leaf tissues. The leaves of the plants that received no treatment were also infiltrated with 50 mM potassium phosphate buffer (pH 7.8) containing only 10 mM NaN3 that served as control.
Measurement of NBT reduction, a method used for the determination of O2.-, was as described by Doke ([1983]). FA-treated as well as control leaves were immersed in 3 ml 10 mM potassium phosphate buffer (pH 7.8) containing 0.05% NBT and 10 mM NaN3 for 1 h. Then the mixture was heated at 85°C for 15 min. and cooled rapidly. The activity of leaf tissues to reduce NBT was expressed as increased absorbance at 580 nm per h per g fresh weight (FW).
Estimation of H2O2 level
Leaves treated with FA were cut with razor blade 1 cm above the base of the petiole and immediately placed in a beaker containing 1 mg/ml 3,3′-diaminobenzidine hydrochloric acid (DAB-HCl), adjusted to pH 5.6 with NaOH and were incubated in a humid growth chamber for 8 hours in the dark. The leaves were kept in the vertical position with 5 mm of the basal part dipped into the DAB solution. After DAB uptake, the leaves were cleared in 96% boiling ethanol, and examined with light microscope. H2O2 is visualized as a reddish-brown coloration.
The H2O2 produced was determined according to Sagisaka ([1976]). The FA-treated and control plant leaf tissue were homogenized in 5% cold trichloro acetic acid (TCA) and the homogenate was centrifuged at 17000 g for 10 min at 0°C. The reaction mixture contained 1.6 ml of supernatant, 0.4 ml of 50% TCA, 0.4 ml of ferrous ammonium sulphate and 0.2 ml of potassium thiocynate. The absorbance was recorded at 480 nm after 15 min of incubation. The amount of H2O2 was estimated by a calibration curve prepared with known concentrations of H2O2.
Determination of lipid peroxidation
Lipid peroxidation was determined by measuring the amount of MDA (Heath and Packer [1968]), the end product of peroxidation of polyunsaturated fatty acids after treatment with various cell death inducers. The FA-treated and control leaf tissues were cut into small pieces and homogenized by the addition of 1 ml of 0.1% cold TCA. The homogenates were then transferred into fresh tubes and centrifuged at 10000 g for 20 min at room temperature. To 1 ml supernatant, 1 ml 20% TCA containing 0.5% thiobarbituric acid (TBA) (freshly prepared) and 0.01 ml butylated hydroxyl toluene (BHT) prepared 4% solution in ethanol were added into a new tube and incubated at 96°C for 35 min. The tubes were then transferred into ice bath and kept for 5 min followed by centrifugation at 10000 g for 5 min. The absorbance of the supernatant was recorded at 532 nm and corrected for non-specific turbidity by substracting the absorbance at 600 nm. The concentration of MDA was calculated from its molar extinction coefficient 156 mM/cm.
Ethylene production assay
For ethylene production, the FA-treated and control leaf tissues of tomato plants were excised and placed in test tubes for 30 min to allow the escape of wound ethylene (Lund et al. [1998]). After this the tubes were sealed for 2 h. Gaseous samples were then analysed for ethylene content on gas chromatograph (Model CP-3800 GC, Varian, Inc. CA, USA) equipped with an flame ionization detector (FID) and a column (CP-Pora PLOT Q, 25 m × 0.32 mm) packed with fused silica. For measuring ethylene content 1 ml gas sample was injected into the GC column using a Hamilton gastight syringe (Model: 701 RN). The column, injector and detector temperatures were set at 110°C, 130°C and 130°C respectively. Nitrogen gas at a flow rate of 30 ml/min was used as the carrier. The ethylene content was quantified by comparison of peak areas with standard curve constructed from known amounts of ethylene gas (SSG, Alltech Asso. Inc. USA).
Assays of antioxidative enzyme activities
The leaves were homogenized (1:5 w/v) in 1 M NaCl in 50 mM potassium phosphate buffer (pH 7.0) containing 1% polyvinyl pyrrolidone (PVP) and 1 mM EDTA. For assay of APX, leaf extracts were prepared in the same medium containing 1 mM sodium ascorbate. The homogenate was centrifuged and the supernatant was used as an enzyme extract to assay SOD, CAT and APX activities.
