
Abstract
Background
The design, construction and application of a Pinus microarray platform are described. The oligonucleotide microarray was developed using publicly available Pinus cDNA sequences mostly derived from Pinus taeda to test whether heterologous hybridisation of microarray probes will generate useful data when hybridised with cRNA constructed from the dominant New Zealand forestry species Pinus radiata.
Methods
A comprehensive consensus sequence collection of Pinus cDNA sequences was collated into a non-redundant database used for automated design of 60-mer oligonucleotide microarray probes. The microarray slides, manufactured by Agilent Technologies (Palo Alto, California), were used to monitor gene expression in an induction experiment using 2-chloroethylphosphonic acid, common name ethephon and the active ingredient of the plant growth regulator Ethrel® (Bayer Crop Science). The transcriptomes from tissues of 2-year old Pinus radiata saplings +/− ethephon treatment were compared by hybridisation onto the Pinus microarray slides.
Results
Statistically significant differentially expressed genes identified by heterologous hybridisation to the Pinus microarray following ethephon induction included the up-regulation of genes in the xylem that were related to the metabolism of phenylpropanoids and flavonoids, and also defence responses, specifically against fungal/insect attack and oxidative stress. Bark, mucilaginous xylem and xylem generated largely mutually exclusive cohorts of genes and Gene Ontology (GO) classes. The results are also interpreted in reference to gross and microscopic morphological changes. Samples of gene responses were validated by quantitative RT-PCR.
Conclusion
These results confirm the successful development of a Pinus microarray and demonstrate the utility of the microarray for transcriptomic research in Pinus radiata through heterologous hybridisation.
Similar content being viewed by others


Background
Microarray technology has been extensively used to profile gene expression for more than a decade. The ability to perform up to a million parallel hybridisations on a single microarray provides researchers with a very efficient platform for characterisation of genetic processes in target organisms. In addition to characterising the transcriptomic differences generated from an experiment involving one species, the same microarray platform can be used for research involving related species through heterologous hybridisation (Davey et al. [2009]). Microarrays can also be used for studies involving methylation and epigenetics by hybridisation with bisulfite-treated genomic DNA (Reinders et al. [2008]). This versatility in other downstream applications helps to ensure that microarray technology continues to be relevant in crop research, despite the availability and widely recognised superiority of RNA-Seq for transcriptomic analyses.
Pinus radiata D. Don is grown in 1.6 M ha in New Zealand, making it the premier species for silviculture (New Zealand Forest Owners Association [2011]). Microarrays based on comprehensive transcriptomic data can be used to advance pine research, however most examples of Pinus microarrays published in the last 10 years are based on very modest collections containing less than 3,500 sequenced genes (Heath et al. [2002]; Yang et al. [2004]; Myburg et al. [2006]; Adomas et al. [2007]; Balk et al. [2007]; Perdiguero et al. [2012]; Li et al. [2012]). A cDNA microarray based on 26,496 sequences for P. taeda was recently used to assess the effects of drought in P. taeda L. (Lorenz et al. [2011]).
As of July 2013, there were approximately 13,000 publicly available nucleotide sequences from P. radiata curated at GenBank (http://www.ncbi.nlm.nih.gov/), but much larger collections of DNA sequences exist from other Pinus species, principally P. taeda, which had approximately 140,000 sequences. Microarrays can be used for comparative genomic hybridisation among near relatives via heterologous hybridisation (Davey et al. [2009]). Heterologous hybridisation between cDNA probes from P. taeda, immobilised on nylon membrane, with cDNA from P. sylvestris L. and Picea abies (L.) H. Karst has been successful (Van Zyl et al. [2002]). Heterologous hybridisation requires microarray probes that are capable of tolerating a certain degree of mismatch with the target transcripts. Single nucleotide deletions in targets of 60-mer probes can reduce signal intensity by 20-50% whereas single deletions in targets of 20-mer probes essentially eliminated any signal (LeProust [2008]). Probes of 60-mer can still generate useful signals even with 10% mismatched bases between the probe and target gene (Hughes et al. [2001]).
The utility of a microarray is best appreciated by the degree to which the results from this technology confirm or clarify well-characterised biological phenomena. The effect of long-term exposure to an ethylene-generating paste containing 2-chloroethylphosphonic acid (ethephon) on the stem anatomy of Pinus radiata saplings was used as a physiological model to test heterologous hybridisation to the Pinus microarray. The response of xylogenesis to ethylene has not previously been studied adequately at the molecular level in Pinus species. Ethephon breaks down in plant tissues to form ethylene, a major plant hormone with diverse effects on the plant's physiology (Warner and Leopold [1969]). It is known that different Pinus species show clear responses to ethephon treatment. Previously, the application of an ethylene-generating paste has been found to induce localised radial stem growth in white pine (Pinus strobus L.) (Brown and Leopold [1973]). Ethylene also stimulated growth in bark and xylem tissues of Pinus halepensis Mill. (Yamamoto and Kozlowski [1987a]). Ethephon reduced lignification of tracheids in compression wood that developed on the underside of tilted stems of Pinus densiflora Siebold & Zucc. (Yamamoto and Kozlowski [1987b]). In P. halepensis, ethephon application implicated ethylene in the control of resin duct formation (Yamamoto and Kozlowski [1987a]). Ethephon sprays of 1–2 g L−1 effectively reduced height and radial growth of jack pine (Pinus banksiana Lamb.) (Schnurr et al. [1996]).
The central assumption of this study is that heterologous microarray hybridisation can be used to generate useful data with probes from closely related species. Microarray probes were designed primarily from P. taeda hybridised with cRNA constructed from the close relative P. radiata. The creation of a comprehensive consensus sequence database for Pinus species, the design and construction of a microarray platform from this database, and the use of this microarray platform to monitor and characterise the transcriptomic response of P. radiata saplings to ethephon will be described. The results of heterologous hybridisation were verified by comparing signal data reported from specific probes in the array with physiological observations and, for a limited number of probes, by quantitative reverse transcription polymerase chain reaction (RT-PCR). The lists of differentially expressed genes were analysed using the Database for Annotation, Visualisation and Integrated Discovery (DAVID) Bioinformatics Resources (Huang et al. [2008]) and are discussed in relation to gross and microscopic changes induced by ethephon application.
