
Abstract
In the last decades, numerous post-mortem case series have documented chronic traumatic encephalopathy (CTE) in former contact-sport athletes, though reports of CTE pathology in former soccer players are scarce. This study presents a clinicopathological case of a former professional soccer player with young-onset dementia. The patient experienced early onset progressive cognitive decline and developed dementia in his mid-50 s, after playing soccer for 12 years at a professional level. While the clinical picture mimicked Alzheimer’s disease, amyloid PET imaging did not provide evidence of elevated beta-amyloid plaque density. After he died in his mid-60 s, brain autopsy showed severe phosphorylated tau (p-tau) abnormalities fulfilling the neuropathological criteria for high-stage CTE, as well as astrocytic and oligodendroglial tau pathology in terms of tufted astrocytes, thorn-shaped astrocytes, and coiled bodies. Additionally, there were TAR DNA-binding protein 43 (TDP-43) positive cytoplasmic inclusions in the frontal lobe and hippocampus, and Amyloid Precursor Protein (APP) positivity in the axons of the white matter. A systematic review of the literature revealed only 13 other soccer players with postmortem diagnosis of CTE. Our report illustrates the complex clinicopathological correlation of CTE and the need for disease-specific biomarkers.
Similar content being viewed by others


Introduction
With more than 250 million professional and recreational participants, soccer is the most popular sport worldwide. [1] As soccer involves multiple sources of head impact, such as collisions and heading, there are growing concerns regarding the long-term brain health of soccer players. In 2019, Mackay et al. reported a 3.5 times higher mortality risk of neurodegenerative diseases among former professional Scottish soccer players. [2] These results were later corroborated by findings from Russell et al. (2021), that further connected the risk for neurodegenerative diseases to outfield player position and career duration. [3] This relationship might be linked to soccer-related symptomatic concussive, and the more common asymptomatic ‘subconcussive’ head impacts and these results were consistent with mortality studies among athletes performing other contact sports. [4,5,6,7]
The link between repetitive head impacts (RHI) in sports and late-life progressive cognitive, neuropsychiatric, and motor impairments has been recognized for decades. [8] Post-mortem neuropathological evaluation has revealed unique neurodegenerative changes, known as chronic traumatic encephalopathy (CTE), in hundreds of former contact sport athletes. The unique pathological features of CTE are depositions of phosphorylated tau (p-tau) deep in the cortical sulci around small blood vessels and have been detected most frequently in the brains of American football players and boxers. [9,10,11,12,13] CTE in former soccer players, however, has been reported to a lesser extent and, as such, the burden of CTE in this population remains unknown. [14, 15] Given the worldwide popularity of soccer, and the fact that head impacts in soccer are different and unique compared to other contact or collision sports, it is important to investigate the occurrence of CTE and associated pathology in the brains of former soccer players.
Well-documented clinical and neuropathological information about soccer players with CTE is scarce yet important for a better understanding of this neurodegenerative disease. In the current study, we present a case report of a former professional Dutch soccer player with young-onset dementia and pathology-confirmed CTE, supported by comprehensive longitudinal clinical, neuroimaging, and fluid biomarker data. We also provide a literature overview of current evidence of CTE among soccer players.
Case presentation
Clinical description
This male patient played soccer for 24 years, including 12 years at a professional level in the top league of the Netherlands (the 1980s), and retired at the age of 32. As a forward, he was a skilled header, and he was praised for his header goals. He also mentioned having experienced multiple collisions that involved head impact playing soccer, at least once leading to loss of consciousness. He was referred to the memory clinic of the Alzheimer Center Amsterdam at the age of 54. The referral center considered that the patient suffered from young-onset Alzheimer’s disease (AD). According to his family, the problems started around the age of 50 with progressive short-term memory complaints, leading to forgetfulness, and difficulties in household activities and financial administration. The patient was also increasingly disorientated to place. Apathy was reported, but there were no other psychiatric features, such as explosivity, impulsive behavior, emotional lability, or mood symptoms. Subsequent to his soccer career, there was no known history of traumatic brain injury. The patient had no history of any medical disease, drug abuse, or alcoholism and did not use any medication. He smoked more than 30 pack years. He reported that his father experienced mild, non-progressive memory complaints at the age of 60, but he did not have functional limitations and he never received a formal diagnosis. Further family history was negative for neurological disorders. Physical examination revealed no abnormalities. Neuropsychological assessment at baseline showed severe memory deficits, decreased processing speed, and deficits in executive functioning. The Mini-Mental State Examination (MMSE) was 23 out of 30 and the Clinical Dementia Rating (CDR) was 1, indicative of mild dementia. Neuropsychiatric symptoms were assessed via the Neuropsychiatry Inventory Questionnaire (NPI-Q) [16], revealing the presence of apathy and loss of appetite (NPI-Q total score: 12). A T1 brain MRI scan showed moderate to severe atrophy, with a bilateral medial temporal lobe atrophy score of 3, a global cortical atrophy score of 1–2 and a bilateral parietal atrophy score of 2 [17,18,19]. The fluid-attenuated inversion recovery (FLAIR) showed some punctate