SOD activity assay was based on the method of Beauchamp and Fridovich ([1971]) which measures the inhibition of the photochemical reduction of NBT. In the spectrophotometric assay, the 3 ml mixture contained 50 mM potassium phosphate buffer (pH 7.8), 13 mM L-methionine, 75 μM NBT, 0.1 mM EDTA, 2 μM riboflavin and enzyme extract. The riboflavin was added last and the reaction was started by placing the tubes under two 15 W fluorescent lamps. The reaction was terminated after 10 min by removal from the light source. Non-illuminated and illuminated tubes without enzyme extract served as control. The absorbance was measured at 560 nm. The volume of enzyme extract corresponding to 50% inhibition of the reaction was considered as one unit of enzyme activity.
CAT activity was measured spectrophotometrically according to Dhindsa et al. ([1981]). The assay mixture contained 50 mM potassium phosphate buffer (pH 7.0), 15 mM H2O2 and enzyme extract. The decomposition of H2O2 (absorbance coefficient of 45.2/mM cm) was measured at 240 nm at 25°C.
APX was assayed following the oxidation of ascorbate to dehydroascorbate at 265 nm (an absorbance coefficient of 13.7/mM cm) by a modified method of Nakano and Asada ([1981]). The assay mixture contained 50 mM potassium phosphate buffer (pH 7.0), 0.25 mM sodium ascorbate, 25 μM H2O2 and enzyme extract. Addition of H2O2 started the reaction. The rates were corrected for the non-enzymatic oxidation of ascorbate by the inclusion of reaction mixture without enzyme extract.
Protein was determined by the method of Bradford ([1976]), with standard curves prepared using bovine serum albumin (Sigma).
Plant genomic DNA extraction and analysis
Genomic DNA from the FA-treated and control plant leaf tissue was isolated by Doyle and Doyle CTAB method (Doyle and Doyle [1987]). For DNA extraction, 7.5 ml of CTAB isolation buffer was preheated in a 30 ml centrifuge tube to 60°C in a water bath. 1.0 g leaf tissue was ground in a 60°C CTAB isolation buffer in a preheated mortar. The sample was then incubated at 60°C for 30 min with occasional gentle swirling. The extraction was done once with equal volume of chloroform: isoamyl alcohol in a ratio of 24:1 by mixing gently but thoroughly. The solution was then centrifuged at room temperature at 10000 g for 10 min. Aqueous phase of the solution was aspirated with wide bore pipette and transferred to a clean centrifuge tube. Then 2/3rd volumes of cold isopropanol was added and mixed gently and left for 12 h at 4°C to precipitate nucleic acid. It was then spinned at room temperature for 5 min at 10000 g and the supernatant was gently poured off as much as possible without losing the precipitate. To the pellet 5 ml wash buffer was added and swirled gently to resuspend DNA. The pellet was washed with wash buffer for 25 min at room temperature. It was followed by a brief spin at room temperature. The supernatant was poured off carefully and the pellet was allowed to air dry briefly at room temperature. Dried DNA was resuspended in 1 ml TE solution.
To check the quality and effect of FA treatment, the extracted DNA was run on 1.2% agarose gel along with molecular weight marker. The gel was run in 0.5X TBE at 100 V.
Statistical analyses
Experimental design was completely randomized. All the experiments were carried out in triplicates and repeated three times. The mean, standard error and One-way ANOVA were calculated using the average data of the experiment. Analysis of variance (ANOVA) was calculated at 5% probability level according to the method described by Gomez and Gomez ([1984]). The mean seperations were carried out using Duncan’s multiple range tests (Duncan [1955]) and significance was determined at p < 0.05.