Methods
Plant materials
One-year-old rooted clonal saplings of a single genotype of Pinus radiata were purchased from ArborGen Australasia (Whakatane, New Zealand) for this experiment. These were placed in 500 mL pots containing standard potting mix that were then arrayed on an unroofed concrete pavement for another year prior to their use in this research. A total of 60 saplings were required to provide 15 plants per treatment for 4 treatments.
Ethephon application
Lanolin pastes (0.2 to 0.5 g) containing 0, 1, 5 and 25 mg of ethephon supplied from ETHREL (48% Ethephon, Bayer Crop Science, NZ) were dispensed onto 3 × 6 cm aluminium foil sheets that were wrapped around the basal stem of each experimental sapling, 5–7.5 cm from the soil level. The 1 mg per plant dosage corresponded to the 1% in lanolin dose used in P. halepensis (Yamamoto and Kozlowski [1987a]).
RNA extraction
RNA was extracted from bark, mucilaginous xylem (containing the cambium) and xylem samples. Bark refers to the outermost layer of tissues of the stem beyond the mucilaginous developing xylem. Xylem refers to the woody fibrous tissue left after scraping off the mucilaginous developing xylem. RNA was extracted from the 0 and 1 mg treatments for transcriptomic analysis. The 5 and 25 mg ethephon treatments at 8 weeks after treatment (WAT) elicited the greatest physiological responses but transcriptomic analyses were not attempted for these treatments due to inconsistent RNA extraction possibly due to the adverse effect of high resin content. Three replicates consisting of 5 saplings per replicate were destructively sampled 1 week after ethephon treatment and an identical batch was sampled 8 weeks after treatment. Thus each tissue was sampled from each of 15 saplings for a given treatment. After wiping off the residual lanolin from the bark surface with tissue paper, the bark was cut and peeled off, the mucilaginous xylem was scraped off with the blunt edge of a box cutter, and the remaining woody xylem was cut off from the rest of the stem. The respective tissues were wrapped in water-proof paper, flash-frozen in liquid nitrogen and stored at −80°C prior to RNA extraction. Total RNA from xylem was extracted using Concert reagent (Invitrogen; Carlsbad, CA) whilst RNA extracted from bark was obtained using Concert reagent followed by purification with the Plant RNA Isolation kit (Agilent). RNA from the mucilaginous xylem was extracted using the Plant RNA Isolation kit.
Assembly and annotation of the heterologous Pinus Consensus Transcript database
In mid-2010, there were 18,900 Unigene entries for P. taeda in the US National Center for Biotechnology Information (NCBI) database (ftp://ftp.ncbi.nih.gov/repository/UniGene/Pinus_taeda/Pta.seq.uniq.gz). The Plant Genome Database (http://www.plantgdb.org/) had 77,500 Putative Unique Transcripts (PUT), (http://www.plantgdb.org/download/download.php?dir=/Sequence/ESTcontig/Pinus_taeda/current_version/Pinus_taeda.mRNA.PUT.fasta.bz2). Harvard's Computational Biology and Functional Genomics Laboratory (http://compbio.dfci.harvard.edu/cgi-bin/tgi/gimain.pl?gudb=pine) had 39,800 tentative consensus (TC) sequences (ftp://occams.dfci.harvard.edu/pub/bio/tgi/data/Pinus/ PGI.release_8.zip). These sequences were combined into a FASTA file generated using BioEdit software (Hall [1999]) and then reassembled using the default parameters for sequence assembly using CAP3 software (http://seq.cs.iastate.edu/CAP3/cap3.linux.i686_xeon64.tar) (Huang and Madan [1999]). The final database consisted of 68,225 sequences, with 39,869 assembled contigs in common between at least two of the three databases and 28,356 'singletons' unique to one database. The assembled sequences ranged from 50 to 6175 base pairs (bp), with an average length of 811 bp. The distribution of the assembled sequences is shown in Figure 1. The reassembled database, henceforth referred to as the All-Pinus database, was used for the design of 60-mer probes for the Pinus microarray.

Sequence size distribution in the All- Pinus database.
The most similar protein for each sequence assembly was determined using the BLASTX algorithm as implemented with the BioEdit software, using the default criteria and a maximum E-value set at 0.001. The closest protein was identified as the one with the most significant E-value among those identified by BLASTX. The actual average E-value for the whole database was 1.9 × 10−5. With these criteria, 40,238 (59%) sequences were assigned a putative protein ID, of which 14,838 were unique. A cut-off E-value set at 1 × 10−5 was used in the annotation of Conifer DBMagic (Lorenz et al. [2012]) and a smaller number of putative protein IDs (36,995 sequences (54%)) within the All-Pinus database sequences were identified when this cut-off E-value was used. The annotations for these protein IDs were downloaded from DAVID v6.7, (http://david.abcc.ncifcrf.gov/) (Huang et al. [2009]). The top 40 sequences identified by comparison with the Swiss-Prot database of annotated protein sequences and the numbers of transcript variants that code for essentially the same protein are shown in Table 1.
Pre-testing the All-Pinus sequence database
The 7,538 expressed sequence tags (ESTs) from P. radiata found in the US National Center for Biotechnology Information (NCBI) database as of 2010 were used to query the 68,225 sequences in the All-Pinus database via BLASTN algorithm (Altschul et al. [1997]). The BLASTN algorithm was set at a cut-off value of 1.0 × E−10. The alignments that generated less than the highest score for each EST were eliminated. The average data from BLASTN (630 bp aligned length, 99.2% identity, 619 bp actual length, 12.7 unaligned bases, 3.7 mismatched bases and 0.8 gaps) reinforced the potential of heterologous hybridisation of P. radiata sequences to a Pinus microarray developed from the All-Pinus database.