foci of white matter hyperintensities (WMH) consistent with Fazekas grade 1. [20] In addition, a cavum septum pellucidum (CSP) was visible (Fig. 1). Based on the clinical presentation, neuropsychological examination, and MRI scan, the patient was diagnosed with probable AD dementia, according to the 2011 National Institute on Aging—Alzheimer’s Association diagnostic criteria. [21] To support this diagnosis, a lumbar puncture was performed to measure AD-specific biomarkers in cerebrospinal fluid (CSF). CSF revealed normal levels of Aβ 1–42, total tau and p-tau-181; Aβ 1–42: 979 pg/ml (ref > 550 pg/ml), total tau: 324 pg/ml (ref < 375 pg/ml), p-tau-181: 40 pg/ml (ref < 52 pg/ml), measured with Innotest© assays. [22] Additionally, a negative 18F-Flutametamol positron emission tomography (PET) did not support a clinical diagnosis of Alzheimer’s disease. The patient’s APOE genotype was APOE E2/E3 and the microtubule-associated protein tau (MAPT) haplotype was H1/H1. His DNA was sequenced for the most common autosomal dominant causes of dementia, including pathogenic MAPT gene mutations and repeat expansions of chromosome 9 open reading frame 72 (C9orf72); the results were negative. Because of the unlikelihood of Alzheimer’s pathology as the underlying cause, and because no other disorders or conditions could fully account for the patient’s clinical presentation, a diagnosis of dementia due to CTE was suggested and the patient was followed clinically. In retrospect, using the 2021 National Institute of Neurological Disorders and Stroke (NINDS) Consensus Diagnostic Criteria for Traumatic Encephalopathy Syndrome (TES; meant to represent the clinical manifestation of CTE pathology, to be used in research settings), the patient would meet the criteria for TES with probable CTE level of certainty, because of the extensive exposure to RHI, the progressive cognitive impairment (stage mild dementia), the delayed onset, and the presence of a moderate level of apathy. [23]

Brain MRI. Images were acquired at baseline and after two years. The coronal T1 images on the left show global atrophy (global cortical atrophy score 1–2), and severe bilateral hippocampal atrophy (medial temporal atrophy score 3, red triangle). The axial T2 FLAIR images in the middle and on the right display the white matter hyperintensities. The red arrows indicate the cavum septum pellucidum (CSP)
After his initial visit, there was a slow deterioration over the years. A follow-up MRI scan after two years showed similar atrophy scores but increased ventricular dilatation (Fig. 1). Follow-up neuropsychological examination after two, three, and four years since the diagnosis of dementia demonstrated a further decline in memory, executive functions, and visuospatial abilities, whereas his processing speed and attention remained relatively stable. The MMSE decreased by 5 points over a four-year period (latest score: 17/30). According to the NPI-Q, more neuropsychiatric symptoms were reported during the course of the disease. The NPI-Q scores were 8 and 13 respectively two and three years after baseline, including low scores in a variety of symptoms: apathy, delusions, disinhibition, irritability, sleep disturbances, loss of appetite, and aberrant motor behavior. He had to be admitted to a nursing home, experiencing further disease progression. There was full dependency in basic activities of daily living (CDR 3). He died due to a COVID-19 infection at the age of 63.
Additional fluid biomarkers
At the time of diagnostic examination as described above, venous blood was collected, and serum aliquots were stored at the biobank of Amsterdam Dementia Cohort. [24] The patient and family gave consent to use this body material for future research purposes. Nine years later, p-tau-181, glial fibrillary acidic protein (GFAP), and neurofilament light (NfL) were analyzed in serum with commercially available Simoa® Assay Kits from Quanterix (analysis performed in 2022). Serum assays resulted in a low p-tau-181 concentration of 0.33 pg/ml (below the lower limit of quantification). The NfL concentration was 12.81 pg/ml and the GFAP concentration was 52.93 pg/ml. Both NfL and GFAP levels were compared to age-dependent reference ranges established by the Neurochemistry laboratory of the Amsterdam UMC. The concentration of NfL was found between the 75th and 90.th percentile for the control reference group, between the 10th and 25th percentile for frontotemporal dementia, and between the 25th and 50th percentile for AD. [25, 26] The level of GFAP was within the 25th and 50th percentile for the control group and below the 5th percentile for AD. [27]
Post-mortem investigation
Before the patient’s death, relatives gave written informed consent for a post-mortem examination and pathological assessment as part of the NEurodegeneration: Traumatic brain injury as Origin of the Neuropathology (NEwTON) brain bank cohort. [28] Autopsy was performed within 8 h after death. Tissue was fixed in 4% formaldehyde for four weeks until macroscopic evaluation and dissection of multiple regions. Tissue blocks were embedded in paraffin and 5 μm sections were prepared for staining with Hematoxylin and Eosin (H&E), p-tau (AT8), anti-Aβ/amyloid precursor protein (APP), phosphorylated TAR DNA-binding protein (pTDP-43), alpha-synuclein, 3-repeat (3R) and 4-repeat (4R) tau (RD3, RD4), and p62. Additional staining for microglial activation (iba1 and CD68) was performed with tissue from the frontal cortex. Detailed methods of immunostaining are included in the "Additional file 1". The neuropathological evaluation was performed by an experienced neuropathologist and a pathological diagnosis was made according to international consensus guidelines [29, 30]. The preliminary NINDS/National Institute of Biomedical Imaging and Bioengineering (NIBIB) diagnostic neuropathological criteria for CTE were applied, including the recently revised staging method. [9, 12] The extent of immunoreactivity for each protein was visually assessed by two authors (SA, AJJR) and scored as none (-), minimal ( +), moderate (+ +), and extensive (+++).