Results
Fusaric acid induced cell death
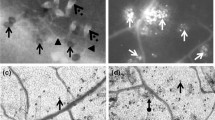
The phytotoxic effect of FA was observed on the leaves of tomato plant. The leaves treated with FA exhibited visible necrotic lesions (Figure 1A, B) as a result of cell death. Evans blue staining was used as a marker of cell death (Figure 1C, D). The dead cells were stained blue whereas the untreated ones appeared unstained under the light microscope (Figure 1E, F, G). The cell death was observed to increase with increasing concentration of FA (Figure 2). Maximum cell death was induced by 250 μg/ml of FA as observed by Evans blue uptake. FA at higher concentration of 300 μg/ml also showed similar amount of cell death and the cell death induced by 250 μg/ml and 300 μg/ml of FA was almost similar. Hence, the concentration of 250 μg/ml for FA was selected for further experiments to induce cell death in the leaves of tomato plant.

Effect of FA (250 μg/ml concentration) on the cell death of tomato leaves. (A) Control leaves, (B) FA treated leaves showing necrotic lesions, (C and D) Evans blue staining of control and FA-treated leaves showing cell death, (E) Microscopic observation of unstained control leaf tissue, (F and G) Microscopic observation of stained tissues of FA-treated leaves at 24 h and 72 h after treatment respectively.

Cell death in the leaves of tomato plant treated with various concentrations of FA after 72 h (A, Optical density). Bars represent ± standard error. Different letters indicate that values are significantly different (p < 0.05).
FA induced oxidative burst
The production of O2.- was studied by NBT reduction which gave rise to a purple formazan precipitate (Figure 3A). Intense staining of leaves by NBT confirmed the presence of high amount of superoxide anion radical in the FA treated leaves after 12 h. The accumulation of H2O2 was visualized by DAB staining where reddish-brown coloration was observed (Figure 3B).

Detection of ROS generation in the tomato leaves treated with FA. (A) Accumulation of O2.- in the tomato leaves by NBT staining method at 12 h of FA treatment. (B) Localization of H2O2 in the leaves of tomato by DAB staining method at 24 h of FA treatment.
In FA-treated leaves, an increase in NBT reducing activity was detected as early as 4 h after the treatment and sustained for 2 days. The result indicated that FA treatment evoked burst of O2.- in tomato leaves. The elevated peak was observed at 48 h after the FA treatment, then after the O2.- level declined (Figure 4A).

Measurement of ROS generation in the tomato leaves treated with FA. (A) O2.- production in the leaves of tomato plant treated with FA. Bars represent ± standard error. Different letters indicate that values are significantly different (p < 0.05). (B) H2O2 production in the leaves of tomato plant treated with FA. Bars represent ± standard error. Different letters indicate that values are significantly different (p < 0.05).
The leaves treated with FA showed enhanced H2O2 release. The amount of H2O2 started to increase from 8 h after FA application that reached to maximum at 48 h and decreased thereafter (Figure 4B). The control leaves had a relatively low basal levels of ROS.
Effect of FA on lipid peroxidation
The level of MDA, the final decomposition product of lipid peroxidation in the leaves of tomato plants treated with FA was significantly different from control in all sampling. MDA accumulation in leaves was significant after 8 h of treatment and increased gradually and peaked at 48 h, then declined afterwards (Figure 5).

Lipid peroxidation in the leaves of tomato plant treated with FA. Bars represent ± standard error. Different letters indicate that values are significantly different (p < 0.05).
Ethylene evolution
A significant increase in ethylene production was observed following the treatment of FA in comparison to untreated tomato leaves. FA triggered ethylene evolution from tomato leaves within 4 h of treatment. With FA treatment ethylene evolution was initially slower but increased gradually with time, reached at peak at 12 h and declined afterwards (Figure 6).

Ethylene production in the leaves of tomato plant treated with FA. Bars represent ± standard error. Different letters indicate that values are significantly different (p < 0.05).
Activity of antioxidative enzymes
Minimal increase in total SOD activity was observed in the early stages of the experiment, but after 12 h of the FA application an evident increase in SOD activity was observed as compared with the control (Figure 7A).

Activities of antioxidative enzymes in the leaves of tomato plant treated with FA. (A) Effect of FA treatment on enzymatic activity of SOD in tomato leaves. Bars represent ± standard error. Different letters indicate that values are significantly different (p < 0.05). (B) Effect of FA treatment on enzymatic activity of catalase in tomato leaves. Bars represent ± standard error. Different letters indicate that values are significantly different (p < 0.05). (C) Effect of FA treatment on enzymatic activity of APX in tomato leaves. Bars represent ± standard error. Different letters indicate that values are significantly different (p < 0.05).