Microarray Probe design and in silico validation
Candidate probes were selected based on base-composition profiles optimised through eArray (https://earray.chem.agilent.com/earray/). Three 60-mer sense probes were selected across the length of the template sequence. The 191,500 candidate probes were trimmed to 175,614 to fit the 180 k format required by the Agilent software. Probes were eliminated based on high similarity and hybridisation quality as predicted by eArray. The remaining probes were used to query the original All-Pinus database by BLASTN, which was set at a cut-off value of 1.0 × E−10. The results indicated that 123,311 probes (70.2%) for 49,081 genes (71.9%) would give a target-specific signal. The remaining probes were expected to hybridise to sequences such as paralogues and splice variants that have different accession numbers but are essentially different versions of the same protein. The probe sequences were used to query the 7538 ESTs from P. radiata in the GenBank sequence database provided by the NCBI. The average identity, aligned length, non-identical bases and gaps were 99.6%, 59.9 bp, 0.2 bp and 0.0 bp respectively. These results affirmed the potential utility of the All-Pinus database for generating heterologous Pinus microarray probes for analysing transcriptomes derived from P. radiata.
Microarray analysis
Paired hybridisation for each microarray comprised treatment versus control of each tissue from the same time point, for example; 'bark, 0 mg ethephon, 1 WAT' versus 'bark, 1 mg ethephon, 1 WAT', and so forth. Total RNA samples of 50–100 ng were used for labelling with the fluorescent cyanine dyes Cy3 or Cy5 using the Agilent Low input Quick Amp labelling kit. After labelling, unincorporated dye was removed using RNeasy (Qiagen, Valencia, CA) mini-spin columns. Purified samples were assessed for yield and specific activity using a Nanodrop ND-1000 UV–VIS spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). Samples that gave yields of >0.8 μg cRNA and specific activity (Concentration of Cy3/Concentration of cRNA ×1000) >10 were used for arrays. Labelled samples were hybridised to arrays at 65°C for 17 hours at 10 rpm in an Agilent G2545A hybridisation oven using 2× GEx Agilent hybridisation buffer. Slides were washed using Agilent wash buffers. Array scanning and feature extraction was performed using the Agilent G2565CA microarray scanner as outlined in the Agilent protocol for Two-colour Microarray-Based Gene Expression Analysis, Version 6.5, May 2010.
Data were imported into R software using the limma package (Smyth [2005]), transformed to the log scale (base 2), and normalized via loess smoothing with no background correction. Statistical analysis of data was performed using the linear models approach implemented in limma, using the M values for each array as the dependent data. The empirical Bayes shrinkage-based approach to per-probe variance estimation was used to generate a moderated t-statistic for each probe (Smyth [2005]). This statistic was then used to determine the statistical significance of the differences for each gene at each time/tissue combination. Probability (P) values were calculated for each comparison, and used to define the False Discovery Rate threshold by the Benjamin-Hochberg method (Benjamini and Hochberg [1995]).
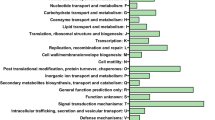
Probes with adjusted P-values greater than 0.01, log2 fold changes between −1 and 1, and probes with no Uniprot homologs were eliminated. The remaining significantly expressed probes, divided into up- and down-regulated subgroups, were uploaded onto the DAVID portal (http://david.abcc.ncifcrf.gov/home.jsp) for singular enrichment analysis in comparison with the list of protein sequences in the All-Pinus database. The number of protein IDs associated with each gene ontology classification that were significantly different at p < 0.01 and displayed a conservative False Discovery Rate less than 10% are shown in Table 2.
Quantitative RT-PCR verification of microarray signals
Ten probes were selected that generated stable signals greater than background across all treatment combinations after heterologous hybridisation of the Pinus microarray; the probes included Contig12730c, which showed strong stable signals that did not change with ethephon/sampling time for use as a control. Forward and reverse primers were designed to amplify >45 bp fragments within the 60-mer probe using OligoAnalyzer 3.1 (http://www.idtdna.com/analyzer/Applications/OligoAnalyzer/). Primer details are shown in Table 3 for the successful primer pairs. Aliquots from the original samples that contained 200 to 1300 ng RNA were reverse transcribed using qScript (Quanta Biosciences, MD, USA) in 20 μl reactions. A 1:20 dilution was added as template for a qPCR mix using Lightcycler® Faststart DNA Masterplus SYBR Green i (Roche, USA). A Lightcycler® Carousel-based system (Roche, USA) was used for thermal cycling. A 10 min denaturation step was followed by 45 thermal cycles consisting of a 10 sec denaturation step at 95°C, annealing step of 4 sec at 60°C and an extension step of 3 sec at 72°C. This was followed by melting point analysis from 65°C to 95°C at a ramp rate of 0.2°C sec−1. Fold-change calculation, using the expression level of Contig12730c as the constant divisor, was performed with a formula (3.5) for relative quantification (Pfaffl [2004]).
Microscopy
The gross physiological effects of ethephon were analysed by microscopic analysis of sectioned stems of all treatments. Stem cross-sections were prepared with a sledge microtome and images of the cut ends of stems were obtained using a stereo microscope. Stems were fixed in FAA (5% formalin, 5% glacial acetic acid, 90% ethanol v/v) fixative for 1 month and then examined via confocal microscopy and autofluorescence. Images were obtained with simultaneous excitation at 488 and 561 nm.
Results
Transcriptomic analysis of ethephon induction in Pinus radiata saplings
Pinus radiata transcriptomes were analysed by heterologous hybridisation to the Pinus microarray with cRNA generated from different tissues extracted from ethephon-treated and control plants. The transcriptomes in bark, mucilaginous xylem and xylem 1 and 8 WAT with 0.2 g of lanolin containing 0 or 1 mg ethephon were compared by two-colour hybridisation. Limma analysis of the data revealed that the mucilaginous xylem had 66 and 23,084 significantly different signals (relative to the pertinent control) 1 and 8 WAT with ethephon, respectively; similarly, bark had 127 and 1761 (1 and 8 WAT) whilst xylem had 1,352 and 12,718 probes exhibiting significantly different signals (1 and 8 WAT).
The relationships between the differentially expressed probes among tissues and across time are shown in the Venn diagrams in Figure 2. At 1 WAT, the mucilaginous xylem and the bark displayed comparable numbers of up- and down-regulated genes, but only one of these was co-expressed. More genes in the xylem showed significant down-regulation due to ethephon but only a very small fraction were co-expressed in the mucilaginous xylem (12 or 3.9%) or bark (5 or 1.6%). Despite the very low numbers of differentially expressed genes in the bark at 1 WAT, at least 50% of these were still expressed in the bark at 8 WAT. A lower proportion (25-34%) of genes that were differentially expressed in the xylem at 1 WAT was also found seven weeks later.