Postmortem findings
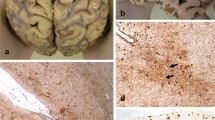
The fresh brain had a slightly low weight of 1220 g (excluding CSF). [31] Macroscopic examination of the whole brain (Fig. 2) showed slight external atrophy and severely widened ventricles upon dissection, especially in the frontal and temporal regions. There was atrophy of the caudate nuclei, the substantia nigra showed loss of pigment and the hippocampi appeared small. Septum abnormalities were noted as CSP and a fenestrated septum. There was only very mild atherosclerosis in the carotid arteries, without any further abnormalities in the large vessels. No macroscopic infarcts were detected. H&E staining demonstrated disturbed architecture of the second cortical layer, severe gliosis, depigmentation of the locus coeruleus, and loss of neurons in multiple cortical areas, basal ganglia, hippocampus, and dentate nucleus. The pattern of neuronal loss in the hippocampus was recognized as hippocampal sclerosis; a pathological condition that is associated with temporal lobe epilepsy but that often coexists with neurodegenerative diseases. [32] One small microscopic infarct was found in the occipital lobe, but no other evidence of cerebrovascular disease or arteriolosclerosis. P-tau staining showed widely distributed, moderate-to-extensive abnormal tau pathology throughout multiple brain regions. P-tau inclusions, including neurofibrillary tangles (NFTs), neuropil threads, thorn-shaped astrocytes, tufted astrocytes, and coiled bodies, were found in cortical and subcortical areas, as well as the cerebellum, brain stem, and cervical spine. Especially in the frontal and parietal cortex, neuronal tau pathology was found with predilection of sulcal depths and perivascular regions, according to the pathological criteria of CTE. (Fig. 3) [9] Thorn-shaped astrocytes were frequently found, located subpial, subependymal, and perivascular (Fig. 4A), as opposed to the granular/fuzzy astrocytes that were rarely present. In the hippocampus, tau positive neurons and glial cells were detected throughout all four areas (CA1-CA4). CA3 and CA4 seems to be most severely affected, but the severe neuronal loss in CA1 and CA2 complicates this assessment. Also notable were the p-tau-positive Purkinje cells in the cerebellum. According to the second NINDS/NIBIB research criteria for the diagnosis of CTE, we staged this case as high CTE, based on the additional presence of neuronal p-tau in more than 5 regions of interest (at least NFTs in gyral side and crest adjacent to CTE lesion, NFTs in CA2 and CA4 of the hippocampus, amygdala, thalamus, and cerebellar dentate nucleus). We are aware, however, of the complexity of staging in the context of mixed pathologies. [9] RD3 and RD4 staining was performed in the temporal cortex, thalamus, basal ganglia, and cerebellum, which showed both 3R and 4R tau positive neurons and coiled bodies. The glial cells in these areas were all 4R positive. There was also a moderate amount of pTDP-43 positivity in all layers of the frontal cortex, and the hippocampus including the parahippocampal gyrus with predilection of the granular layer (Fig. 5A-B). There was minimal positivity in the amygdala, the basal ganglia, and medulla. All pTDP-43 positive structures were recognized as cytoplasmic inclusions and threads, but not as neuronal intranuclear inclusions. Only a few Aβ diffuse plaques were observed in the frontal cortex but none in the hippocampus and no neuritic plaques were observed (Fig. 5C). Axonal APP positivity was found in the white matter of various regions (frontal, cerebellum, basal ganglia) (Fig. 5D-E). In addition to the AT8 positivity in the depth of the frontal cortical sulci, there was also a high density of pTDP-43, Iba1, and CD68 positive cells found in these areas, as depicted in Fig. 6. No structures immunoreactive for p62 were observed in the granular layer of the cerebellum, which makes an underlying C9orf72 hexanucleotide repeat expansion less likely. [33] In addition, no positive alpha-synuclein structures were detected in the hippocampus, amygdala, or mesencephalon. The distribution and extent of immunoreactivity for different markers are displayed in Table 1.