The H2O2 scavenging enzymes CAT and APX showed different responses to FA. The activities of CAT and APX decreased before 24 h of FA treatment. After the initial decrease CAT and APX activities increased gradually. Three days after FA application their activities were similar to those as observed in untreated leaves (Figure 7B, C).
FA-induced DNA degradation
FA infiltration in the leaves of tomato caused damage to DNA by producing cleavages which was visible in the form of degraded DNA on agarose gel, whereas, the DNA degradation was completely absent in the control leaf tissues (Figure 8).

DNA quality of control as well as FA-treated tomato leaf tissues. Lane M: Molecular weight marker (100 bp ladder), Lane 1: Control leaf tissue, Lane 2: FA-treated leaf tissue after 24 h, Lane 3: FA-treated leaf tissue after 48 h, Lane 4: FA-treated leaf tissue after 72 h.
Discussion
Research has established that mycotoxin-induced cell death in plants resembles with animal apoptosis (Gilchrist [1997]; Gilchrist [1998]; Krishnamurthy et al. [2000]; Zhang et al. [2009]; Doorn [2011]). Our result demonstrates that FA infiltration/treatment can induce necrotic lesions in treated leaves of tomato. These lesions are reminiscent of the HR lesions (a type of programmed cell death) that are induced by plant-pathogen interactions and other abiotic stress factors. This type of necrotic lesions was also observed when Arabidopsis was treated with other mycotoxins such as Fumonisin B1 (Stone et al. [2000]) and AAL-toxin (Gechev et al. [2004]). After infiltration of tomato leaves with FA, symptoms of cell death were observed by Evans blue staining (Figure 1) and also by measuring amount of enhanced ROS levels (Figure 4A, B) as well as by DNA degradation (Figure 8). We estimated the cell death that occurred 24 h after treatment with 250 μg/ml FA as appearance of necrotic lesions on tomato leaves. At 72 h after the incubation period, more number of leaf cells were strongly and positively stained by Evans blue (Figure 1F, G). The dead cells were mostly found in the interior of the lesions.
Reactive oxygen species (ROS) such as H2O2 and O2.- are known toxic metabolic products in plants and other aerobic organisms (Gechev et al. [2006]) and stress factors in plant cells have in common the generation of ROS. The massive, rapid and transient activation of oxidative metabolism immediately after exposure to certain abiotic and biotic stress factors is termed the `Oxidative burst’ in analogy to the `Respiratory burst’, a primary response of mammalian macrophages and neutrophils to invading pathogens (Babior et al. [1997]; Schraudner et al. [1998]; Wohlgemuth et al. [2002]).
In the present study, it was found that O2.- generation, measured by means of NBT reduction, was visibly stimulated up to 2 days after FA treatment (Figure 4A). Treatment of FA also enhanced the accumulation of H2O2. The rate of H2O2 began to increase later than that of O2.- generation and it stayed high between 12 h and 48 h after FA treatment (Figure 4B). Unlike some pathosystems the FA-induced time course of H2O2 accumulation was not fully coincident with that of O2.- generation. Moreover, the timing and intensity of FA-induced O2.- and H2O2 production were distinct from a typical two phase oxidative burst reported for infected plant cells (Baker et al. [1995]; Low and Merida [1996]).
Ethylene is a gaseous phytohormone that has been reported to play an important role in regulating and modulating plant responses including cell death, to both biotic and abiotic stresses (de Jong et al. [2002]). In the present study, ethylene production was highly stimulated by FA treatment (Figure 6) which suggests that ethylene is another major player to bring out the process of cell death in tomato plant. Results demonstrate that ethylene potentiate the oxidative burst, while ROS increases ethylene production. In this way, these signalling molecules stimulate each other’s production, thus amplifying the initial signal, ultimately leading towards plant cell death.