Number of differentially expressed gene cohorts in the various sample tissues. The value in the overlapping portions of the circles corresponds to the number of co-expressed gene cohorts in each treatment pair. Legend: UR, upregulated; DR, downregulated; Mxylem, mucilaginous xylem; 1 and 8 weeks after treatment.
Differentially expressed (fold change >2, adj-p #x003C; 0.05) genes were systematically mapped to the associated biological annotation (e.g., Gene Ontology Terms, KEGG pathways) using DAVID, and the most over-represented (enriched) biological annotation were statistically highlighted. Enrichment analysis increases the likelihood of identifying the biological processes most relevant to the experiment (Huang et al. [2008]). The Functional Annotation Table of Gene Ontologies with False Discovery Rates less than 0.1, generated through the DAVID portal, are shown in Table 2. Ethephon had no significant effect on the bark transcriptome one week after application. The xylem transcriptome had a greater response to ethephon than the bark transcriptome resulting in an increase in the activity of genes associated with cellular amino acid derivatives and phenylpropanoid biosynthesis in xylem at 1 WAT and 8 WAT. Ethephon initially depressed defence responses in the xylem but, after eight weeks, ethephon application activated a wide range of genes related to plant defence. These included genes involved in phenylpropanoid and flavonoid biosynthesis, chitin detection and degradation, oxidation-reduction, and toxin catabolism. Lignin metabolic processes were significantly down-regulated by ethephon treatment.
In the bark, genes associated with structural constituents of the cell wall (GO:0005199) were activated by ethephon. The down-regulation of light-responsive genes in the bark was expected, but the suppression of genes associated with abiotic stress was unexpected.
Validation of microarray signals by quantitative RT-PCR
To test the validity of signal quantitation by the Pinus microarray, 60-mer probes were selected and corresponding transcripts in cDNA transcribed from the original total RNA extracts used for microarray hybridisation were quantified by RT-PCR. Five primer pairs designed to amplify a 45–60 bp segment within each probe (Table 4) generated fold change estimates that were in general agreement with the direction (up- or down-regulation) of the raw signal data from the microarrays (Figure 3). The sixth probe used as a constitutive background control (contig 12730c) gave a consistent signal. Four primer pairs failed to amplify a corresponding target which is a consequence of the higher specificity required for PCR amplification which will not always be compatible for the detection of heterologous targets.

Quantitative RT-PCR verification of Pinus microarray signals. Log fold-change values are indicated in the Y axis; negative values correspond to down-regulated transcripts. Bars correspond to standard deviation. Legend: LogFC, Log Fold Change; MA, microarray.
Anatomical changes after long-term ethephon exposure
Most of the visible response to ethephon was limited to the treated area on the bark. There was little effect on stem girth and appearance 1 cm above or below the lanolin band (Figure 4). After 8 weeks of treatment, the 1 mg dose induced an increase in girth (16%) relative to the girth immediately above and below the treated area. Increased stem diameter was evident in the ethephon-treated area but not in the xylem. Most of the increased growth was manifested in increased amounts of phloem and cortex in the bark that displayed deep fissures and cracks, and sap bleeding that turned the normally colourless lanolin white. Higher ethephon dosages (5 mg and 25 mg treatments) induced more resinification and generated greater levels of opacity and whitening of the lanolin due to reaction with bark exudates, bark splitting and necrosis and reduced stem growth (data not shown). The ethephon-treated stems exhibited an abnormal layer of unlignified tissue in the outer xylem, which is indicated by an asterisk in Figure 4B. The number and size of the resin canals were unaffected by ethephon treatment. After 1 month of fixation in FAA, unstained sections examined for autofluorescence indicated the presence of strongly red fluorescent extractives in both wood and bark from ethephon-treated plants (Figures 4C and 5). The red fluorescent parenchymatous cells, scattered throughout the outer xylem, are poorly lignified (Figure 5).

Sapling morphology 8 weeks after application of ethephon. A. Resin leakage from cracks in the bark of stems treated with ethephon, causing some of the lanolin to turn white. B. increased girth and cracked bark induced by ethephon. Arrows indicate the mucilaginous xylem in each stem while the asterisk indicates an unlignified layer of parenchymatous tissue in the outer xylem in the 1 mg ethephon treatment. Note no appreciable difference in diameter of the lignified xylem or in the number and size of resin ducts (round light-brown spots) was observed between different treatments. Scale bars = 2 mm. C. Comparison of resin canals (RC) after FAA fixation of xylem exposed to 0 mg or 1 mg ethephon. Resin canals and tracheids in xylem of ethephon-treated stem are suffused with unidentified extractives that show red fluorescence on excitation at 488 and 561 nm. Scale bars = 100 μm.

Comparison of outer stem anatomy between control (A) and 5 mg ethephon treatment (B). Both Phloem (Ph) and Traumatic xylem (Tx) appear abnormal and contain increased extractives. The extractives are concentrated in poorly lignified parenchymatous cells in the xylem. Cortex (Cx) and Cambium (Ca) are as indicated. The images are at the same scale. Scale bar = 1 mm.
Discussion
Microarray construction and validation
Next Generation Sequencing (NGS) techniques can be used to produce gene libraries for the construction of microarrays (Hiremath et al. [2011]), however generating numerous fully sequenced and annotated transcriptomes is expensive and time consuming. It is more efficient to capture a comprehensive picture of gene activity of an organism through the combining and normalisation of existing transcriptomes derived from various tissues sampled temporally and from a wide variety of natural and experimental conditions. Extensive sequence databases exist for Pinus taeda that were generated from diverse experimental conditions by many research groups; less extensive databases are also available for other Pinus species. Pre-existing public gene databases for Pinus species were used to construct the Pinus microarray described in this paper. The Pinus data were processed using CAP3 for reassembly of contigs, to remove redundancy and to generate consensus contigs (Huang and Madan [1999]; Min et al. [2009]; Feldmesser et al. [2013]; Huang et al. [2012]) for the All-Pinus database. In silico validation by BLASTN analysis of the All-Pinus database and the probes constructed from it revealed >99% identity of aligned sequences with reference to a set of publicly available P. radiata sequences. These in silico results were corroborated by the results obtained after P. radiata cRNA hybridisation.
