
Abstract
TAR DNA-binding protein 43 (TDP-43) and Fused in Sarcoma/Translocated in Sarcoma (FUS) are ribonucleoproteins associated with pathogenesis of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). Under physiological conditions, TDP-43 and FUS are predominantly localized in the nucleus, where they participate in transcriptional regulation, RNA splicing and metabolism. In disease, however, they are typically mislocalized to the cytoplasm where they form aggregated inclusions. A number of shared cellular pathways have been identified that contribute to TDP-43 and FUS toxicity in neurodegeneration. In the present study, we report a novel pathogenic mechanism shared by these two proteins. We found that pathological FUS co-aggregates with a ribosomal protein, the Receptor for Activated C-Kinase 1 (RACK1), in the cytoplasm of spinal cord motor neurons of ALS, as previously reported for pathological TDP-43. In HEK293T cells transiently transfected with TDP-43 or FUS mutant lacking a functional nuclear localization signal (NLS; TDP-43ΔNLS and FUSΔNLS), cytoplasmic TDP-43 and FUS induced co-aggregation with endogenous RACK1. These co-aggregates sequestered the translational machinery through interaction with the polyribosome, accompanied by a significant reduction of global protein translation. RACK1 knockdown decreased cytoplasmic aggregation of TDP-43ΔNLS or FUSΔNLS and alleviated associated global translational suppression. Surprisingly, RACK1 knockdown also led to partial nuclear localization of TDP-43ΔNLS and FUSΔNLS in some transfected cells, despite the absence of NLS. In vivo, RACK1 knockdown alleviated retinal neuronal degeneration in transgenic Drosophila melanogaster expressing hTDP-43WT or hTDP-43Q331K and improved motor function of hTDP-43WT flies, with no observed adverse effects on neuronal health in control knockdown flies. In conclusion, our results revealed a novel shared mechanism of pathogenesis for misfolded aggregates of TDP-43 and FUS mediated by interference with protein translation in a RACK1-dependent manner. We provide proof-of-concept evidence for targeting RACK1 as a potential therapeutic approach for TDP-43 or FUS proteinopathy associated with ALS and FTLD.
Similar content being viewed by others
Introduction
The Receptor for Activated C-Kinase 1 (RACK1) is a member of the tryptophan, aspartic acid repeat (WD-repeat) protein family. It was first identified as a receptor for protein kinase C (PKC) βII to activate the translational activity of Eukaryotic Translation Initiation Factor 6 (eIF6), and regulates a variety of cellular events, such as cell growth, motility, differentiation, microRNA biogenesis and function, and protein synthesis [7, 8, 10, 21, 25, 33, 41, 42, 61, 66].
Similar to many other WD-repeat proteins, RACK1 is highly conserved throughout evolution from prokaryotes to eukaryotes [70], and adopts a seven-bladed β-propeller structure as confirmed by X-ray crystallography studies [13]. Also as with other WD-repeat proteins, RACK1 itself lacks catalytic enzymatic activities, but plays a crucial role in scaffolding numerous proteins, such as kinases, transmembrane receptors, ion channels, and ribosomal proteins that are involved in a wide range of cellular functions [1]. As such, RACK1 serves as a hub connecting a complex network of cellular signaling events.
To date, at least 80 proteins have been identified to interact with RACK1 [1], including the RACK1-ribosome association, which has drawn intense research interest in the past two decades since first identified by mass spectrometry [60]. It is now established that RACK1 is a key constituent of the eukaryotic small (40S) subunit of the ribosome and is located in the vicinity of the mRNA exit channel [60]. Association of RACK1 with the ribosome plays a crucial role in cap-dependent translation and the downstream signaling events involved [20, 45].
Given the diverse roles of RACK1 in cellular function mentioned above, it is not surprising that RACK1 has been implicated in many human diseases, such as cancer, developmental disorders, addiction, and cardiovascular diseases [1]. However, little is known about the role of RACK1 in the pathogenesis of neurodegenerative disorders, such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD).
ALS is the most common motor neuron disease characterized by the ultimately fatal progressive degeneration of motor neurons in the spinal cord and motor cortex of the brain, which leads to paralysis of muscles that control limb movement, speech, swallowing, and breathing. While only ~ 10% of ALS is familial and associated with various genetic mutations (fALS), the remaining ~ 90% is sporadic (sALS). Frontotemporal lobar degeneration (FTLD), a pathological process occurring in clinical syndrome FTD, is characterized by neuronal degeneration in the frontal and temporal lobes of the brain. The most common clinical presentations include abnormal behavior, language disorders, and disorders of movement. ALS and FTD are considered to belong to a common disease spectrum (ALS–FTD) because of overlaps in genetic, clinical symptoms, and pathologies [12], and ~ 20% of ALS patients fall into the ALS–FTD spectrum based on clinical criteria [47, 49, 56].
Among the pathological similarities between ALS and FTLD, one major hallmark is the accumulation of cytoplasmic inclusions of TAR DNA-binding protein 43 (TDP-43) or Fused in Sarcoma/Translocated in Sarcoma (FUS/TLS), which are both ubiquitously expressed in most cell types and tissues. TDP-43 proteinopathy occurs in 60–70% of fALS and 90–95% of sALS, whereas TDP-43 pathology is observed in 50% of FTD. FUS pathology accounts for 10% of fALS and 10% of FTD [38, 46]. FUS inclusions are associated only with mutations in ALS and wild-type FUS in a small percentage of FTLD with neuronal inclusions composed of an unidentified ubiquitinated protein (atypical FTLD-U; aFTLD-U) [43]. TDP-43 and FUS proteins share many similarities in their cellular functions, protein structures, and subcellular distribution. TDP-43 and FUS are both RNA-binding proteins that play a pivotal role in multiple aspects of RNA processing, such as alternative splicing, metabolism, transport, and transcriptional and translational regulation [29, 46, 55]. Structurally, TDP-43 and FUS are both heterogeneous nuclear ribonucleoproteins (hnRNPs) that possess RNA-recognition motifs (RRMs), glycine-rich domains, a prion-like low complexity domain (PrLCD), and a nuclear localization signal (NLS) [19, 36]. Under physiological conditions, TDP-43 and FUS shuttle between the nucleus and cytoplasm, with ~ 90% being localized to the nucleus at any given time [29, 46, 55]. Under pathological conditions, such as ALS and FTLD, TDP-43 or FUS can be depleted from the nucleus and mislocalized to the cytoplasm where they can form inclusions in affected neurons and glial cells.
To date, a number of mechanistic pathways have been identified to explain the pathogenic activities of TDP-43 and FUS in ALS and FTLD, including disruption of mitochondrial function, disturbance of proteostasis, pro-inflammatory responses, and oxidative stress [55]. Importantly, a recent study has reported that RACK1 co-aggregates with cytoplasmically mislocalized TDP-43 in both a transfected cell line and ALS spinal cord motor neurons [57]. In addition, this study shows that TDP-43 cytoplasmic aggregates significantly suppress global translation, a process that requires the association of TDP-43 with ribosome-bound RACK1 [57], suggesting a novel pathogenic mechanism underlying TDP-43-associated neurodegeneration through RACK1-mediated global translational suppression.
Considering the many similarities between TDP-43 and FUS, in the present study we sought to: (1) investigate whether TDP-43 and FUS may utilize a similar pathogenic pathway through co-aggregation with RACK1 and suppression of global translation; (2) better understand the molecular mechanism by which interaction of RACK1 with pathological TDP-43 or FUS alters the homeostasis of global translation; (3) explore an RNAi-based approach targeting RACK1 that could potentially ameliorate the adverse effects of pathogenic TDP-43 and FUS on cellular function and neurodegeneration in vitro and in vivo.
Results
Pathological TDP-43 and FUS co-aggregate with RACK1 in the cytoplasm of spinal cord motor neurons of ALS
Using post-mortem human tissues, it has been shown that TDP-43 co-aggregates with RACK1 in the cytoplasm of spinal cord motor neurons from ALS spinal cords [57], a finding that we have confirmed (Additional file 1: Fig. S1). This led us to test whether pathological FUS may also co-aggregate with RACK1 in situ. Immunohistochemical analysis was performed on spinal cord sections from a fALS-FUS case carrying a R521C mutation. As shown in Fig. 1, in contrast to normal control spinal cords, where FUS and RACK1 were predominantly localized in the nucleus and cytoplasm, respectively, pathological FUS co-aggregated with RACK1 in the cytoplasm, similar to what was observed with TDP-43 [57].