Interestingly, in FA-treated leaves a transient decrease in activity of H2O2 scavenging enzymes namely CAT and APX was observed (Figure 7B, C). Similar observations were also found in the previous work of Kuzniak et al. ([1999]). The strongest fall in CAT and APX activities was visible within the first day after FA treatment. Similarly, a decline in CAT activity was described in bean leaves following inoculation with Pseudomonas syringae pv. phaseolicola (Adam et al. [1995]) and lower level of H2O2 scavenging activity was correlated with increased H2O2 production during interaction between Pseudomonas syringae pv. glycinea and soyabean suspension cells (Baker et al. [1995]). The observed decline in CAT and APX activity might have resulted from both an inhibitory effect of FA and an activated down regulation mechanism. The present results suggest that in FA-treated tomato leaves, SOD activity increased and transient decline in both CAT and APX activities may at least partially accounted for the enhanced H2O2 accumulation.
FA produces oxidative stress which is evident from enhancement in lipid peroxidation (Figure 5). FA may cause molecular damage to plant cells either directly or indirectly through the formation of ROS, which include free radicals as well as non-radical molecules of high reactivity. Membrane lipid especially prone to attack by free radicals (.OH, H2O2), which can convert fatty acids to toxic lipid peroxides, destroying the biological membranes (Foyer et al. [1994]). Since lipid peroxidation is ascribed to oxidative damage (Zenk [1996]), measurement of MDA levels, a common product of lipid peroxidation, is routinely used as sensitive index of oxidative stress (Smirnoff [1993]; Metwally et al. [2005]; Choudhary et al. [2007]). In this study, MDA level rose significantly high when plants were subjected to FA treatment. This suggests that superoxide radicals are produced on FA treatment leading to increased peroxidation.
Conclusion
Results from experiments are reproducible with respect to the morphological effects and cell death. In our study, a concomitant increase in ROS production, the down regulation of the antioxidant enzymes activities and a change of lipid peroxidation were crucial for the onset of cell death. The present investigations demonstrated that FA-induced lesions on tomato leaves are similar to pathogen-induced lesions (HR) in many respects, such as the occurrence of an oxidative burst, lipid peroxidation, and DNA degradation. FA-induced oxidative burst is evident from enhancement in lipid peroxidation, H2O2 and O2.- levels. However, to cope with the FA toxicity, tomato plants develop a cellular strategy involving activation of various enzymatic antioxidants. This modulation of antioxidative system could reflect a defence response to the cellular damage provoked by FA treatment. The study suggests that the interaction between FA and tomato provides a useful system to study plant cell death.
Authors’ contributions
In this work, author (VKS) designed and performed experiments, analysed data, and wrote the paper. Another author (RSU) supervised the whole work. Both authors have read and approved the final manuscript.
Abbreviations
- APX:
-
Ascorbate peroxidase
- ANOVA:
-
Analysis of variance
- BHT:
-
Butylated hydroxyl toluene
- CAT:
-
Catalase
- DAB-HCl:
-
3,3′-Diaminobenzidine hydrochloric acid
- FA:
-
Fusaric acid
- FW:
-
Fresh weight
- HR:
-
Hypersensitive response
- MDA:
-
Malondialdehyde
- NBT:
-
Nitroblue tetrazolium
- PDB:
-
Potato dextrose broth
- PVP:
-
Polyvinyl pyrrolidone
- ROS:
-
Reactive oxygen species
- SA:
-
Salicylic acid
- SDS:
-
Sodium dodecyl sulphate
- SOD:
-
Superoxide dismutase
- TBA-RS:
-
Thiobarbituric acid-reactive substances
- TCA:
-
Trichloro acetic acid
References
Adam AL, Bestwick CS, Barna B, Mansfield JW: Enzymes regulating the accumulation of active oxygen species during the hypersensitive reaction of bean to Pseudomonas syringae pv. phaseolicola . Planta 1995, 197: 240–249. 10.1007/BF00202643
Apel K, Hirt H: Reactive oxygen species : metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 2004, 55: 373–399. 10.1146/annurev.arplant.55.031903.141701
Arias JA: Secretory organelle and mitochondrial alterations induced by fusaric acid in root cells of Zea mays . Physiol Plant Pathol 1985, 27: 149–158. 10.1016/0048-4059(85)90063-3
Babior BM, Benna JE, Chanock SJ, Smith RM: The NADPH Oxidase of Leukocytes: The Respiratory Burst Oxidase. In Oxidative Stress and the Molecular Biology of Antioxidant Defences. Edited by: Scandalios JG. Cold Spring Harbour Press, Plainview; 1997:737–783.