General observations on the transcriptomes induced by ethephon
Large numbers of genes were identified with highly significant levels (adjusted P-value < 0.01) of differential expression induced by ethephon treatment, and these were dependent on both tissue type and duration of ethephon exposure (Figure 2). Ethephon is a chemical that diffuses into the underlying plant tissues following application then generates the potent plant hormone ethylene in slightly acid or basic conditions in the cytoplasm (Lieberman [1979]) where the effect of ethephon on endogenous ethylene biosynthesis is transient. This effect occurred in less than 6 days in the case of apple cuttings (Sun and Bassuk [1993]) although the effect of ethephon on P. radiata after 1 week was minimal. Once formed, however, ethylene triggered gene expression from each type of tissue at 8 WAT, and these were different in composition (Figure 2) and function (Table 2). Some (25-50%) of the genes expressed at 1 WAT were found to be differentially expressed at 8 WAT, indicating that these genes may form the core response of these tissues to ethephon. Amongst these were at least 8 gene ontology classes that showed bidirectional responses (both up- and down-regulated) to ethephon in the xylem (Table 2). Overall, ethephon acted as a suppressor rather than as a promoter of gene transcription.
In P. radiata saplings, the mucilaginous xylem exists as a thin layer located between bark and xylem and includes the undifferentiated 1–2 cell thick cambium layer, expanding cells and a few early secondary walls classifiable as xylem but not phloem (Cato et al. [2006]). This mucilaginous layer was practically absent in ethephon-treated stems by 8 WAT such that the recovered tissues contained proportionally more fibrous material comprising young xylem. At 8 WAT, extensive contamination of the 'tissue scrapes' with more mature xylem tissues from ethephon-treated plants is the most probable reason for the unusually high number (>23,000) of probes showing significantly different signals when compared with corresponding control plants from which 'tissue scrapes' uncontaminated by xylem were obtained. For this reason, the data from this treatment will not be discussed further, but serve as a reminder of the need for robust tissue typing and tissue collection if between-tissue comparisons are to be attempted. When comparing different studies it is important to understand the influence of the composition of the woody portion of the stem from which the RNA is extracted. This RNA from woody tissues is expected to comprise primarily of RNA from the outermost layer of woody differentiating xylem, since the final autolysis of cellular contents is slow in conifer tracheids (Bollhöner et al. [2012]) but it will also contain RNA from rays and resin duct parenchyma, as the latter can live for more than 30 years in Pinus (Nakaba et al. [2008]). The xylem transcriptomes obtained from the 2-year-old saplings used in this study likely reflect developmentally young, but woody, outer xylem seen immediately below the gelatinous 'developing xylem' of older trees. An expressed sequence tagged library has previously been generated from such older xylem tissues in 10-year-old P. taeda (Kirst et al. [2003]). Differential gene expression among samples obtained from different organs (xylem, embryo, megagametophytes) have also been reported in P. taeda (Loopstra and Sederoff [1995]).
Ethephon induction of secondary metabolism
As expected, ethephon treatment triggered a chain of events that manifested as bark cracking and necrosis, resinification, and chlorosis, and led to significantly different transcriptomes (Table 2) in all sampled tissues. Ethephon increased the transcription of genes associated with secondary metabolism, specifically of phenylpropanoids and flavonoids in xylem tissues (Table 2). Some of these secondary metabolites may be responsible for the red-fluorescent extractives that developed in ethephon-treated xylem after fixation with FAA (Figure 4C). These extractives seem to be limited to the young outer xylem nearest to the mucilaginous xylem. This is consistent with the hypothesis that, in conifers, the ethylene signal is transported to the mucilaginous xylem which is subsequently reprogrammed to generate cells with altered physiology to produce metabolites that counter the stress factor identified by the signalling pathway (Hudgins and Franceschi [2004]). Resinification was more pronounced at the higher 5 and 25 mg ethephon dosages (data not shown); transcriptomic analyses were not attempted for these treatments due to inconsistent RNA extraction possibly due to the adverse effect of high resin content.
Ethephon and plant defence
Ethylene is involved in the regulation of genes involved in resistance, defence and wounding response (Van Zhong and Burns [2003]; Berrocal-Lobo and Molina [2004]; Harfouche et al. [2006]). Ethephon elicited greater transcript activity from genes implicated in defence against fungal and insect attack and oxidative stress (Table 2). Genes associated with binding (i.e. recognising) chitin, a primary component of fungal cell walls and insect exoskeletons, and chitinases, enzymes designed to degrade chitins, were up-regulated in the xylem. Ethephon exposure also led to the up-regulation of genes related to oxidative stress; active oxygen species, such as the superoxide radical generated during pathogen attack and abiotic stress, are recognised by plants as a signal for triggering defence responses (Vranova et al. [2002]).
Ethephon and anatomical structure of P. radiata
An increase in girth within the treated area (Figure 4) was similar to that reported for balsam fir Abies balsamea (L.) Mill. (Eklund and Little [1996]), indicating that the effect of the ethylene generated from ethephon is limited to the area of application. The thickened cortex displayed deep fissures that did not extend to the xylem. Stereomicroscopic examination of the cut stem revealed that most of the increased growth was due to increased amounts of phloem and cortex rather than the woody portion of the stem. This confirms a previous report (Yamamoto and Kozlowski [1987a]) where a 1% ethephon dose, which is approximately equal to the 1 mg ethephon treatment in this study, was shown to elicit the most bark growth. No significant increase in diameter in the xylem portion of the stem cross sections was observed (see Figure 4) and the increased growth on the xylem side of the mucilaginous xylem consisted of traumatic parenchyma rather than xylem tissue. There was no evidence of increased lignification or increased volume in the woody cross sections. In fact, stems exposed to 1 mg ethephon exhibited an abnormal layer of unlignified tissue in the outer xylem (asterisk in Figure 4B). This corroborates the down-regulation of a cohort of genes identified via the microarray that are associated with lignin biosynthesis in the xylem which is consistent with a previous claim that ethephon reduced lignification of tracheids in compression wood that developed on the underside of tilted stems of Pinus densiflora (Yamamoto and Kozlowski [1987b]).