Immunohistochemical analysis shows co-aggregation of RACK1 with FUS in the cytoplasm (arrows) in spinal cord sections of a fALS-FUS-R521C case, in contrast to normal nuclear expression of FUS in control (Ctrl). Nuclei stained with DAPI in merged images. Scale bar: 10 μm
Cytoplasmic aggregates of nuclear localization signal-deficient mutants of TDP-43 and FUS are associated with RACK1 co-aggregation in HEK293T cells
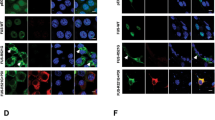
To confirm and extend a previous finding that cytoplasmic TDP-43 co-aggregates with RACK1 in the neuroblastoma cell line SH-SY5Y [57], we transiently transfected HEK293T cells with HA-tagged wild-type (WT) TDP-43 or a nuclear localization signal (NLS)-deficient mutant of TDP-43, TDP-43ΔNLS. As shown in Fig. 2a, in WT TDP-43 over-expressing cells, where HA-WT TDP-43 was almost exclusively localized in the nucleus, endogenous RACK1 was diffusely expressed in the cytoplasm. In contrast, expression of the engineered mutant TDP-43ΔNLS, which aggregates in the cytoplasm, apparently induced RACK1 co-aggregation in the cytoplasm of HEK293T cells, consistent with the report in SH-SY5Y cells [57]. Similar experiments were performed using two variants of HA-tagged mutants of FUS that aggregate in the cytoplasm: natural fALS mutations R495x- and P525L-FUS, collectively referred to as FUSΔNLS herein. While RACK1 was diffusely expressed in the cytoplasm of WT FUS over-expressing cells, both FUSΔNLS mutants were associated with RACK1 co-aggregation in the cytoplasm (Fig. 2b). In contrast, expression of a ribosome binding deficient mutant of RACK1, R36D/K38E-RACK1 (DE-RACK1) [13], which forms large aggregates in both the cytoplasm and the nucleus, did not alter the normal nuclear distribution of endogenous TDP-43 and FUS (Additional file 1: Fig. S2), suggesting that it is TDP-43ΔNLS and FUSΔNLS that played an initiating role in the formation of RACK1 co-aggregates and not the reverse. Transfection of HEK293T cells with a mutant disrupted in schizophrenia 1 (DISC1) with all tryptophan residues mutated to serines, 10W/S-DISC1, which forms large aggregates in the cytoplasm, displayed minimal co-aggregation with RACK1 (Additional file 1: Fig. S3), indicating that RACK1 aggregation is unlikely to be a generic consequence of cytoplasmic protein aggregation.

Immunocytochemical analysis demonstrates expression of HA-tagged TDP-43ΔNLS (a, bottom row) or FUSΔNLS mutants, R495x- and P525L-FUS (b, middle & bottom rows), but not WT TDP-43 (a, top row) or WT-FUS (b, top row), which are primarily localized to the nucleus, is associated with co-aggregation of endogenous RACK1 in HEK293T cells. Nuclei stained with DAPI in merged images. Scale bars: 20 μm
RACK1 is misfolded when co-aggregated with pathological TDP-43 and FUS
Using a computational modeling method we have developed, “Collective Coordinates” [48], we generated a rabbit monoclonal antibody, “RACK1mis”, against a misfolding-specific conformational epitope of RACK1. As shown in Fig. 3, RACK1mis did not react with normal cytoplasmic RACK1 in un-transfected cells but specifically reacted with the species of RACK1 present in TDP-43ΔNLS or FUSΔNLS co-aggregates, indicating that RACK1 is misfolded when co-aggregated with pathological TDP-43 or FUS. Furthermore, reactivity with misfolded RACK1 was also observed in post-mortem FTLD-TDP brain and sALS spinal cord tissue, but not in normal controls (Additional file 1: Fig. S4).

Immunocytochemical analysis demonstrates reactivity of RACK1 misfolding specific antibody “RACK1mis” with RACK1 in cytoplasmic aggregates of HA-tagged TDP-43ΔNLS (middle row) or R495x-FUS (bottom row) transfected HEK293T cells but not with diffuse, non-aggregated RACK1 in the cytoplasm (stained by a Pan RACK1 antibody). RACK1mis shows no reactivity with endogenous, physiological RACK1 in un-transfected (UT, top row) cells. Nuclei stained with DAPI in merged images. Scale bars: 10 μm
Aggregates of TDP-43ΔNLS and FUSΔNLS mutants suppress global translation
A body of evidence has suggested a role for RACK1 in translational regulation in either a ribosome-bound or a ribosome-free form [1, 45]. In particular, TDP-43ΔNLS has been shown to suppress global translation through interaction with ribosome-bound RACK1 in neuroblastoma SH-SY5Y cells [57]. The in vitro and in situ results described above led us to hypothesize that the activities of RACK1-associated translational machinery may be similarly impaired by TDP-43ΔNLS and FUSΔNLS in HEK293T cells. Utilizing Surface Sensing of Translation (SUnSET) (a non-radioactive method utilizing puromycin to tag newly synthesized proteins in live cells) [58], followed by western blotting (SUnSET-WB), we found that TDP-43ΔNLS did suppress global translation in HEK293T cells (Fig. 4a, b). Similarly, both FUSΔNLS mutants also significantly suppressed global translation (Fig. 5a, b).

RACK1 knockdown alleviates aggregation and global translational suppression by TDP-43ΔNLS and WT over-expression in HEK293T cells. a Representative SUnSET-Western Blot (WB) demonstrates that compared to control, expression of HA-tagged TDP-43ΔNLS and to a lesser extent over-expression of WT TDP-43, induces a significant reduction in puromycin (PMY) incorporation. b RACK1 siRNA KD alleviates global translational levels in both cases as determined by quantification of PMY band intensities normalized to loading control β-actin. c SUnSET-ICC shows inhibition of PMY incorporation by TDP-43ΔNLS (HA) expression preferably occurs in cells containing distinctive cytoplasmic aggregates (asterisks, top row). In cells where TDP-43ΔNLS displays a filamentary expression pattern (arrows, bottom row), PMY incorporation is comparable to neighbouring HA negative UT cells. d RACK1 KD alleviates aggregation, resulting in diffuse cytoplasmic expression (top row) or predominantly nuclear localization (bottom row) of TDP-43ΔNLS in a sub-population of transfected cells and correlating with normal PMY incorporation compared to neighbouring HA-negative UT cells. Nuclei stained with DAPI in merged images. Statistics: Ordinary one-way ANOVA Tukey multiple comparisons. n = 4 *p < 0.05; ***p < 0.001; ****p < 0.0001. Scale bars: 20 μm

RACK1 knockdown alleviates aggregation and restores global translational suppression by FUSΔNLS mutants. a Representative SUnSET-Western Blot (WB) demonstrates that compared to control empty vector (EV) transfected cells, expression of HA-tagged FUSΔNLS mutants, R495x- and P525L-FUS, and to a lesser extent over-expression of WT FUS, induces a significant reduction in puromycin (PMY) incorporation. b siRNA KD alleviates global translational levels in all cases as determined by quantification of PMY band intensities normalized to loading control α-tubulin. c SUnSET-ICC demonstrates HA-tagged FUSΔNLS mutants, R495x- and P525L-FUS, cause aggregation and completely inhibit PMY incorporation in transfected cells (asterisks). d RACK1 KD alleviates aggregation, resulting in diffuse cytoplasmic expression or predominantly nuclear localization of HA-tagged FUSΔNLS mutants, R495x- and P525L-FUS, in a sub-population of transfected cells and correlating with normal PMY incorporation compared to neighbouring HA-negative UT cells. Nuclei stained with DAPI in merged images. Statistics: Student’s t-test, unpaired, two-tailed. *p < 0.05 and **p < 0.005. n = 5 Error bars: SEM. Scale bars: 20 μm
To validate these findings and visualize global translational changes in individual cells, SUnSET followed by immunocytochemistry (SUnSET-ICC) was performed in cells transfected with TDP-43ΔNLS or FUSΔNLS. As illustrated in Fig. 4c, TDP-43ΔNLS aggregate-containing cells showed undetectable level of puromycin staining, in sharp contrast to neighbouring un-transfected (UT) HA-negative cells, consistent with strong translational suppression by TDP-43ΔNLS aggregates. However, in cells where TDP-43ΔNLS displayed a diffuse filamentary cytoplasmic expression pattern, translational level was comparable to neighbouring UT cells (Fig. 4c), suggesting that global translational suppression is predominantly caused by consolidated larger size TDP-43ΔNLS/RACK1 co-aggregates. Similar inhibition of puromycin incorporation was observed in FUSΔNLS aggregate-containing cells (Fig. 5c). Together, these results strongly suggest that co-aggregation of RACK1 with TDP-43ΔNLS or FUSΔNLS significantly inhibits normal function of the translational machinery.