Baker CJ, Harmon GL, Glazener JA, Orlandi EW: A noninvasive technique for monitoring peroxidative and H 2 O 2 -scavenging activities during interactions between bacterial plant pathogens and suspension cells. Plant Physiol 1995, 108: 353–359.
Baker CJ, Mock NM: An improved method for monitoring cell death in cell suspension and leaf disc assays using Evans blue. Plant Cell Tissue Org Cult 1994, 39: 7–12. 10.1007/BF00037585
Baker CJ, Orlandi EW: Active oxygen in plant pathogenesis. Annu Rev Phytopathol 1995, 33: 299–321. 10.1146/annurev.py.33.090195.001503
Beauchamp C, Fridovich I: Superoxide dismutase: improved assays and assay applicable to acrylamide gels. Annu Rev Plant Physiol Plant Mol Biol 1971, 44: 276–287.
Bouizgarne B, El-Maarouf-Bouteau H, Frankart C, Reboutier D, Madiona K, Pennarun AM, Monestiez M, Trouverie J, Amiar Z, Briand J, Brault M, Rona JP, Ouhdouch Y, El Hadrami I, Bouteau F: Early physiological responses of Arabidopsis thaliana cells to fusaric acid: toxic and signalling effects. New Phytol 2006, 169: 209–218. 10.1111/j.1469-8137.2005.01561.x
Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976, 72: 248–254. 10.1016/0003-2697(76)90527-3
Breusegem FV, Dat JF: Reactive oxygen species in plant cell death. Plant Physiol 2006, 141: 384–390. 10.1104/pp.106.078295
Choudhary M, Jetley UK, Khan MA, Zutshi S, Fatma T: Effect of heavy metal stress on proline, malondialdehyde and superoxide dismutase activity in the cyanobacterium Spirulina platensis -55. Ecotoxicol Environ Saf 2007, 66: 204–209. 10.1016/j.ecoenv.2006.02.002
D’Alton A, Etherton B: Effects of fusaric acid on tomato root hair membrane potentials and ATP levels. Plant Physiol 1984, 74: 39–42. 10.1104/pp.74.1.39
de Jong AJ, Yakimova ET, Kapchina VM, Woltering EJ: A critical role for ethylene in hydrogen peroxide release during programmed cell death in tomato suspension cells. Planta 2002, 214: 537–545. 10.1007/s004250100654
Dhindsa RS, Plumb-Dhindsa P, Thorpe TA: Leaf senescence: correlated with increased levels of membrane permeability and lipid peroxidation, and decreased levels of superoxide dismutase and catalase. J Exp Bot 1981, 32: 93–101. 10.1093/jxb/32.1.93
Doke N: Involvement of superoxide anion generation in the hypersensitive response of potato tuber tissues to infection with an incompatible race of Phytophthora infestans and to hyphal wall components. Physiol Plant Pathol 1983, 23: 354–357.
Doorn WG: Classes of programmed cell death in plants compared to those in animals. J Exp Botany 2011, 62: 4749–4761. 10.1093/jxb/err196
Doyle JJ, Doyle JL: A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 1987, 19: 11–15.
Duncan DB: Multiple range and multiple F tests. Biometrics 1955, 11: 1–42. 10.2307/3001478
Foyer CH, Deascouveries P, Kunert KJ: Protection against oxygen radicals: important defence mechanisms studied in transgenic plants. Plant Cell Environ 1994, 17: 507–523. 10.1111/j.1365-3040.1994.tb00146.x
Gäumann E: Fusaric acid as a wilt toxin. Phytopathology 1957, 47: 342–357.
Gäumann E: The mechanism of fusaric acid injury. Phytopathology 1958, 48: 670–686.