Macroscopic images. The macroscopic evaluation revealed, amongst other things, external atrophy (a), atrophy of the medial temporal lobe (b), as well as the cavum septum pellucidum (c, red triangle)

Unique CTE p-tau pathology in the frontal lobe. Overview of p-tau depositions (AT8 immunostaining) preferentially located in the sulcal depths (a, b) perivascular distribution of neuronal lesions (red triangle) and glial lesions (red asterix) (c)

Other p-tau pathology in the frontal lobe. Thorn-shaped astrocytes immunostained for AT8, subpial and perivascular located, typical of ARTAG (a), neurofibrillary tangles (b), tufted astrocytes (c), coiled body in the white matter (d)

Non-tau pathology. Lesions immunostained for pTDP-43 in the hippocampus (a) and frontal lobe (b). Some diffuse plaques immunostained for Aβ in the frontal lobe (c). APP positivity in the white matter of the frontal lobe (d-e)

Non-tau pathology in the sulcal depths of the frontal lobe. Lesions immunostained for pTDP-43 (a, b), CD68 (c, d), Iba1 (e, f)
Literature review
We systematically reviewed the current literature for all confirmed CTE cases in former soccer players. We searched for literature in the PubMed and Embase databases, including the search queries: chronic traumatic encephalopathy, dementia pugilistica, neurodegeneration, soccer, (association) football, and heading (see Additional file 1). Cases were included when they fulfilled the NINDS/NIBIB preliminary neuropathological criteria for CTE and when they had a history of playing soccer. We only included papers that have been written in English. All hits (Pubmed N = 676, Embase N = 879) were independently screened for title and abstract by two authors (SA, SK). From this search, we identified 6 peer-reviewed papers and one conference abstract that involved neuropathological evaluation in soccer player(s) (total number of cases: 18). [14, 15, 34,35,36,37,38] We excluded four cases that lacked CTE-specific tau depositions and one case where trauma-related tau deposits were suggested but with too few NFTs to determine any preferential location. [34] This led to the identification of 13 deceased soccer players with confirmed CTE pathology. The clinical and post-mortem characteristics of these 13 individuals and the case described in this manuscript (total N = 14) are further evaluated below and summarized in Tables 2 and 3.
All cases were male and the age at death ranged between 24 and 83, with a mean symptom duration of 9.1 years (range 2–16). Most of them played soccer at a professional level for at least 16 years, as a defender (N = 6) or forward player (N = 5). Twelve out of 14 cases were clinically diagnosed with dementia and showed progressive cognitive impairment, one case presented with progressive weakness fitting a clinical diagnosis of amyotrophic lateral sclerosis and one case had a clinical diagnosis of bipolar disorder. Eight cases presented with behavioral symptoms early in their disease, two cases developed behavioral symptoms later in their disease, and only three cases described no prominent behavioral changes. Structural imaging findings (brain MRI or CT scan) were reported in three other cases, apart from the case described in this study. The CT-scan of case 2 revealed no abnormalities, case 3 demonstrated temporal atrophy and white matter hypoattenuation. The brain MRI scan of case 8 showed dilated temporal and frontal horns of the lateral ventricle and a small anterior CSP. None of the other cases had available blood and/or CSF biomarker results, or amyloid/tau PET scans. Upon post-mortem examination, 11 cases were reported to have septum pellucidum abnormalities. Severe CTE pathology was described in 6 cases, however, the severity information was missing in 6 other cases. TDP-43 proteinopathy was reported in 11 cases, including one meeting criteria for frontotemporal lobar degeneration (FTLD)-TDP and one for motor-neuron disease. Other pathology was found in terms of AD neuropathologic changes (intermediate level N = 6, high-level N = 2), cerebral amyloid angiopathy (N = 6), cardiovascular disease (N = 5), hippocampal sclerosis (N = 4), aging-related tau astrogliopathy (ARTAG) (N = 3), α-synucleinopathy/Lewy Body Disease (N = 2), corticobasal degeneration/progressive supranuclear palsy (PSP) tauopathies (N = 2), and primary age-related tauopathy (N = 1).
Discussion
We present a comprehensive case description and post-mortem evaluation of a former Dutch professional soccer player with dementia, and we contextualize our findings through a systematic literature overview of current evidence regarding CTE and soccer. We found that, even though the clinical phenotype and the MRI scan were suggestive of AD, fluid and imaging biomarkers were unsupportive of amyloid pathology as the underlying cause of his young-onset progressive dementia. The lack of amyloid neuritic plaque pathology was confirmed at neuropathological investigation. However, the patient showed severe p-tau pathology mostly localized towards the depths of cortical sulci, fitting the NINDS/NIBIB consensus criteria of CTE [9, 12] and is suggested to be related to his extensive soccer career and exposure to RHI. A literature review revealed another 13 soccer cases with neuropathologically confirmed CTE, many with similar cognitive changes, septum abnormalities, and coexisting pathologies.
Pathological mechanisms
Chronic traumatic changes in the brains of former athletes have been characterized for multiple decades. However, there remain many uncertainties regarding the exact pathophysiological mechanisms, the neuropathology, and its associations with RHI. Pathophysiological processes, such as diffuse axonal injury, neuroinflammation, microglial activation, and blood–brain barrier disruption all have been suggested to contribute to the onset of CTE pathology as a consequence of RHI exposure. [39] In this case, we found remarkable APP positivity, a marker for axonal injury, in the white matter. Although APP leakage has been described in brains of subjects that died after hypoxic or ischemic events, the typical pattern of hypoxic APP leakage is not observed in this case [40]. Post-mortem indications for axonal injury (including positive APP staining) have also been described in three cases with a recent COVID-19 infection, but we did not observe other acute pathologies related to COVID-19, such as hemorrhagic lesions or microvascular injury [41, 42]. APP positivity has also been reported after traumatic diffuse axonal injury, or exposure to multiple blast injuries, even years after the injury [40, 43,44,45]. It is possible that the APP finding in this case could be related to past RHI exposure. This needs further evaluation in future studies. We also found evidence for a close relationship between CTE pathology in the frontal sulcal depths and markers for microglial activation based on their similar preferential location (Fig. 6C-F). This is in line with previous studies, that demonstrated a direct link between elevated markers of neuroinflammation and microglial activation and p-tau pathology in brains with CTE [46, 47]. Still, we assessed the elevated protein levels only visually, and it is complicated to determine whether the activity is a consequence of RHI or neurodegeneration in general.