TDP-43ΔNLS and FUSΔNLS/RACK1 co-aggregates sequester the polyribosome to form large aggregated complexes
We next tested whether TDP-43ΔNLS or FUSΔNLS inhibited global translation in our HEK293T cell system through sequestration of the polyribosome into RACK1 co-aggregates, as has been reported in SH-SY5Y cells [57]. Immunocytochemical analysis was performed to detect the association of TDP-43ΔNLS or FUSΔNLS/RACK1 aggregates with Rps6 and RPL14, two established markers for the eukaryotic small (40S) and large (60S) ribosomal subunits, respectively. As shown in Fig. 6, UT cells displayed the expected pattern of diffuse expression of Rps6 and RPL14 in the cytoplasm. In contrast, transfection with either TDP-43ΔNLS or FUSΔNLS resulted in prominent co-localization with RACK1 and both Rps6 and RPL14. Together, these results demonstrate that TDP-43ΔNLS and FUSΔNLS cytoplasmic aggregates may disrupt normal function of the translational machinery through aberrant sequestration of RACK1 and associated polyribosome into large aggregated complexes.

Immunocytochemical analysis shows co-aggregation of HA-tagged TDP-43ΔNLS (a, b) or R495x-FUS (c, d) with RACK1 and eukaryotic 40S (Rps6) and 60S (RPL14) ribosome subunits (arrows). In neighbouring HA-negative UT cells, RACK1 is diffusely localized in the cytoplasm. Nuclei stained with DAPI in merged images. Scale bars: 10 μm
RACK1 knockdown diminishes TDP-43ΔNLS and FUSΔNLS cytoplasmic aggregation and leads to nuclear localization in a sub-population of cells
In light of the above results, we considered whether RACK1 knockdown (KD) would render a beneficial effect on TDP-43ΔNLS or FUSΔNLS induced RACK1 aggregation. To this end, HEK293T cells were treated with human-specific anti-RACK1 siRNA prior to TDP-43ΔNLS or FUSΔNLS transfections. As shown in Fig. 7a and b, KD of endogenous RACK1 significantly reduced the average aggregate size of TDP-43ΔNLS. A similar phenomenon was observed in FUSΔNLS transfected cells (Additional file 1: Fig. S5).

RACK1 knockdown not only reduces cytoplasmic aggregation but also increases nuclear localization of TDP-43ΔNLS in HEK293T cells. a Representative images showing that in contrast to control cells where distinctive aggregates of HA-tagged TDP-43ΔNLS are associated with RACK1 co-aggregation in the cytoplasm (arrows), RACK1 siRNA KD not only alleviates cytoplasmic aggregation (white arrowheads) but also leads to nuclear localization (yellow arrowheads) of TDP-43ΔNLS in a sub-population of transfected cells. Nuclei stained with DAPI in merged images. Scale bar: 10 μm. b Quantification of the average aggregate size in each individual TDP-43ΔNLS transfected cells shows a decrease following RACK1 KD. c Quantification of TDP-43ΔNLS nuclear localization from 3 biological repeats shows an increase as a result of RACK1 KD. d Western blot analysis confirms the quality of nucleocytoplasmic fractionation using nuclear and cytosolic protein markers, lamin B1 and β-Actin, respectively (top). TDP-43ΔNLS (HA) or total TDP-43 (pan TDP-43) from either fraction was normalized to β-actin or lamin B1. Quantification by densitometry shows that RACK1 KD decreases the cytoplasmic to nuclear ratio, fragmentation (red arrows, observed in 3 independent experiments), and phosphorylation of TDP-43ΔNLS (quantitated in one experiment). Statistics: Student’s t-test unpaired two-tailed. Error bars: SEM
Surprisingly, RACK1 KD also resulted in partial redistribution of TDP-43ΔNLS from being predominantly cytoplasmic to nuclear in a sub-population of transfected cells, despite its lack of nuclear localization signal (Figs. 4d, 7a and c). This observation was confirmed by biochemical analyses demonstrating a decreased cytoplasmic/nuclear ratio of TDP-43ΔNLS (by ~ 42%) upon RACK1 KD (Fig. 7d). In addition, RACK1 KD led to a reduction of low molecular weight fragments and phosphorylated-TDP-43 in the cytoplasm by ~ 50% and ~ 21%, respectively (Fig. 7d), which are both hallmark biochemical features of TDP-43 pathology in affected CNS regions of ALS/FTLD patients [3, 11, 15, 44, 55, 63, 67]. Similar reversal from predominantly cytoplasmic to nuclear localization was observed with FUSΔNLS upon RACK1 KD (Fig. 5d, Additional file 1: Fig. S5a). It is notable that no detectable changes were observed with the morphology or viability of HEK293T cells upon RACK1 KD, and that endogenous TDP-43 and FUS retained their normal nuclear localization as in control RACK1 expressing cells (Additional file 1: Fig. S6).
RACK1 KD restores global translational suppression by TDP-43ΔNLS and FUSΔNLS
In light of the above results showing that RACK1 KD reduced cytoplasmic aggregation and increased nuclear/cytoplasmic ratio of TDP-43ΔNLS and FUSΔNLS, we hypothesized that RACK1 KD may mitigate sequestration of polyribosomes into aggregates and thereby alleviate global translational suppression by both mutants. Utilizing SUnSET-WB and SUnSET-ICC, we showed that global translational suppression by either mutant was indeed significantly restored to levels comparable to those of controls by RACK1 KD (Figs. 4, 5).
RACK1 KD inhibits TDP-43ΔNLS intercellular transmission
A body of evidence has suggested that misfolded/aggregated TDP-43 can be transmitted from cell to cell in a prion-like fashion and contributes to the spreading of neurodegeneration across CNS regions [9, 11, 18, 24, 28, 30, 53,54,55]. We therefore tested whether RACK1 KD might have an impact on cell-to-cell transmission of misfolded TDP-43ΔNLS. We employed a previously established procedure [23, 50], in which conditioned medium from donor cells expressing TDP-43ΔNLS without or with RACK1 KD was added to naïve recipient cells for quantitation of transmitted TDP-43ΔNLS (Fig. 8a). Compared to control donor cells, RACK1 KD reduced both total TDP-43ΔNLS in recipient cells and the ratio of recipient/donor TDP-43ΔNLS (Fig. 8b–d), indicating that RACK1 KD significantly inhibited the intercellular transmission of misfolded TDP-43ΔNLS.

RACK1 knockdown inhibits intercellular transmission of TDP-43ΔNLS in HEK293T cells. a Schematic illustration of the intercellular transmission procedure. Representative western blot (WB) b and densitometric quantification show reduced total HA-tagged TDP-43ΔNLS transmitted to naïve recipient cells c as well as a reduction in the recipient/donor ratio of TDP-43ΔNLS upon RACK1 KD. Statistics: Student’s t-test, unpaired two-tailed. **p < 0.005; ****p < 0.0001. n = 5
RACK1 KD alleviates retinal degeneration and delays the decline of motor function in human WT and mutant TDP-43 transgenic D. melanogaster
Finally, we sought to investigate whether the aforementioned beneficial effects of RACK1 KD on TDP-43ΔNLS associated phenotypes in HEK293T cells could be translated into protective activity for neurons in vivo. To this end, we employed an established D. melanogaster UAS-Gal4 expression system to target expression of human wild-type TDP-43 and ALS-associated mutant TDP-43Q331K (hTDP-43WT and hTDP-43Q331K) [17] in retinal (driven by the GMR promotor) or motor (driven by the D42 promotor) neurons with or without knocking down of endogenous RACK1 by RACK1-RNAi co-expression (Fig. 9a), with mCherry-RNAi serving as a control.