Gechev TS, Gadjev IZ, Hille J: An extensive microarray analysis of AAL-toxin-induced cell death in Arabidopsis thaliana brings new insights into the complexity of programmed cell death in plants. Cell Mol Life Sci 2004, 61: 1185–1197. 10.1007/s00018-004-4067-2
Gechev TS, Van Breusegem F, Stone JM, Denev I, Laloi C: Reactive oxygen species as signals that modulate plant stress responses and programmed cell death. Bioessays 2006, 28: 1091–1101. 10.1002/bies.20493
Gilchrist DG: Mycotoxins reveal connections between plants and animals in apoptosis and ceramide signalling. Cell Death Differ 1997, 4: 689–698. 10.1038/sj.cdd.4400312
Gilchrist DG: Programmed cell death in plant disease: the purpose and promise of cellular suicide. Annu Rev Phytopathol 1998, 36: 393–414. 10.1146/annurev.phyto.36.1.393
Gomez KA, Gomez AA: Statistical Procedures for Agricultural Research. John Wiley and sons, Inc, London, UK; 1984.
Harborne JB: Introduction to Ecological Biochemistry . Academic Press, London, UK; 1989.
Heath RL, Packer L: Photoperoxidation in isolated chloroplasts. I. Kinetics and stoichiometry of fatty acid peroxidation. Arch Biochem Biophys 1968, 125: 189–198. 10.1016/0003-9861(68)90654-1
Heiser I, Obwald W, Elstner EF: The formation of reactive oxygen species by fungal and bacterial phytotoxins. Plant Physiol Biochem 1998, 36: 703–713. 10.1016/S0981-9428(98)80020-X
Jabs T: Reactive oxygen intermediates as mediators of programmed cell death in plants and animals. Biochem Pharmacol 1999, 57: 231–245. 10.1016/S0006-2952(98)00227-5
Karuppanapandian T, Moon JC, Kim C, Manoharan K, Kim W: Reactive oxygen species in plants: their generation, signal transduction, and scavenging mechanisms. Aust J Crop Sci 2011, 5(6):709–725.
Krishnamurthy KV, Krishnaraj R, Chozhavendan R, Christopher FS: The programme of cell death in plants and animals: a comparison. Curr Sci 2000, 79: 1169–1181.
Kuzniak E, Patykowski J, Urbanek H: Involvement of the antioxidative system in tomato response to fusaric acid treatment. J Phytopathol 1999, 147: 385–390. 10.1111/j.1439-0434.1999.tb03838.x
Low PS, Merida JR: The oxidation burst in plant defence function and signal transduction. Physiol Plant 1996, 96: 533–542. 10.1111/j.1399-3054.1996.tb00469.x
Lund ST, Stall RE, Klee HJ: Ethylene regulates the susceptible response to pathogen infection in tomato. Plant Cell 1998, 10: 371–382. 10.1105/tpc.10.3.371
Maksymiec W, Krupa Z: The effects of short term exposition to Cd, excess Cu ions and jasmonate on oxidative stress appearing in Arabidopsis thaliana . Environ Exp Bot 2006, 57: 187–194. 10.1016/j.envexpbot.2005.05.006
Marre MT, Vergani P, Albergoni FG: Relationship between fusaric acid uptake and its binding to cell structures in leaves of Egeria densa and its toxic effects on membrane permeability and respiration. Physiol Mol Plant Pathol 1993, 42: 141–157. 10.1006/pmpp.1993.1012
Mehdy MC, Sharma YK, Sathasivan K, Bays NW: The role of activated oxygen species in plant disease resistance. Physiol Plant 1996, 98: 365–374. 10.1034/j.1399-3054.1996.980219.x
Metwally A, Safronova AB, Dietz KJ: Genotypic variation of the response to cadmium toxicity in Pisum sativum L. J Exp Bot 2005, 56: 167–178.
Nakano Y, Asada K: Hydrogen peroxide is scavenged by ascorbate-specific peroxidase in spinach chloroplasts. Plant Cell Physiol 1981, 22: 867–880.