Oleoresin and resin canals
In response to biotic stress, conifers secrete an oleoresin that is composed mainly of mono- and diterpenes (Schmidt et al. [2010]). Oleoresin production may be associated with resin duct formation in the cambium zone and developing xylem in response to insect attack or experimentally following methyl jasmonate treatment (Zulak and Bohlmann [2010]). In this study, applications of >1 mg of ethephon induced significant resin secretion although there were no clear differences in resin duct formation (Figure 4) over the duration of the experiment. The traumatic xylem that developed in stems treated with ethephon showed poorly lignified parenchymatous cells suffused with extractives (Figure 5). The lack of change in the size and number of the resin canals indicates that the effect of ethephon did not extend to the type of resin-duct formation that is associated with terpene synthesis over the time course of this experiment. By contrast, a similar treatment (1% ethephon in lanolin) had been found previously to induce significant resinification as well as terpene-duct formation in P. halepensis (Yamamoto and Kozlowski [1987a]). Traumatic resin-duct formation after ethephon injection has also been observed in four-year-old Douglas-fir (Pseudotsuga menziesii (Mirb.) Franco) and giant redwood trees (Sequoiadendron giganteum Lindl. Buchholz) (Hudgins and Franceschi [2004]). The higher levels of ethephon (5 and 25 mg) used in this study induced greater resin bleeding and bark splitting, and a reduction in stem girth and plant vitality leading to chlorosis.
Microarray and quantitative RT-PCR
Heterologous hybridisation of Pinus radiata derived probes to the Pinus microarray may produce spurious hybridisation results due to imperfect annealing that can lead to incorrect interpretations of gene expression levels; quantitative RT-PCR is therefore necessary to verify the microarray results. Quantitative real-time PCR is commonly used to validate gene expression results obtained from microarray analysis however there are factors inherent in these two methods that can lead to contradictory data (Morey et al. [2006]). Microarrays are relatively insensitive in detecting < 2-fold changes (Wang et al. [2006]). For example, 28% of the rice genes selected for validation gave non-significant correlations between microarray and quantitative RT-PCR signals (Dubouzet et al. [2007]), which was taken to be within normal expectations for microarray experiments. In this study, five probes were identified with consistent fold-change values by quantitative RT-PCR that confirmed the direction of expression revealed by the microarray and three of the genes had almost identical fold change estimates. Four of the probes tested could not be amplified, indicating a difference in the ability to detect target transcripts between the heterologous microarray and the PCR platforms. A greater level of probe mismatching is tolerated within a microarray hybridisation (1–6 base differences over 60-mer probe can reduce signal intensity by at least 50% (LeProust [2008]; Hughes et al. [2001])) compared with lower levels of mismatch that are permissible within a quantitative PCR experimental design. Mismatches may prevent successful RT-PCR validation with particular PCR primers or for the more dissimilar heterologous genes. In some cases, successful RT-PCR validation may be overcome through the fine tuning of PCR conditions including the re-design of an appropriate target within the microarray probe or target gene on which to base primer design so that it avoids areas of mismatch in the heterologous gene while still generating >45 bp fragments. Experimental factors such as using different reverse transcriptase-PCR kits or thermal cycling conditions may also need to be explored. It is important to note that a microarray is a tool to uncover genes of interest under particular experimental designs, and that those identified need to be confirmed using other approaches including quantitative RT-PCR. An important experimental consideration therefore is to ensure that sufficient material is archived to allow the required testing that may be required for validation.
Conclusion
It can be concluded that an annotated database of Pinus cDNA sequences can provide a sound basis for the design of a Pinus microarray to study gene expression in P. radiata by heterologous hybridisation. In silico comparisons indicated a very high similarity between publicly available transcripts derived from P. radiata and the microarray probes that were predominantly derived from P. taeda. Further genome sequencing initiatives within the Pinaceae have the potential to further expand the database upon which microarrays can be designed to interrogate transcriptome data across related species. The Pinus microarray was tested by heterologous hybridisation with cRNA produced from bark, mucilaginous xylem and xylem of P. radiata clonal saplings that were subjected to an ethephon induction trial. The microarray was able to highlight that ethephon initiated very different transcriptomes for all tissues sampled at 1 and 8 WAT and these could be associated with an expected cascade of physiological responses. As is common practice with microarray experiments validation of gene expression highlighted by the Pinus microarray via quantitative RT-PCR is required; a caveat for validation of heterologous hybridisation is that primer design needs to overcome sequence differences that can exist between the heterologous test probes and the microarray 60-mer sequences. While not further tested here, the Pinus microarray developed may be useful for P. taeda and other closely related species in the Pinaceae since the majority (>98%) of sequences originate from P. taeda. Further development of the All-Pinus gene database is also valuable as more sequence data from related species in the Pinaceae becomes available.
Authors' contributions
All authors contributed to this paper. JDG contributed initial idea and concept development, microarray design, experimental manipulations and manuscript writing; LD provided microscopy and interpretation; MAB and LM implemented microarray hybridisation and analysis; VL contributed database construction; and GLl-J contributed to concept development and manuscript writing. All authors read and approved the final manuscript.
References
Adomas A, Heller G, Li G, Olson A, Chu T-M, Osborne J, Craig D, van Zyl L, Wolfinger R, Sederoff R, Dean RA, Stenlid J, Finlay R, Asiegbu FO: Transcript profiling of a conifer pathosystem: response of Pinus sylvestris root tissues to pathogen ( Heterobasidion annosum ) invasion. Tree Physiology 2007,27(10):1441–1458. 10.1093/treephys/27.10.1441
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 1997,25(17):3389–3402. 10.1093/nar/25.17.3389
Balk P, Bronnum P, Perks M, Stattin E, van der Geest L, van Wordragen M: Innovative Cold Tolerance Test for Conifer Seedlings. In National Proceedings: Forest and Conservation Nursery Associations-2006. RMRS-P-50. USDA Forest Service, Rocky Mountain Research Station, Fort Collins, CO; 2007:9–14.
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995,57(1):289–300.