RACK1 knockdown significantly reduces hTDP-43-induced neuronal death and dysfunction in transgenic D. melanogaster. a Schematic illustration for the construction of transgenic D. melanogaster lines specifically targeting retinal (GMR-driven) or motor (D42-driven) neurons. b Representative images of fly eyes. Expression of hTDP-43WT (A–D) or hTDP-43Q331K (E–H) in retinal neurons cause neuronal death in both males and females, detected as darkened ommatidia. RACK1-RNAi reduces the appearance of dead ommatidia in all four conditions (B, D, F, H), compared to mCherry-RNAi control (A, C, E, G). Control flies with no dead ommatidia include those expressing RACK1-RNAi alone (I), and those harbouring unexpressed (undriven) transgenes (J–L). Scale bar: 250 mm. c Quantification of retinal neuronal death. Mann–Whitney test indicates a significant reduction in ommatidia death upon RACK1-RNAi KD in both males and females expressing either hTDP-43WT or hTDP-43Q331K. 21–33 flies were scored per condition. Statistics: Mann–Whitney test, *p < 0.05, ***p < 0.001. ****p < 0.0001. d Histological analysis of retina from flies expressing hTDP-43WT (A, B) or hTDP-43Q331K (C, D) in conjunction with control mCherry-RNAi (A, C) or RACK1-RNAi (B, D) show preservation of retinal architecture by RACK1 KD. Scale bar: 50 mm. e Both male and female hTDP-43WT transgenic flies show reduced climbing ability compared to mCherry-RNAi controls, which was significantly improved by RACK1-RNAi. Statistics: 2-way ANOVA. **p < 0.01, ****p < 0.0001. 60–100 flies were scored per condition. Error bars: SD
Firstly, retinal degeneration was assessed for the loss of ommatidia and structural organization of the retina. As shown in Fig. 9b, compared to undriven controls (J–L), flies co-expressing mCherry-RNAi with hTDP-43WT displayed mild neurodegeneration (A & C), consistent with a previous report [37], and the phenotype was more severe with expression of mutant hTDP-43Q331K (E & G). Quantification of retinal degeneration (Fig. 9c) showed that in both male and female flies, hTDP-43Q331K caused more neuronal cell death than hTDP-43WT (Males: mean of 14.8 dead ommatidia out of ~ 700 per eye for hTDP-43Q331K vs. 8.2 for hTDP-43WT; Females: 18.5 vs. 9.2). In contrast, flies co-expressing RACK1-RNAi with either hTDP-43WT or hTDP-43Q331K displayed significantly less degeneration in all four of these populations (B, D, F, H). Note that in several individuals expressing hTDP-43WT with RACK1-RNAi, the number of dead ommatidia was reduced to zero. In contrast, in flies expressing either allele of hTDP-43 with control mCherry-RNAi, neuronal death was always observed (Fig. 9c). Additional evidence of neurodegeneration was observed in both hTDP-43WT and hTDP-43Q331K expressing flies, which displayed structural disorganization of the retina. These phenotypes were also significantly restored by RACK1-RNAi (Fig. 9d). Interestingly, flies expressing GMR-RACK1-RNAi alone (Fig. 9b, I) were comparable to undriven controls (Fig. 9b, J–L), suggesting that RACK1 may be dispensable in mature fly retinal neurons.
Secondly, the motor function of transgenic flies was assessed using a previously established climbing assay [59]. As shown in Fig. 9e, both male and female flies expressing hTDP-43WT in motor neurons displayed reduced climbing ability compared to controls, which was significantly improved by RACK1-RNAi in both sexes, suggesting that knocking down of endogenous RACK1 delayed the decline of motor function caused by human TDP-43.
Together, these results demonstrate that knocking down of RACK1 significantly alleviated retinal degeneration and delayed motor neuron dysfunction caused by expression of hTDP-43WT or hTDP-43Q331K in D. melanogaster.
Discussion
TDP-43 and FUS have drawn great interest in neuroscience since first identified as pathogenic proteins associated with ALS and FTLD. Because TDP-43 and FUS share many similarities in terms of their protein structure, nucleocytoplasmic distribution, physiological function, and histopathology in diseased human tissues, it is not surprising that a number of common cellular pathways have been identified to participate in the pathogenic activities of TDP-43 and FUS cytoplasmic aggregates. In the present study, we report our findings that support an additional shared pathway underlying the detrimental effects of TDP-43 and FUS aggregation in the cytoplasm, and explore a potential therapeutic avenue for the treatment of neurodegenerative diseases with TDP-43 and FUS proteinopathies.
RACK1, a key player in pathogenic TDP-43 and FUS aggregation-induced global translational suppression
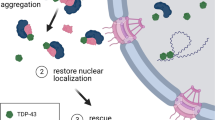
RACK1 can come in proximity and interact with TDP-43 and FUS under physiological conditions existing in normal cells, and in stress granules, which can transiently repress protein translation as a protective activity [68]. However, the interaction of RACK1 with protein aggregates in diseased cells may be pathologically significant and contribute to disruption of protein expression. In a Drosophila model of ALS, mutant TDP-43 was shown to repress translation of Futsch/Map1B, a key protein regulating axonal and dendritic development and microtubule organization at the neuromuscular junction [14]. In mouse primary neurons, mutant TDP-43, together with Fragile X Syndrome protein (FMRP), represses translation of neuronal proteins Rac1 and GluR1 [39]. In Drosophila and rat primary neurons, mutant TDP-43 induces a prolonged translational suppression by stimulating eIF2α phosphorylation [34]. Importantly, more recent studies using transfected human neuroblastoma cell lines or fibroblasts derived from ALS-FUS patients have provided strong evidence that TDP-43 and FUS cytoplasmic aggregates suppress global translation, and that mutant TDP-43 exerts this effect through direct interaction with the ribosome scaffold protein RACK1 [32, 57]. This work is consistent with our current findings that TDP-43ΔNLS and FUSΔNLS mutants co-aggregate with RACK1 and suppress global translation in transfected HEK293T cells. Furthermore, we show that RACK1 KD not only reduced aggregation of TDP-43ΔNLS and FUSΔNLS, but also caused a shift of both mutants from being predominantly cytoplasmic to nuclear in a sub-population of transfected cells (discussed further below), accompanied by restoration of global translation, strongly suggesting that RACK1 is a key player in this aggregation/translational suppression process.
RACK1—a scaffold linking pathogenic TDP-43 and FUS to ribosomes in formation of large aggregate complexes
To decipher the mechanism underlying the above observation, it is worthwhile to recapitulate the fundamental biological function of ribosomes. The ribosome is well appreciated as the central apparatus of protein synthesis, with the small (40S in eukaryotes) subunit initiating the mRNA to amino acid decoding/translational process while the large (60S in eukaryotes) subunit catalyzes the peptide transferase activity to generate nascent polypeptide chains. RACK1 is widely recognized to be an integral constituent of the 40S ribosomal subunit, which in turn is stably associated with the 60S subunit. Our confocal microscopic studies have demonstrated that TDP-43ΔNLS and FUSΔNLS recruited both 40S and 60S subunits, in addition to RACK1, to the same aggregate complex in the cytoplasm (Fig. 6), resulting in global suppression of translation.
Aggregate inhibition of global translation: size matters?
An interesting phenomenon observed from our SUnSET-ICC studies is that global translational suppression appears to be predominantly correlated with large distinctive aggregates of TDP-43ΔNLS or FUSΔNLS but not the diffuse pool in the cytoplasm (Figs. 4c, 5c). This implies that global translation may not be significantly impaired unless the optical area of aggregated TDP-43 or FUS reaches a certain threshold. This is in line with previous findings using a motoneuron-like neuroblastoma cell line, NSC-34, which show that neuronal toxicity is attributed to the largest cytoplasmic aggregates of TDP-43 [6].
Redistribution of NLS-deficient TDP-43 and FUS from predominantly cytoplasmic to nuclear expression upon RACK1 knockdown
Perhaps the most unexpected observation from the present study was that RACK1 KD changed the predominantly cytoplasmic mislocalization of TDP-43ΔNLS and FUSΔNLS to a normal nuclear localization in a sub-population of transfected cells despite their lack of a functional NLS. One potential explanation is that the anti-aggregation activity of RACK1 KD reduced formation of compact large aggregates of TDP-43ΔNLS and FUSΔNLS with the polyribosome, consequently allowing non-aggregated free molecules to diffuse from the cytoplasm to the nucleus. Given the current knowledge that cytosolic proteins are capable of crossing the nuclear pore complexes (NPC) through passive diffusion (as well as active transport), and that the likelihood of this process is not only inversely correlated with the molecular mass but also can be influenced by protein conformation [64], we speculate that TDP-43ΔNLS and FUSΔNLS may have partly entered the nucleus through passive diffusion, as opposed to the classical NLS-dependent active nuclear import pathway for physiological TDP-43 and FUS [4]. Moreover, the cutoff molecular mass for passive diffusion across NPC can be up to 90-110kD [69], which encompasses the molecular weights of monomer/dimer TDP-43 and monomer FUS.