Our case also underscores the complexity of coexisting pathology in cases with CTE. In addition to the distinct p-tau lesions matching pathological criteria for CTE, we found multiple other tau and non-tau pathologies. The subpial, subependymal, and perivascular thorn-shaped astrocytes fit the criteria for aging-related tau astrogliopathy (ARTAG) [48] and the neuronal tau in the subthalamic nucleus and substantia nigra in combination with tufted astrocytes and coiled bodies fulfill the criteria for PSP [49,50,51]. Aiming to differentiate between these multiple tauopathies, we performed staining for microtubule-binding repeat domains in the temporal cortex and specific PSP regions of interest. The pattern of neuronal 3R/4R tau isomers was consistent with CTE pathology studies and not with PSP (predominant 4R tauopathy), although this could not fully exclude the (co-)presence of neuronal PSP pathology. The RD4 positive glial tau lesions were consistent with previous findings of glial tau in CTE, ARTAG as well as PSP [52,53,54]. We also noted cytoplasmic inclusions of TDP-43 in multiple layers of the frontal cortex, the hippocampus, basal ganglia and medulla; this distribution is consistent with the criteria of FTLD type B [55, 56]. The pattern of hippocampal and frontal TDP-43 positivity in combination with hippocampal sclerosis resembles limbic-predominant age-related TDP-43 encephalopathy (LATE), but the extensive frontal depositions and the young age makes this diagnosis unlikely. [57, 58]
Similar to other neurodegenerative diseases such as AD, mixed pathologies in CTE are common. Mez et al. [2017] reported co-pathology to be present in 45% of all CTE cases, with greater prevalence in high-stage CTE. [13] The two largest case series of CTE in soccer players revealed concomitant pathologies in all cases, recognized as AD-related changes, ARTAG, alpha-synuclein, and TDP-43 proteinopathy. [14, 15] Recently, Nicks et al. [59] demonstrated that TDP-43 inclusions (43.3%) and hippocampal sclerosis (23.4%) were prevalent in cases with CTE and that FTLD-TDP may also be present as co-pathology (6%). [59] With coexisting pathologies in neurodegenerative diseases, the question remains how to distinguish primary pathologies from secondary pathologies, and specifically in this case whether non-CTE pathologies are related to CTE p-tau pathology or whether they reflect separate processes, including those associated with RHI exposure. Nicks et al. [59] suggested that CTE may be a risk factor for the development of hippocampal sclerosis and hippocampal TDP-43, and mentioned that TDP-43 inclusions with a predilection for the sulcal depths might be a part of CTE pathology, [59] which was also noted in this case (Fig. 6 A-B). The link between RHI exposure and both ARTAG and PSP, with or without CTE p-tau pathology, has also been suggested in the past but lacks solid evidence. [60, 61] It is possible that CTE pathology accelerates the development of other pathologies, or that CTE involves a wider spectrum of pathological mechanisms and that trauma-induced processes may activate multiple pathological cascades that potentially lead to numerous neurodegenerative proteinopathies, apart from the specific p-tau lesions in the depths of the cortical sulci. Future research is necessary to disentangle these hypotheses.
Clinicopathological correlation
The occurrence of multiple pathologies in this case complicates the clinicopathological correlation. The clinical presentation (progressive cognitive decline) resembled Alzheimer’s dementia, but this diagnosis was not supported by biomarkers and eventually excluded after neuropathological assessment. Cognitive symptoms were the most frequently reported features in RHI-exposed cases with CTE pathology, and dementia was reported in more than 50% of the CTE cases. However, formal neuropsychological test results were often unavailable, and reports mostly relied on retrospective interviews. [23, 62] Nevertheless, other major pathologies lacked strong clinicopathological correlation. The patient did not fulfil the clinical diagnostic criteria for PSP, due to the absence of ocular motor dysfunction, postural instability, and akinesia. [63] Neither did the patient meet the criteria for possible or probable behavioral variant of FTD [64], or for non-fluent variant primary progressive aphasia (the most important clinical syndromes of FTLD-TDP type B). [65, 66] ARTAG is a common pathology primarily described in the tissue of elderly brain donors and it has been suggested that ARTAG lowers the threshold for other pathologies (and related clinical impairment) to develop. [60] Still, the clinical importance of ARTAG is elusive, with studies demonstrating the lack of deficits associated with ARTAG pathology alone. [67, 68] So, it is unlikely that a single pathology fully accounted for the clinical presentation and the progressive course in this patient. Therefore, it is possible that the non-CTE pathologies (i.e., TDP-43 proteinopathy, hippocampal sclerosis), in addition to the CTE pathology, collectively contributed to the clinical disorder. Further research is needed to better understand the clinicopathological correlation of CTE and coexisting pathology in RHI-exposed individuals.