Overmyer K, Brosche M, Pellinen R, Kuittinen T, Tuominen H, Ahlfors R, Keinanen M, Saarma M, Scheel D, Kangasjarvi J: Ozone-induced programmed cell death in the Arabidopsis radical-induced cell death 1 mutant. Plant Physiol 2005, 137: 1092–1104. 10.1104/pp.104.055681
Paciolla C, Dipierro N, Mule G, Logrieco A, Dipierro S: The mycotoxins beauvericin and T-2 induce cell death and alteration to the ascorbate metabolism in tomato protoplasts. Physiol Mol Plant Pathol 2004, 65: 49–56. 10.1016/j.pmpp.2004.07.006
Sagisaka S: The occurence of peroxide in a perennial plant, Populus gelrica . Plant Physiol 1976, 57: 308–309. 10.1104/pp.57.2.308
Sapko OA, Utarbaeva AS, Makulbek S: Effect of fusaric acid on prooxidant and antioxidant properties of the potato cell suspension culture. Russ J Plant Physiol 2011, 58(5):828–835. 10.1134/S1021443711050190
Schraudner M, Moeder W, Wiese C, Camp WV, Inze D, Langebartels C, Sandermann H Jr: Ozone-induced oxidative burst in the ozone biomonitor plant, tobacco Bel W3. Plant J 1998, 16: 235–245. 10.1046/j.1365-313x.1998.00294.x
Smirnoff N: The role of active oxygen in the response of plants to water deficit and dessication. New Phytol 1993, 125: 27–58. 10.1111/j.1469-8137.1993.tb03863.x
Stefan E: Thin layer chromatography- an appropriate method for fusaric acid estimation. Biologia Bratislava 2005, 60(1):104.
Stone JM, Heard JE, Asai T, Ausubel FM: Simulation of fungal-mediated cell death by fumonisin B1 and selection of fumonisin B1-resistant (fbr) Arabidopsis mutants. Plant Cell 2000, 12: 1811–1822. 10.1105/tpc.12.10.1811
Sutherland ML, Pegg GF: The basis of host recognition on Fusarium oxysporum f.sp. lycopersici . Physiol Mol Plant Pathol 1992, 40: 423–436. 10.1016/0885-5765(92)90033-R
Venter S, Steyn PJ: Correlation between fusaric acid production and virulence of isolates of Fusarium oxysporum that causes potato dry rot in South Africa. Potato Res 1998, 41: 289–294. 10.1007/BF02358198
Wohlgemuth H, Mittelstrass K, Kschieschan S, Bender J, Weigel HJ, Overmyer K, Kangasjarvi J, Sandermann H, Langebartels C: Activation of an oxidative burst is a general feature of sensitive plants exposed to the air pollutant ozone. Plant Cell Environ 2002, 25: 717–726. 10.1046/j.1365-3040.2002.00859.x
Wu H, Yin X, Liu D, Ling N, Bao W, Ying R, Zhu Y, Guo S, Shen Q: Effect of fungal fusaric acid on the root and leaf physiology of watermelon ( Citrullus lanatus ) seedlings. Plant Soil 2008, 308: 255–266. 10.1007/s11104-008-9627-z
Zenk MH: Heavy metal detoxification in higher plants-a review. Gene 1996, 179: 21–30. 10.1016/S0378-1119(96)00422-2
Zhang X, Boesch-Saadatmandi C, Lou Y, Wolffram S, Huebbe P, Rimbach G: Ochratoxin A induces apoptosis in neuronal cells. Genes Nutr 2009, 4: 41–48. 10.1007/s12263-008-0109-y
Acknowledgements
The author (VKS) is thankful to UGC, New Delhi for providing financial support during this work and Indian Institute of Vegetable Research, Varanasi for providing tomato seeds. The authors gratefully acknowledge the Head of the Botany Department, Banaras Hindu University, Varanasi, India for providing necessary facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Singh, V.K., Upadhyay, R.S. Fusaric acid induced cell death and changes in oxidative metabolism of Solanum lycopersicum L. Bot Stud 55, 66 (2014). https://doi.org/10.1186/s40529-014-0066-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40529-014-0066-2