RACK1 knockdown rescues TDP-43-associated neurodegeneration in vivo
A most important question arising from the present study is whether the healthy nuclear TDP-43 and FUS are capable of carrying out their physiological functions in cells in which RACK1 KD attenuates the toxic effects of TDP-43ΔNLS or FUSΔNLS. Healthy TDP-43 and FUS, as nuclear ribonucleoproteins, are well-known for their role in alternative splicing regulation of specific mRNA targets as well as mRNA transport and stability [2, 5, 16, 22, 26, 27, 52, 65]. In the present study, we directly tested RACK1 KD strategy in Drosophila models transgenic for human WT or mutant TDP-43 exclusively in retinal or motor neurons in vivo. We found that RACK1 KD significantly alleviated retinal neurodegeneration and delayed locomotion deficit of TDP-43 transgenic flies, indicating that RACK1 KD allows normal healthy fly TDP-43 to perform its physiological functions. Moreover, flies with RACK1 KD alone were not only viable, but also exhibited retinal health comparable to control flies, suggesting that physiological level of RACK1 may not be required for the survival and function of differentiated neurons in flies (Fig. 9). This is in agreement with previous findings that knocking down of RACK1 does not affect mature cell viability and proliferation in Drosophila [40], despite lethality when expressed in early development, particularly oogenesis [31]. Similar strategies can be employed in future studies to characterize the effect of RACK1 KD on transgenic flies expressing human FUS mutations [35, 62].
Overall, our results reveal a novel shared mechanism of pathogenesis for misfolded aggregates of TDP-43 and FUS mediated by interference with protein translation in a RACK1-dependent manner and provide proof-of-concept evidence for targeting RACK1 as a therapeutic approach for TDP-43 or FUS proteinopathy associated with ALS and FTLD.
Materials and methods
Cell culture, plasmids, and transfection
Cell culture
Human embryonic kidney 293T (HEK293T) cell line was purchased from American Type Culture Collection (ATCC, Rockville, MD, USA), and maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), GlutaMax™ (2 mM) and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin) at 37 °C in 5% CO2.
Plasmids
HA-tagged WT TDP-43, TDP-43ΔNLS (K82A/R83A/K84A), WT FUS, R495x-FUS, and P525L-FUS FUS (collectively designated FUSΔNLS) were generated as previously described [51]. His-Myc-tagged R38D/K40E-RACK1 (DE-RACK1) was a kind gift from Marcello Ceci lab (Tuscia University, Italy). Flag-tagged human long variant (Lv) isoform of DISC1, 10W/S-DISC1, was constructed by GenScript Biotech (Piscataway, NJ, USA).
Transfection
cDNA plasmids were delivered into HEK293T cells using Lipofectamine LTX reagent (ThermoFisher Scientific, Waltham, MA, USA) following the manufacturer’s instruction, and cells were analyzed 48 h post-transfection. RACK1 KD was achieved by transfection of a pool of 3 target-specific siRNA plasmids against human RACK1 (Santa Cruz Biotechnology, Dallas, TX, USA, sc-36354) using Lipofectamine RNAiMAX reagent (ThermoFisher) according to the manufacturer’s instruction, and incubated for 72 h prior to cDNA plasmid transfection where indicated.
SUnSET
To monitor global translation, cells were treated with 5 µg/ml of puromycin (ThermoFisher) for 10 min at 37 °C in conditioned media, and lysed in 2% SDS for total protein extraction, electrophoresis, and western blotting quantification or fixed in 4% paraformaldehyde (PFA) for immunocytochemical and microscopic analysis.
Immunocytochemistry
HEK293T cells were washed twice with Phosphate Buffered Saline (PBS) and fixed in 4% PFA for 15 min at room temperature (RT), followed by wash with 20 mM glycine for 10 min at RT with constant rocking to quench residual PFA. Cells were then incubated with blocking buffer containing PBS, 1% Bovine Serum Albumin (BSA), 10% normal goat serum, and 0.1% Triton-X-100 for 30 min at RT. The following primary antibodies were incubated for 1 h at RT or overnight at 4 °C: rabbit polyclonal anti-HA (Abcam, Cambridge, UK, ab9110, 1:1,000), chicken polyclonal anti-HA (Abcam, ab9111, 1:10,000), rabbit polyclonal anti-c-Myc (Abcam, ab9106, 1:1,000), mouse monoclonal anti-RACK1 (BD Biosciences, San Jose, CA, USA, 610178, 1:500), rabbit monoclonal RACK1mis (ProMIS Neurosciences, see below, 1 μg/ml), rabbit monoclonal anti-RpS6 (Cell Signaling, Danvers, MA, USA, 5G10, 1:100), rabbit polyclonal anti-RPL14 (Bethyl Laboratories, Montgomery, TX, USA, A305-052, 1:1,000), mouse monoclonal anti-puromycin (ThermoFisher, clone 12D10, 1:1,000), rabbit monoclonal anti-Flag (Cell Signaling, 2368S, 1:2,000). Cells were then washed with PBS/0.1%Triton-X-100 three times for 10 min with constant rocking, followed by incubation with Alexa Fluor® goat anti-rabbit, -mouse, or -chicken secondary antibody (ThermoFisher, 1:1,000) for 30 min at RT in the dark. Cells were then washed with PBS/0.1%Triton-X-100 three times for 10 min, dipped in 5% PBS, and mounted with ProLong Gold Anti-fade Reagent with DAPI (ThermoFisher, P36931). Cells were analyzed by confocal microscopy (Leica TCS SP8 MP, Wetzlar, Germany).
The RACK1mis monoclonal antibody was generated by ProMIS Neurosciences using a proprietary computational modeling method, “Collective Coordinates” [48], to identify regions of RACK1 thermodynamically likely to be exposed on misfolded/unfolded RACK1 but not on the properly folded form of the protein. Rabbits were immunized with the predicted epitopes to generate monoclonal antibodies that were screened by ICC for recognition of RACK1 present in cytoplasmic aggregates of HEK293T cells transfected with TDP-43ΔNLS or R495x-FUS, and without detectable staining of diffuse, physiological RACK1 present in the cytoplasm. The staining properties of the RACK1mis antibody are shown in Additional file 1: Fig. S7.
Immunoblotting
Cells were washed twice with ice-cold PBS and lysed in 2% SDS, followed by sonication at 30% power for 15 s to extract total protein. Protein content was determined by BCA assay (ThermoFisher). 10 µg of protein was separated on 4–12% NuPAGE Bis–Tris SDS-PAGE (ThermoFisher), transferred onto a PVDF membrane, and blocked in Tris buffered saline (TBS) containing 5% skim milk and 0.1% Tween-20 for 1 h at RT. The following primary antibodies were incubated overnight at 4 °C: rabbit anti-HA (Abcam, ab9110, 1:1,000), mouse anti-RACK1 (BD Biosciences, 610178, 1:2,000), mouse anti-puromycin (ThermoFisher, clone 12D10, 1:10,000), mouse anti-α-tubulin (ProteinTech, Rosemont, IL, USA, 66031-1-Ig, 1:20,000), mouse anti-β-actin (Applied Biological Materials, Richmond, BC, Canada, G043, 1:1,000), rabbit anti-lamin B1 (abcam, ab16048, 1:1,000), mouse anti-TDP-43 (ProteinTech, 60019-2-Ig, 1:5,000), rabbit anti-phospho TDP-43 pS403/404 (Cosmo Bio, Carlsbad, CA, USA, TIP-PTD-P05, 1:3,000), mouse anti-GAPDH (ThermoFisher, AM4300, 1:100,000). Membranes were washed with TBS/0.1%Tween (TBST) three times for 10 min at RT with constant rocking, followed by horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Sigma, St. Louis, MI, USA, AP181P, 1:5,000) or donkey anti-rabbit secondary IgG secondary antibody (Sigma, AP182P, 1:5,000) incubation for 30 min at RT. Membranes were then washed with TBST three times for 10 min, and developed with SuperSignal™ West Femto Maximum Sensitivity Substrate (ThermoFisher).
Immunohistochemistry
For fluorescence-based IHC, paraffin-embedded spinal cord sections (6 μm) from healthy control, fALS-C9orf72, fALS-FUS-R521C, or sALS (kindly gifted by Drs. Ian Mackenzie and Janice Robertson) were de-paraffinized for 20 min at 55 °C, followed by rehydration in xylene, xylene/ethanol (1:1), 100%, 95%, 75%, 50% of ethanol, and TBS. The sections were then exposed to sub-boiling antigen retrieval buffer containing 20 mM sodium citrate pH 6.0 for 10 min, cooled at RT for 30 min, and incubated in blocking buffer containing TBS, 10% normal goat serum (NGS), 3% BSA, and 0.3% Triton-X-100 for 2 h, prior to incubation with primary antibodies diluted in background-reducing antibody diluent (Agilent, Santa Clara, CA, USA) overnight at 4 °C in a humidified chamber. The following antibodies were used: mouse anti-RACK1 (BD Biosciences, 610178, 1:100), rabbit anti-FUS (Sigma, HPA008784, 1:1,000), rabbit monoclonal RACK1mis (ProMIS Neurosciences, 1 μg/ml). Sections were then washed in TBS/0.3% Triton-X-100 (TBS-T) three times for 5 min with constant rocking, followed by incubation with Alexa Fluor® goat anti-rabbit, or -mouse IgG secondary antibody (ThermoFisher, 1:500) for 45 min at RT in the dark, and washed in TBS/0.3% Triton-X-100 3 X 5 min. Auto-fluorescence was then quenched by 0.1% Sudan Black B in 70% ethanol. The sections were finally mounted with ProLong Gold Anti-fade Reagent with DAPI (ThermoFisher), and analyzed by confocal microscopy (Leica TCS SP8 MP, Wetzlar, Germany).