Biomarkers
This case study once again reaffirms that research involving CTE biomarkers is still in its early stages. Amyloid PET scan results were used to evaluate the presence of neuritic amyloid plaque pathology. The negative scan was in line with previous work that demonstrated a lack of elevated amyloid plaque density in cognitively impaired American football players, [69] but had no role in demonstrating CTE or other pathology. Still, determining AD specific biomarkers in patients that have participated in contact sports may be helpful with differential diagnosis. PET scans with p-tau radiotracers may have greater potential for detecting CTE neuropathology. Flortaucipir PET scans have shown some preliminary promise, especially in late stage CTE, but there is a need for tau tracer development that is more specific to CTE tau isoforms. [69, 70] Other potential neuroimaging biomarkers are often restricted to low specificity. Like in this case, although the structural MRI scan demonstrated definite signs of neurodegeneration, the atrophy patterns were not distinguishable from other neurodegenerative diagnoses such as AD. The WMH that appeared on FLAIR MRI may be cerebrovascular-related but may also be related to RHI. Previous work demonstrated greater WMH in former football players compared to unexposed controls but the underlying etiology of these WMH needs to be unraveled in future work. [71] The CSP is also associated with RHI [72,73,74], frequently noted in autopsy-confirmed CTE brain donors [11], but this finding is not specific as it may also be found in the healthy population or asymptomatic contact-sport athletes. [75, 76] Consequently, the aforementioned MRI findings are potentially helpful as a marker for neurodegeneration or previous injuries rather than a specific diagnostic feature for CTE. Concerning the blood biomarkers, the low concentration of plasma p-tau-181 is consistent with recent work on two other autopsy-confirmed CTE cases that showed comparable low values of plasma p-tau-181. [77] The levels of NfL and GFAP were also relatively low, particularly in comparison with the AD population. These findings suggest that p-tau-181, and plasma NfL and GFAP may have less potential for assessing tau and non-tau-related pathologies in CTE and that other tau epitopes may have greater potential. This needs further evaluation in a larger sample of pathology confirmed CTE cases.
Conclusions
The relationship between soccer and neurodegeneration is increasingly recognized in clinical studies, but less supported by pathological studies with very few post-mortem confirmed CTE cases in the literature. This study adds to the current literature and will hopefully increase awareness and appreciation of the complexity of the clinicopathological correlation and diagnosis of CTE during life. Large-scale clinicopathological research among former soccer players is necessary for a better understanding and to find out the exact prevalence and risk factors for CTE in this population.
Availability of data and materials
Data sharing does not apply to this article as no datasets were generated or analyzed during the current study. Material may be available upon reasonable request.
Abbreviations
- 3R:
-
3-Repeat
- 4R:
-
4-Repeat
- AD:
-
Alzheimer's disease
- ALS:
-
Amyotrophic lateral sclerosis
- APP:
-
Amyloid precursor protein
- ARTAG:
-
Aging-related tau astrogliopathy
- Aβ:
-
Amyloid-beta
- α-syn:
-
Alpha-synuclein
- C9orf72:
-
Chromosome 9 open reading frame 72
- CAA:
-
Cerebral amyloid angiopathy
- CB:
-
Coiled bodies
- CBD:
-
Corticobasal degeneration
- CDR:
-
Clinical Dementia Rating
- CSF:
-
Cerebrospinal fluid
- CSP:
-
Cavum septum pellucidum
- CTE:
-
Chronic traumatic encephalopathy
- CVD:
-
Cardiovascular disease
- DAB:
-
3,3′-Diaminobenzidine tetrahydrochloride
- DLB:
-
Dementia with Lewy Bodies
- FLAIR:
-
Fluid-attenuated inversion recovery
- FTLD:
-
Frontotemporal lobar degeneration
- GFAP:
-
Glial fibrillary acidic protein
- GFA:
-
Granular/fuzzy astrocytes
- H&E:
-
Hematoxylin and Eosin
- IHC:
-
Immunohistochemistry
- LATE:
-
Limbic-predominant age-related TDP-43 encephalopathy
- LC:
-
Locus coeruleus
- MAPT:
-
Microtubule-associated protein tau
- MMSE:
-
Mini-mental state examination
- MND:
-
Motor neuron disease
- MRI:
-
Magnetic resonance imaging
- NBB:
-
Netherlands brain bank
- neu:
-
Neuronal
- NEwTON:
-
Neurodegeneration: Traumatic brain injury as Origin of the Neuropathology
- NfL:
-
Neurofilament light
- NFTs:
-
Neurofibrillary tangles
- NIBIB:
-
National Institute of Biomedical Imaging and Bioengineering
- NINDS:
-
National Institute on Neurological Disorders and Stroke
- NPI-Q:
-
Neuropsychiatry Inventory Questionnaire
- NT:
-
Neuropil threads
- oligo:
-
Oligodendrocytes
- PART:
-
Primary age-related tauopathy
- PD:
-
Parkinson's disease
- PET:
-
Positron emission tomography
- PSP:
-
Progressive supranuclear palsy
- p-tau:
-
Phosphorylated tau
- RHI:
-
Repetitive head impacts
- STN:
-
Subthalamic nucleus
- TA:
-
Tufted astrocytes
- (p)TDP(-43):
-
(Phosphorylated) TAR DNA-binding protein (43)
- TES:
-
Traumatic Encephalopathy Syndrome
- TSA:
-
Thorn-shaped astrocytes
- UMC:
-
University Medical Centers
- VaD:
-
Vascular dementia
- VU:
-
Vrije Universiteit
- WMH:
-
White matter hyperintensities
- α-syn:
-
Alpha-synuclein
References
FIFA Communications Division Information Services. FIFA Big Count 2006. https://digitalhub.fifa.com/m/55621f9fdc8ea7b4/original/mzid0qmguixkcmruvema-pdf.pdf. Accessed 06 Dec 2022.