Berrocal-Lobo M, Molina A: Ethylene response factor 1 mediates Arabidopsis resistance to the soilborne fungus Fusarium oxysporum . Molecular Plant-Microbe Interactions 2004,17(7):763–770. 10.1094/MPMI.2004.17.7.763
Bollhöner B, Prestele J, Tuominen H: Xylem cell death: emerging understanding of regulation and function. Journal of Experimental Botany 2012,63(3):1081–1094. 10.1093/jxb/err438
Brown K, Leopold A: Ethylene and the regulation of growth in pine. Canadian Journal of Forest Research 1973,3(1):143–145. 10.1139/x73-019
Cato S, McMillan L, Donaldson L, Richardson T, Echt C, Gardner R: Wood formation from the base to the crown in Pinus radiata : gradients of tracheid wall thickness, wood density, radial growth rate and gene expression. Plant Molecular Biology 2006,60(4):565–581. 10.1007/s11103-005-5022-9
Davey M, Graham N, Vanholme B, Swennen R, May S, Keulemans J: Heterologous oligonucleotide microarrays for transcriptomics in a non-model species; a proof-of-concept study of drought stress in Musa . BMC Genomics 2009,10(1):436. 10.1186/1471-2164-10-436
Dubouzet JG, Ishihara A, Matsuda F, Miyagawa H, Iwata H, Wakasa K: Integrated metabolomic and transcriptomic analyses of high-tryptophan rice expressing a mutant anthranilate synthase alpha subunit. Journal of Experimental Botany 2007,58(12):3309–3321. 10.1093/jxb/erm179
Eklund L, Little CHA: Laterally applied Ethrel causes local increases in radial growth and indole-3-acetic acid concentration in Abies balsamea shoots. Tree Physiology 1996,16(5):509–513. 10.1093/treephys/16.5.509
Feldmesser E, Rosenwasser S, Vardi A, Ben-Dor S: Improving automated de-novo transcriptome definition in non-model organisms by integrating manually defined gene information. EMBnet. Journal 2013,19(A):45–46. 10.14806/ej.19.A.614
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nuclear Acids Symposium Series 1999, 41: 95–98.
Harfouche AL, Shivaji R, Stocker R, Williams PW, Luthe DS: Ethylene signaling mediates a maize defense response to insect herbivory. Molecular Plant Microbe Interactions: MPMI 2006,19(2):189–199. 10.1094/MPMI-19-0189
Heath LS, Ramakrishnan N, Sederoff RR, Whetten RW, Chevone BI, Struble CA, Jouenne VY, Chen D, van Zyl L, Grene R: Studying the functional genomics of stress responses in loblolly pine with the Expresso microarray experiment management system. Comparative and Functional Genomics 2002,3(3):226–243. 10.1002/cfg.169
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, Tuteja R, Kumar A, Bhanuprakash A, Mulaosmanovic B, Gujaria N, Krishnamurthy L, Gaur PM, Kavikishor PB, Shah T, Srinivasan R, Lohse M, Xiao Y, Town CD, Cook DR, May GD, Varshney RK: Large-scale transcriptome analysis in chickpea ( Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnology Journal 2011,9(8):922–931. 10.1111/j.1467-7652.2011.00625.x
Huang X, Madan A: CAP3: A DNA sequence assembly program. Genome Research 1999,9(9):868. 10.1101/gr.9.9.868
Huang DW, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 2008,4(1):44–57. 10.1038/nprot.2008.211
Huang DW, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research 2009,37(1):1–13. 10.1093/nar/gkn923
Huang Y-L, Fang X-T, Lu L, Yan Y-B, Chen S-F, Hu L, Zhu C-C, Ge X-J, Shi S-H: Transcriptome analysis of an invasive weed Mikania micrantha . Biologia Plantarum 2012,56(1):111–116. 10.1007/s10535-012-0024-3
Hudgins J, Franceschi V: Methyl jasmonate-induced ethylene production is responsible for conifer phloem defense responses and reprogramming of stem cambial zone for traumatic resin duct formation. Plant Physiology 2004,135(4):2134. 10.1104/pp.103.037929
Hughes T, Mao M, Jones A, Burchard J, Marton M, Shannon K, Lefkowitz SM, Ziman M, Schelter JM, Meyer MR, Kobayashi S, Davis C, Dai H, He YD, Stephaniants SB, Cavet G, Walker WL, West A, Coffey E, Shoemaker DD, Stoughton R, Blanchard AP, Friend SH, Linsley PS: Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nature Biotechnology 2001,19(4):342–347. 10.1038/86730
Kirst M, Johnson A, Baucom C, Ulrich E, Hubbard K, Staggs R, Paule C, Retzel E, Whetten R, Sederoff R: Apparent homology of expressed genes from wood-forming tissues of loblolly pine ( Pinus taeda L.) with Arabidopsis thaliana . Proceedings of the National Academy of Sciences 2003,100(12):7383–7388. 10.1073/pnas.1132171100
LeProust, E. (2008). Agilent's Microarray Platform: How High-Fidelity DNA Synthesis Maximizes the Dynamic Range of Gene Expression Measurements Agilent's Application Notes (p. 12), Agilent Technologies ([http://www.agilent.com/chem/dna])
Li X, Wu HX, Southerton SG: Identification of putative candidate genes for juvenile wood density in Pinus radiata . Tree Physiology 2012,32(8):1046–1057. 10.1093/treephys/tps060
Lieberman M: Biosynthesis and action of ethylene. Annual Review of Plant Physiology 1979,30(1):533–591. 10.1146/annurev.pp.30.060179.002533
Loopstra CA, Sederoff RR: Xylem-specific gene expression in loblolly pine. Plant Molecular Biology 1995,27(2):277–291. 10.1007/BF00020183
Lorenz WW, Alba R, Yu YS, Bordeaux J, Simoes M, Dean J: Microarray analysis and scale-free gene networks identify candidate regulators in drought-stressed roots of loblolly pine ( P. taeda L.). BMC Genomics 2011,12(1):264. 10.1186/1471-2164-12-264
Lorenz WW, Ayyampalayam S, Bordeaux JM, Howe GT, Jermstad KD, Neale DB, Rogers DL, Dean JFD: Conifer DBMagic: a database housing multiple de novo transcriptome assemblies for 12 diverse conifer species. Tree Genetics Genomics 2012, 8: 1477–1485. 10.1007/s11295-012-0547-y
Min, XJ, Butler, G, Storms, R, & Tsang, A. (2009). Comparative Assessment of DNA Assemblers for Assembling Expressed Sequence Tags. In Bioinformatics (OCCBIO'09. Ohio Collaborative Conference on, 2009, pp. 79–82). IEEE. doi:10.1109/OCCBIO.2009.19.