For chromogenic IHC, fresh frozen cryo-sections (25 μm) of AD frontal cortex brains and sALS cervical spinal cords (kindly gifted by Dr. Ian Mackenzie) were fixed in 10% NBF for 5 min, washed in TBS three times for 10 min, followed by incubation with 0.3% H2O2 in methanol for 30 min at RT to quench endogenous peroxidase. Sections were washed in TBS-T, three times for 5 min, and incubated in blocking buffer for 2 h at RT. Endogenous biotin was then blocked by incubation with Avidin (Vector Laboratories, Newark, CA, USA) for 15 min at RT and two 5 min washes in TBS-T prior to incubation with primary antibodies diluted in background reducing antibody diluent (Agilent) overnight at 4 °C. The following antibodies were used: mouse anti-RACK1 (BD Biosciences, 1:100), rabbit RACKmis (ProMIS Neurosciences, 1 μg/ml). Sections were then washed in TBS-T three times for 10 min, followed by incubation with biotin F(ab)’2 fragment goat anti-mouse or -rabbit IgG secondary antibody (ThermoFisher) for 1 h at RT, washed in TBS-T, rinsed briefly in TBS, prior to amplification in Avidin–Biotin Complex (Vector) for 45 min at RT. VIP HRP substrate (Vector) was then applied to the sections for 2 min, followed by nuclear counterstaining in Methyl Green (Vector) for 2 min. Sections were cleared by dehydration in ethanol and incubation in Citrasolv (Decon Labs, PA, USA) twice for 10 min. Finally, sections were mounted onto microscopic slides in Permount mounting medium (ThermoFisher).
Nucleocytoplasmic fractionation
Cells were washed twice in ice-cold PBS, followed by centrifugation at 3000 RPM for 5 min at 4 °C. Cell pellets were resuspended in cytoplasm extraction buffer containing 10 mM HEPES (pH 7.9), 10 mM KCl, 1 mM EDTA (pH 8), 1 mM EGTA (pH 8), 0.1% NP-40, 1 mM PMSF, and protease inhibitor cocktail (ThermoFisher) and homogenized 10 times using a 25-gauge needle, followed by rotation for 15 min at 4 °C and centrifugation at 3000 RPM for 5 min at 4 °C. Supernatant was collected as the cytosolic fraction. Pellets were washed three times in cytoplasm extraction buffer, then resuspended in nuclear extraction buffer containing 20 mM HEPES (pH 7.9), 420 mM NaCl, 1 mM EDTA (pH 8), 1 mM EGTA (pH 8), 0.5% NP-40, 0.1% SDS, 10% glycerol, 1 mM PMSF, and protease inhibitor cocktail (ThermoFisher), and homogenized 10 times using a 25-gauge needle. The mixture was sonicated for 3 s at 20% power, followed by centrifugation at 14,000 RPM for 30 min at 4 °C. Supernatant was collected as the nuclear fraction.
Intercellular transmission
Intercellular transmission assay was performed following a previously established procedure and is illustrated in Fig. 8a. Briefly, donor cells were transfected with RACK1 siRNA or left un-transfected as control for 72 h, followed by TDP-43ΔNLS cDNA plasmid transfection for an additional 48 h. Donor cells were then collected and subjected to western blot analysis for confirmation of RACK1 siRNA activity and TDP-43ΔNLS expression. The conditioned medium from donor cells was centrifuged at 1000 RPM for 5 min at RT to remove cell debris. Supernatant was then added to naïve recipient cells and incubated for 72 h, followed by western blot analysis for quantitation of transmitted TDP-43ΔNLS.
Drosophila melanogaster in vivo studies
Source of D. melanogaster
The following publicly available D. melanogaster lines were obtained from Bloomington Drosophila Stock Center (BDSC, Bloomington, IN, USA): GMR-Gal4 (BDSC #9146), D42-Gal4 (BDSC #7009), UAS-hTDP-43WT (BDSC #79587) [17], UAS-hTDP-43Q331K (BDSC #79590) [17], UAS-RACK1-RNAi TRiP.HMS01171 (BDSC #34692), UAS-mCherry-RNAi (BDSC #35785).
External eye imaging
For eye analysis, flies were maintained at 25 °C. Photographs of fly external eyes were obtained using a Zeiss Stemi SV 11 dissection microscope at 6× magnification equipped with an Olympus OM-D E-M1 digital camera controlled by Olympus Image Share app. Dead ommatidia were counted visually using a dissecting microscope, with the experimenter blinded to condition. 21–33 flies were scored per condition. Each data point is the mean of left and right eye for one individual.
Preparation of fly eyes for histology
Flies were anaesthetized with CO2 and decapitated. Heads were fixed in 4% PFA 0.2% Triton-x in PBS for 2 h at RT, and then incubated in 30% sucrose in PBS for 2 h at 4 °C for cryopreservation. Heads were embedded in OCT (Tissue-Tek) in rubber molds and frozen at − 80 °C. Tissue was cryo-sectioned at − 20 C into 10 µm slices, mounted onto slides, and cover-slipped using Vectashield (Vector Laboratories, Newark, CA, USA). No counter-stain was needed as retinal pigments are highly autofluorescent between approximately 470–600 nm excitation.
Imaging and analysis of retina
Retinal sections were viewed on an Olympus FLUOVIEW FV1000 Confocal Laser Scanning Biological Microscope with Fluoview software. Single plane images were captured using 488 nm excitation with a 20 × objective. Image files were code-named in order to blind the experimenter to genotype.
Climbing assay
The locomotion of flies was performed as previously described [59]. Cohorts of 15 adult flies, which had been allowed to mate for 1–2 days after eclosion, were separated by sex and housed in their testing cohorts under controlled conditions of 23 °C, 50–60% humidity, and 12-h on/off light cycle. Each week, 6–9 h into the 12-h light cycle of the flies (coinciding with their most stable activity levels), the climbing assays were conducted. Lighting and environmental conditions were carefully controlled in the testing area. Flies were anaesthetized briefly with CO2 and placed in a 100 ml measuring cylinder. They were given 15 min to recover from anesthesia and to acclimatize to the cylinder. The measuring cylinder was tapped 3 times upon a mouse pad to send the flies to the bottom, and a still photograph was taken 10 s later using an iPhone. Flies were then placed onto fresh food until the following week. From the photographs, the location of each fly in the graduated cylinder and distance climbed was noted. > 60 flies per genotype were measured, and the results analyzed by two-way ANOVA.
Statistics
Statistical analysis was performed using GraphPad Prism (GraphPad Software, San Diego CA, USA). ANOVA, Mann–Whitney, or Student’s t-test was performed where appropriate as indicated to determine the significant differences. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001.
Availability of data and materials
The datasets generated in the current study are available from the corresponding author on reasonable request.