Mackay DF, Russell ER, Stewart K, MacLean JA, Pell JP, Stewart W (2019) Neurodegenerative disease mortality among former professional soccer players. N Engl J Med 381(19):1801–1808
Russell ER, Mackay DF, Stewart K, MacLean JA, Pell JP, Stewart W (2021) Association of field position and career length with risk of neurodegenerative disease in male former professional soccer players. JAMA Neurol 78(9):1057–1063
Daneshvar DH, Mez J, Alosco ML, Baucom ZH, Mahar I, Baugh CM et al (2021) Incidence of and mortality from amyotrophic lateral sclerosis in national football league athletes. JAMA Netw Open 4(12):e2138801
Lehman EJ, Hein MJ, Baron SL, Gersic CM (2012) Neurodegenerative causes of death among retired National Football League players. Neurology 79(19):1970–1974
Nguyen VT, Zafonte RD, Chen JT, Kponee-Shovein KZ, Paganoni S, Pascual-Leone A et al (2019) Mortality among professional American-style football players and professional American baseball players. JAMA Netw Open 2(5):e194223
Russell ER, Mackay DF, Lyall D, Stewart K, MacLean JA, Robson J, et al (2022) Neurodegenerative disease risk among former international rugby union players. J Neurol Neurosurg Psychiatry
Martland HS (1928) Punch drunk. J Am Med Assoc 91(15):1103–1107
Bieniek KF, Cairns NJ, Crary JF, Dickson DW, Folkerth RD, Keene CD et al (2021) The second NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. J Neuropathol Exp Neurol 80(3):210–219
McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE et al (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68(7):709–735
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain: J Neurol 136(Pt 1):43–64
McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I et al (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131(1):75–86
Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR et al (2017) Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 318(4):360–370
Lee EB, Kinch K, Johnson VE, Trojanowski JQ, Smith DH, Stewart W (2019) Chronic traumatic encephalopathy is a common co-morbidity, but less frequent primary dementia in former soccer and rugby players. Acta Neuropathol 138(3):389–399
Ling H, Morris HR, Neal JW, Lees AJ, Hardy J, Holton JL et al (2017) Mixed pathologies including chronic traumatic encephalopathy account for dementia in retired association football (soccer) players. Acta Neuropathol 133(3):337–352
Kaufer DI, Cummings JL, Ketchel P, Smith V, MacMillan A, Shelley T et al (2000) Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci 12(2):233–239
Koedam EL, Lehmann M, van der Flier WM, Scheltens P, Pijnenburg YA, Fox N et al (2011) Visual assessment of posterior atrophy development of a MRI rating scale. Eur Radiol 21(12):2618–2625
Pasquier F, Leys D, Weerts JG, Mounier-Vehier F, Barkhof F, Scheltens P (1996) Inter- and intraobserver reproducibility of cerebral atrophy assessment on MRI scans with hemispheric infarcts. Eur Neurol 36(5):268–272
Scheltens P, Leys D, Barkhof F, Huglo D, Weinstein HC, Vermersch P et al (1992) Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry 55(10):967–972
Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA (1987) MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol 149(2):351–356
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH et al (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia 7(3):263–269
Mulder C, Verwey NA, van der Flier WM, Bouwman FH, Kok A, van Elk EJ et al (2010) Amyloid-beta(1–42), total tau, and phosphorylated tau as cerebrospinal fluid biomarkers for the diagnosis of Alzheimer disease. Clin Chem 56(2):248–253
Katz DI, Bernick C, Dodick DW, Mez J, Mariani ML, Adler CH, et al (2021) National Institute of Neurological Disorders and Stroke Consensus Diagnostic Criteria for Traumatic Encephalopathy Syndrome. Neurology
van der Flier WM, Pijnenburg YA, Prins N, Lemstra AW, Bouwman FH, Teunissen CE et al (2014) Optimizing patient care and research: the Amsterdam Dementia Cohort. J Alzheimer’s Disease JAD 41(1):313–327
NfL interactive manual version 2.5 2022 [updated 20–05–2022]. https://mybiomarkers.shinyapps.io/Neurofilament/. Accessed 06 Dec 2022.