Morey JS, Ryan JC, Van Dolah FM: Microarray validation: factors influencing correlation between oligonucleotide microarrays and real-time PCR. Biological Procedures Online 2006,8(1):175–193. 10.1251/bpo126
Myburg H, Morse A, Amerson H, Kubisiak T, Huber D, Osborne J, Osborne JA, Garcia SA, Nelson CD, Davis JM, Covert SF, van Zyle LM: Differential gene expression in loblolly pine ( Pinus taeda L.) challenged with the fusiform rust fungus, Cronartium quercuum f. sp. fusiforme . Physiological and Molecular Plant Pathology 2006,68(1–3):79–91. 10.1016/j.pmpp.2006.07.002
Nakaba S, Kubo T, Funada R: Differences in patterns of cell death between ray parenchyma cells and ray tracheids in the conifers Pinus densiflora and Pinus rigida . Trees-Structure and Function 2008,22(5):623–630. 10.1007/s00468-008-0220-0
New Zealand Forest Owners Association. (2011). New Zealand Plantation Forest Industry: Facts and Figures. In N. Z. F. O. Association (Ed.), (Vol. 2010/11, p. 4). New Zealand: New Zealand Forest Owners Association.
Perdiguero P, Collada C, Barbero MC, Casado GG, Cervera MT, Soto Á: Identification of water stress genes in Pinus pinaster Ait. by controlled progressive stress and suppression-subtractive hybridization. Plant Physiology and Biochemistry 2012,50(1):44–53. 10.1016/j.plaphy.2011.09.022
Pfaffl MW: Quantification strategies in real-time PCR. AZ of Quantitative PCR 2004, 1: 89–113.
Reinders J, Vivier CD, Theiler G, Chollet D, Descombes P, Paszkowski J: Genome-wide, high-resolution DNA methylation profiling using bisulfite-mediated cytosine conversion. Genome Research 2008,18(3):469–476. 10.1101/gr.7073008
Schmidt A, Wächtler B, Temp U, Krekling T, Séguin A, Gershenzon J: A bifunctional geranyl and geranylgeranyl diphosphate synthase is involved in terpene oleoresin formation in Picea abies . Plant Physiology 2010,152(2):639–655. 10.1104/pp.109.144691
Schnurr JP, Cheng Z, Boe AA: Effects of plant growth regulators on sturdiness of Jack pine seedlings. Journal of Environmental Horticulture 1996, 14: 228–230.
Smyth GK: Limma: Linear Models for Microarray Data. In 'Bioinformatics and Computational Biology Solutions Using R and Bioconductor'. Edited by: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S. Springer Science + Business Media New York, New York; 2005:397–420. 10.1007/0-387-29362-0_23
Sun WQ, Bassuk NL: Auxin-induced Ethylene Synthesis during Rooting and Inhibition of Budbreak ofRoyalty' Rose Cuttings. Journal of the American Society for Horticultural Science 1993,118(5):638–643.
Van Zhong G, Burns JK: Profiling ethylene-regulated gene expression in Arabidopsis thaliana by microarray analysis. Plant Molecular Biology 2003,53(1):117–131. 10.1023/B:PLAN.0000009270.81977.ef
Van Zyl L, Von Arnold S, Bozhkov P, Chen Y, Egertsdotter U, MacKay J, Sederoff RR, Shen J, Zelena L, Clapham DH: Heterologous array analysis in Pinaceae: hybridization of Pinus taeda cDNA arrays with cDNA from needles and embryogenic cultures of P. taeda , P. sylvestris or Picea abies . Comparative and Functional Genomics 2002,3(4):306–318. 10.1002/cfg.199
Vranova E, Inzé D, Van Breusegem F: Signal transduction during oxidative stress. Journal of Experimental Botany 2002,53(372):1227–1236. 10.1093/jexbot/53.372.1227
Wang Y, Barbacioru C, Hyland F, Xiao W, Hunkapiller KL, Blake J, Chan F, Gonzalez C, Zhang L, Samaha RR: Large scale real-time PCR validation on gene expression measurements from two commercial long-oligonucleotide microarrays. BMC Genomics 2006,7(1):59. 10.1186/1471-2164-7-59
Warner H, Leopold A: Ethylene evolution from 2-chloroethylphosphonic acid. Plant Physiology 1969,44(1):156. 10.1104/pp.44.1.156
Yamamoto F, Kozlowski T: Effect of ethrel on growth and stem anatomy of Pinus halepensis seedlings. IAWA Bulletin 1987,8(1):11–19. 10.1163/22941932-90001019
Yamamoto F, Kozlowski T: Effects of flooding, tilting of stems, and ethrel application on growth, stem anatomy and ethylene production of Pinus densiflora seedlings. Journal of Experimental Botany 1987,38(2):293–310. 10.1093/jxb/38.2.293
Yang S-H, van Zyl L, No E-G, Loopstra CA: Microarray analysis of genes preferentially expressed in differentiating xylem of loblolly pine ( Pinus taeda ). Plant Science 2004,166(5):1185–1195. 10.1016/j.plantsci.2003.12.030
Zulak KG, Bohlmann J: Terpenoid biosynthesis and specialized vascular cells of conifer defense. Journal of Integrative Plant Biology 2010,52(1):86–97. 10.1111/j.1744-7909.2010.00910.x
Acknowledgments
The authors wish to express their gratitude to the following Scion staff: Lucy Macdonald, Lisa Stanbra, Lorelle Phillips and, most specially, Heather Flint for their critical support in various technical aspects of this research. Drs. Tim Strabala and Armin Wagner provided invaluable insight and discussions that vastly improved the manuscript. Details of the microarray experiment have been submitted according to the specifications of the National Centre for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) and are available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE42300.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Dubouzet, J.G., Donaldson, L., Black, M.A. et al. Heterologous hybridisation to a Pinus microarray: profiling of gene expression in Pinus radiata saplings exposed to ethephon. N.Z. j. of For. Sci. 44, 21 (2014). https://doi.org/10.1186/s40490-014-0021-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40490-014-0021-8