References
Adams DR, Ron D, Kiely PA (2011) RACK1, A multifaceted scaffolding protein: structure and function. Cell Commun Signal 9:22. https://doi.org/10.1186/1478-811X-9-22
Afroz T, Hock EM, Ernst P, Foglieni C, Jambeau M, Gilhespy LAB, Laferriere F, Maniecka Z, Pluckthun A, Mittl P et al (2017) Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat Commun 8:45. https://doi.org/10.1038/s41467-017-00062-0
Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, Niizato K, Tsuchiya K, Kobayashi Z, Ikeda K et al (2010) Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology 30:170–181. https://doi.org/10.1111/j.1440-1789.2009.01089.x
Benarroch EE (2019) Nucleocytoplasmic transport: Mechanisms and involvement in neurodegenerative disease. Neurology 92:757–764. https://doi.org/10.1212/WNL.0000000000007305
Birsa N, Bentham MP, Fratta P (2020) Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin Cell Dev Biol 99:193–201. https://doi.org/10.1016/j.semcdb.2019.05.023
Cascella R, Bigi A, Riffert DG, Gagliani MC, Ermini E, Moretti M, Cortese K, Cecchi C, Chiti F (2022) A quantitative biology approach correlates neuronal toxicity with the largest inclusions of TDP-43. Sci Adv 8:eabm6376. https://doi.org/10.1126/sciadv.abm6376
Ceci M, Gaviraghi C, Gorrini C, Sala LA, Offenhauser N, Marchisio PC, Biffo S (2003) Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature 426:579–584. https://doi.org/10.1038/nature02160
Chang BY, Conroy KB, Machleder EM, Cartwright CA (1998) RACK1, a receptor for activated C kinase and a homolog of the beta subunit of G proteins, inhibits activity of src tyrosine kinases and growth of NIH 3T3 cells. Mol Cell Biol 18:3245–3256. https://doi.org/10.1128/mcb.18.6.3245
Chen HJ, Mitchell JC (2021) Mechanisms of TDP-43 proteinopathy onset and propagation. Int J Mol Sci. https://doi.org/10.3390/ijms22116004
Cheng ZF, Pai RK, Cartwright CA (2018) Rack1 function in intestinal epithelia: regulating crypt cell proliferation and regeneration and promoting differentiation and apoptosis. Am J Physiol Gastrointest Liver Physiol 314:G1–G13. https://doi.org/10.1152/ajpgi.00240.2017
Chhangani D, Martin-Pena A, Rincon-Limas DE (2021) Molecular, functional, and pathological aspects of TDP-43 fragmentation. iScience 24:102459. https://doi.org/10.1016/j.isci.2021.102459
Couratier P, Corcia P, Lautrette G, Nicol M, Marin B (2017) ALS and frontotemporal dementia belong to a common disease spectrum. Rev Neurol (Paris) 173:273–279. https://doi.org/10.1016/j.neurol.2017.04.001
Coyle SM, Gilbert WV, Doudna JA (2009) Direct link between RACK1 function and localization at the ribosome in vivo. Mol Cell Biol 29:1626–1634. https://doi.org/10.1128/MCB.01718-08
Coyne AN, Siddegowda BB, Estes PS, Johannesmeyer J, Kovalik T, Daniel SG, Pearson A, Bowser R, Zarnescu DC (2014) Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci 34:15962–15974. https://doi.org/10.1523/JNEUROSCI.2526-14.2014
Eck RJ, Kraemer BC, Liachko NF (2021) Regulation of TDP-43 phosphorylation in aging and disease. Geroscience 43:1605–1614. https://doi.org/10.1007/s11357-021-00383-5
Ederle H, Dormann D (2017) TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett 591:1489–1507. https://doi.org/10.1002/1873-3468.12646
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM et al (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466:1069–1075. https://doi.org/10.1038/nature09320
Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, Li D, Thal DR, Walther P, Ludolph AC et al (2015) TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol 211:897–911. https://doi.org/10.1083/jcb.201504057
Francois-Moutal L, Perez-Miller S, Scott DD, Miranda VG, Mollasalehi N, Khanna M (2019) Structural insights Into TDP-43 and effects of post-translational modifications. Front Mol Neurosci 12:301. https://doi.org/10.3389/fnmol.2019.00301
Gallo S, Ricciardi S, Manfrini N, Pesce E, Oliveto S, Calamita P, Mancino M, Maffioli E, Moro M, Crosti M et al (2018) RACK1 specifically regulates translation through its binding to ribosomes. Mol Cell Biol. https://doi.org/10.1128/MCB.00230-18
Gandin V, Senft D, Topisirovic I, Ronai ZA (2013) RACK1 function in cell motility and protein synthesis. Genes Cancer 4:369–377. https://doi.org/10.1177/1947601913486348
Gao J, Wang L, Huntley ML, Perry G, Wang X (2018) Pathomechanisms of TDP-43 in neurodegeneration. J Neurochem. https://doi.org/10.1111/jnc.14327
Grad LI, Pokrishevsky E, Cashman NR (2017) Intercellular prion-like conversion and transmission of Cu/Zn superoxide dismutase (SOD1) in cell culture. Methods Mol Biol 1658:357–367. https://doi.org/10.1007/978-1-4939-7244-9_24
Hanspal MA, Dobson CM, Yerbury JJ, Kumita JR (2017) The relevance of contact-independent cell-to-cell transfer of TDP-43 and SOD1 in amyotrophic lateral sclerosis. Biochim Biophys Acta Mol Basis Dis 1863:2762–2771. https://doi.org/10.1016/j.bbadis.2017.07.007
Hermanto U, Zong CS, Li W, Wang LH (2002) RACK1, an insulin-like growth factor I (IGF-I) receptor-interacting protein, modulates IGF-I-dependent integrin signaling and promotes cell spreading and contact with extracellular matrix. Mol Cell Biol 22:2345–2365. https://doi.org/10.1128/mcb.22.7.2345-2365.2002
Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T (2011) RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol 18:1428–1431. https://doi.org/10.1038/nsmb.2163
Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K (2012) Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep 2:529. https://doi.org/10.1038/srep00529
Ishii T, Kawakami E, Endo K, Misawa H, Watabe K (2017) Formation and spreading of TDP-43 aggregates in cultured neuronal and glial cells demonstrated by time-lapse imaging. PLoS ONE 12:e0179375. https://doi.org/10.1371/journal.pone.0179375
Ito D, Hatano M, Suzuki N (2017) RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aah5436
Jo M, Lee S, Jeon YM, Kim S, Kwon Y, Kim HJ (2020) The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp Mol Med 52:1652–1662. https://doi.org/10.1038/s12276-020-00513-7
Kadrmas JL, Smith MA, Pronovost SM, Beckerle MC (2007) Characterization of RACK1 function in Drosophila development. Dev Dyn 236:2207–2215. https://doi.org/10.1002/dvdy.21217
Kamelgarn M, Chen J, Kuang L, Jin H, Kasarskis EJ, Zhu H (2018) ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc Natl Acad Sci U S A 115:E11904–E11913. https://doi.org/10.1073/pnas.1810413115
Kiely PA, Sant A, O’Connor R (2002) RACK1 is an insulin-like growth factor 1 (IGF-1) receptor-interacting protein that can regulate IGF-1-mediated Akt activation and protection from cell death. J Biol Chem 277:22581–22589. https://doi.org/10.1074/jbc.M201758200
Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM (2014) Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet 46:152–160. https://doi.org/10.1038/ng.2853
Lanson NA Jr, Maltare A, King H, Smith R, Kim JH, Taylor JP, Lloyd TE, Pandey UB (2011) A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum Mol Genet 20:2510–2523. https://doi.org/10.1093/hmg/ddr150
Lee M, Ghosh U, Thurber KR, Kato M, Tycko R (2020) Molecular structure and interactions within amyloid-like fibrils formed by a low-complexity protein sequence from FUS. Nat Commun 11:5735. https://doi.org/10.1038/s41467-020-19512-3
Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, Woodruff EA 3rd, Fushimi K, Wu JY (2010) A Drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci U S A 107:3169–3174. https://doi.org/10.1073/pnas.0913602107
Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9:995–1007. https://doi.org/10.1016/S1474-4422(10)70195-2
Majumder P, Chu JF, Chatterjee B, Swamy KB, Shen CJ (2016) Co-regulation of mRNA translation by TDP-43 and Fragile X Syndrome protein FMRP. Acta Neuropathol 132:721–738. https://doi.org/10.1007/s00401-016-1603-8
Majzoub K, Hafirassou ML, Meignin C, Goto A, Marzi S, Fedorova A, Verdier Y, Vinh J, Hoffmann JA, Martin F et al (2014) RACK1 controls IRES-mediated translation of viruses. Cell 159:1086–1095. https://doi.org/10.1016/j.cell.2014.10.041
McCahill A, Warwicker J, Bolger GB, Houslay MD, Yarwood SJ (2002) The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Mol Pharmacol 62:1261–1273. https://doi.org/10.1124/mol.62.6.1261
Mochly-Rosen D, Smith BL, Chen CH, Disatnik MH, Ron D (1995) Interaction of protein kinase C with RACK1, a receptor for activated C-kinase: a role in beta protein kinase C mediated signal transduction. Biochem Soc Trans 23:596–600. https://doi.org/10.1042/bst0230596
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931. https://doi.org/10.1093/brain/awp214
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. https://doi.org/10.1126/science.1134108
Nilsson J, Sengupta J, Frank J, Nissen P (2004) Regulation of eukaryotic translation by the RACK1 protein: a platform for signalling molecules on the ribosome. EMBO Rep 5:1137–1141. https://doi.org/10.1038/sj.embor.7400291
Nolan M, Talbot K, Ansorge O (2016) Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun 4:99. https://doi.org/10.1186/s40478-016-0358-8
Olney RK, Murphy J, Forshew D, Garwood E, Miller BL, Langmore S, Kohn MA, Lomen-Hoerth C (2005) The effects of executive and behavioral dysfunction on the course of ALS. Neurology 65:1774–1777. https://doi.org/10.1212/01.wnl.0000188759.87240.8b
Peng X, Cashman NR, Plotkin SS (2018) Prediction of misfolding-specific epitopes in SOD1 using collective coordinates. J Phys Chem B 122:11662–11676. https://doi.org/10.1021/acs.jpcb.8b07680
Phukan J, Pender NP, Hardiman O (2007) Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol 6:994–1003. https://doi.org/10.1016/S1474-4422(07)70265-X
Pokrishevsky E, Grad LI, Cashman NR (2016) TDP-43 or FUS-induced misfolded human wild-type SOD1 can propagate intercellularly in a prion-like fashion. Sci Rep 6:22155. https://doi.org/10.1038/srep22155
Pokrishevsky E, Grad LI, Yousefi M, Wang J, Mackenzie IR, Cashman NR (2012) Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS ONE 7:e35050. https://doi.org/10.1371/journal.pone.0035050
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14:459–468. https://doi.org/10.1038/nn.2779
Porta S, Xu Y, Lehr T, Zhang B, Meymand E, Olufemi M, Stieber A, Lee EB, Trojanowski JQ, Lee VM (2021) Distinct brain-derived TDP-43 strains from FTLD-TDP subtypes induce diverse morphological TDP-43 aggregates and spreading patterns in vitro and in vivo. Neuropathol Appl Neurobiol 47:1033–1049. https://doi.org/10.1111/nan.12732
Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ, Lee EB, Trojanowski JQ, Lee VM (2018) Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat Commun 9:4220. https://doi.org/10.1038/s41467-018-06548-9
Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK (2019) Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci 12:25. https://doi.org/10.3389/fnmol.2019.00025
Rodriguez de Rivera FJ, Rambold HA, Diez-Tejedor E (2014) Assessment of cognitive impairment in amyotrophic lateral sclerosis. J Neurol Sci 337:1–2. https://doi.org/10.1016/j.jns.2013.11.030
Russo A, Scardigli R, La Regina F, Murray ME, Romano N, Dickson DW, Wolozin B, Cattaneo A, Ceci M (2017) Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum Mol Genet 26:1407–1418. https://doi.org/10.1093/hmg/ddx035
Schmidt EK, Clavarino G, Ceppi M, Pierre P (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6:275–277. https://doi.org/10.1038/nmeth.1314
Sealey MA, Vourkou E, Cowan CM, Bossing T, Quraishe S, Grammenoudi S, Skoulakis EMC, Mudher A (2017) Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol Dis 105:74–83. https://doi.org/10.1016/j.nbd.2017.05.003
Sengupta J, Nilsson J, Gursky R, Spahn CM, Nissen P, Frank J (2004) Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EM. Nat Struct Mol Biol 11:957–962. https://doi.org/10.1038/nsmb822
Speth C, Willing EM, Rausch S, Schneeberger K, Laubinger S (2013) RACK1 scaffold proteins influence miRNA abundance in Arabidopsis. Plant J 76:433–445. https://doi.org/10.1111/tpj.12308
Steyaert J, Scheveneels W, Vanneste J, Van Damme P, Robberecht W, Callaerts P, Bogaert E, Van Den Bosch L (2018) FUS-induced neurotoxicity in Drosophila is prevented by downregulating nucleocytoplasmic transport proteins. Hum Mol Genet 27:4103–4116. https://doi.org/10.1093/hmg/ddy303
Suk TR, Rousseaux MWC (2020) The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener 15:45. https://doi.org/10.1186/s13024-020-00397-1
Timney BL, Raveh B, Mironska R, Trivedi JM, Kim SJ, Russel D, Wente SR, Sali A, Rout MP (2016) Simple rules for passive diffusion through the nuclear pore complex. J Cell Biol 215:57–76. https://doi.org/10.1083/jcb.201601004
Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V et al (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14:452–458. https://doi.org/10.1038/nn.2778
Vomastek T, Iwanicki MP, Schaeffer HJ, Tarcsafalvi A, Parsons JT, Weber MJ (2007) RACK1 targets the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway to link integrin engagement with focal adhesion disassembly and cell motility. Mol Cell Biol 27:8296–8305. https://doi.org/10.1128/MCB.00598-07
Walker AK, Tripathy K, Restrepo CR, Ge G, Xu Y, Kwong LK, Trojanowski JQ, Lee VM (2015) An insoluble frontotemporal lobar degeneration-associated TDP-43 C-terminal fragment causes neurodegeneration and hippocampus pathology in transgenic mice. Hum Mol Genet 24:7241–7254. https://doi.org/10.1093/hmg/ddv424
Wang L, Yang W, Li B, Yuan S, Wang F (2021) Response to stress in biological disorders: Implications of stress granule assembly and function. Cell Prolif 54:e13086. https://doi.org/10.1111/cpr.13086
Wang R, Brattain MG (2007) The maximal size of protein to diffuse through the nuclear pore is larger than 60kDa. FEBS Lett 581:3164–3170. https://doi.org/10.1016/j.febslet.2007.05.082
Wang S, Chen JZ, Zhang Z, Gu S, Ji C, Tang R, Ying K, Xie Y, Mao Y (2003) Cloning, expression and genomic structure of a novel human GNB2L1 gene, which encodes a receptor of activated protein kinase C (RACK). Mol Biol Rep 30:53–60. https://doi.org/10.1023/a:1022212731316
Acknowledgements
The authors would like to thank Dr. Janice Robertson (University of Toronto, Canada) for providing ALS spinal cord tissue samples, Dr. Marcello Ceci (Tuscia University, Italy) for gifting the His-Myc-tagged DE-RACK1 plasmid, Dr. Ethan Greenblatt (University of British Columbia, Canada) for reviewing the manuscript, and Ali Osouli for sectioning fly retinas.
Funding
This work was sponsored by Canadian Institute of Health Research (PJT-159546 and CIHR-CCNA grant 20R04367), Brain Canada (20R74974), ALS Canada/Brain Canada 2020 Discovery Grant Program (F20-04056), William A. Lambert donation (F17-00928), and ProMIS Neurosciences (SRA F16-05805).
Author information
Authors and Affiliations
Contributions
BZ, CMC, JMK, and NRC designed the experiments. BZ, CMC, JAC, DDC, and AS performed the experiments and analyzed the results. SSP and SCCH performed the computer modeling and epitope prediction. The manuscript was drafted by BZ and edited by JMK, IRM, and NRC. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All of the human post-mortem materials used in this study were obtained from subjects who had been evaluated at the Sunnybrook Health Sciences Centre ALS Clinic and the University of British Columbia Clinic for Alzheimer’s Disease and Related Disorders, and consented for research purposes in accordance with respective ethical review boards.
Consent for publication
Not applicable.
Competing interests
N.R.C., J.M.K., and B.Z. are current employees of ProMIS Neurosciences. S.S.P. and S.C.C.H. have received consultation compensation from ProMIS Neurosciences. S.S.P. and B.Z. possess ProMIS stock options. N.R.C. and J.M.K. possess ProMIS shares and stock options. S.S.P. is the inventor on patent applications for Collective Coordinates computational modeling. N.R.C., S.S.P., J.M.K., and B.Z. are inventors on patent applications relating to conformation-specific epitopes in RACK1, antibodies thereto and methods related thereof.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Fig. S1: RACK1 and TDP-43 co-aggregate in ALS spinal cord motor neurons. Fig. S2: Endogenous TDP-43 and FUS expressions are not affected by mutant RACK1. Fig. S3: RACK1 does not co-aggregate with mutant DISC1. Fig. S4: RACK1 is misfolded in ALS spinal cord and FTLD-TDP brain tissues. Fig. S5: RACK1 knockdown reduces cytoplasmic aggregation of P525L-FUS in HEK293T cells. Fig. S6: Endogenous TDP-43 and FUS expressions are not affected by RACK1 knockdown. Fig. S7: Validation of the RACK1mis antibody.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, B., Cowan, C.M., Coutts, J.A. et al. Targeting RACK1 to alleviate TDP-43 and FUS proteinopathy-mediated suppression of protein translation and neurodegeneration. acta neuropathol commun 11, 200 (2023). https://doi.org/10.1186/s40478-023-01705-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-023-01705-8