Vermunt L, Otte M, Verberk IMW, Killestein J, Lemstra AW, van der Flier WM, et al (2022) Age- and disease-specific reference values for neurofilament light presented in an online interactive support interface. Ann Clin Transl Neurol
Honey MIJ, Teunissen CE, et al (2022) GFAP interactive manual (in preparation)
van Amerongen S, Caton DK, Ossenkoppele R, Barkhof F, Pouwels PJW, Teunissen CE et al (2022) Rationale and design of the “NEurodegeneration: Traumatic brain injury as Origin of the Neuropathology (NEwTON)” study: a prospective cohort study of individuals at risk for chronic traumatic encephalopathy. Alzheimers Res Ther 14(1):119
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123(1):1–11
Schmitt A, Bauer M, Heinsen H, Feiden W, Falkai P, Alafuzoff I et al (2007) How a neuropsychiatric brain bank should be run: a consensus paper of Brainnet Europe II. J Neural Transm (Vienna) 114(5):527–537
Hartmann P, Ramseier A, Gudat F, Mihatsch MJ, Polasek W, Geisenhoff C (1994) Das Normgewicht des Gehirns beim Erwachsenen in Abhängigkeit von Alter, Geschlecht. Körpergröße und Gewicht Der Pathologe 15(3):165–170
Dutra JR, Cortés EP, Vonsattel JP (2015) Update on hippocampal sclerosis. Curr Neurol Neurosci Rep 15(10):67
Pikkarainen M, Hartikainen P, Alafuzoff I (2008) Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J Neuropathol Exp Neurol 67(4):280–298
Geddes JF, Vowles GH, Nicoll JA, Révész T (1999) Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 98(2):171–178
Grinberg LT, Anghinah R, Nascimento CF, Amaro E, Leite RP, Martin Mda G et al (2016) Chronic traumatic encephalopathy presenting as Alzheimer’s disease in a retired soccer player. J Alzheimer’s Disease JAD 54(1):169–174
Hales C, Neill S, Gearing M, Cooper D, Glass J, Lah J (2014) Late-stage CTE pathology in a retired soccer player with dementia. Neurology 83(24):2307–2309
McKee AC, Daneshvar DH, Alvarez VE, Stein TD (2014) The neuropathology of sport. Acta Neuropathol 127(1):29–51
Phalen J, Alosco M, Kiernan P, Montenigro P, Solomon T, Stern R et al (2017) Chronic traumatic encephalopathy in a 24-year-old former soccer player. Neurology 88(16 Suppl):P6.165
Lucke-Wold BP, Turner RC, Logsdon AF, Bailes JE, Huber JD, Rosen CL (2014) Linking traumatic brain injury to chronic traumatic encephalopathy: identification of potential mechanisms leading to neurofibrillary tangle development. J Neurotrauma 31(13):1129–1138
Rahaman P, Del Bigio MR (2018) Histology of brain trauma and hypoxia-ischemia. Acad Forensic Pathol 8(3):539–554
Eschbacher KL, Larsen RA, Moyer AM, Majumdar R, Reichard RR (2022) Neuropathological findings in COVID-19: an autopsy cohort. J Neuropathol Exp Neurol 82(1):21–28
Thakur KT, Miller EH, Glendinning MD, Al-Dalahmah O, Banu MA, Boehme AK et al (2021) COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain 144(9):2696–2708
Benjamini D, Iacono D, Komlosh ME, Perl DP, Brody DL, Basser PJ (2021) Diffuse axonal injury has a characteristic multidimensional MRI signature in the human brain. Brain 144(3):800–816
Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W (2013) Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136(Pt 1):28–42
Ryu J, Horkayne-Szakaly I, Xu L, Pletnikova O, Leri F, Eberhart C et al (2014) The problem of axonal injury in the brains of veterans with histories of blast exposure. Acta Neuropathol Commun 2(1):153
Cherry JD, Meng G, Daley S, Xia W, Svirsky S, Alvarez VE et al (2020) CCL2 is associated with microglia and macrophage recruitment in chronic traumatic encephalopathy. J Neuroinflammation 17(1):370
Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH et al (2016) Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 4(1):112
Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H et al (2016) Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 131(1):87–102
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA (2010) Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 23(4):394–400
Litvan I, Hauw J, Bartko J, Lantos P, Daniel S, Horoupian D et al (1996) Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 55(1):97–105
Roemer SF, Grinberg LT, Crary JF, Seeley WW, McKee AC, Kovacs GG et al (2022) Rainwater Charitable Foundation criteria for the neuropathologic diagnosis of progressive supranuclear palsy. Acta Neuropathol 144(4):603–614
Arena JD, Smith DH, Lee EB, Gibbons GS, Irwin DJ, Robinson JL et al (2020) Tau immunophenotypes in chronic traumatic encephalopathy recapitulate those of ageing and Alzheimer’s disease. Brain 143(5):1572–1587
Arendt T, Stieler JT, Holzer M (2016) Tau and tauopathies. Brain Res Bull 126(Pt 3):238–292
Cherry JD, Kim SH, Stein TD, Pothast MJ, Nicks R, Meng G et al (2020) Evolution of neuronal and glial tau isoforms in chronic traumatic encephalopathy. Brain Pathol 30(5):913–925
Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D et al (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol 113(5):521–533
Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E et al (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122(1):111–113
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K et al (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142(6):1503–1527
Nelson PT, Lee EB, Cykowski MD, Alafuzoff I, Arfanakis K, Attems J et al (2023) LATE-NC staging in routine neuropathologic diagnosis: an update. Acta Neuropathol 145(2):159–173
Nicks R, Clement NF, Alvarez VE, Tripodis Y, Baucom ZH, Huber BR, et al (2023) Repetitive head impacts and chronic traumatic encephalopathy are associated with TDP-43 inclusions and hippocampal sclerosis. Acta Neuropathol
Kovacs GG (2020) Astroglia and Tau: new perspectives. Front Aging Neurosci 12:96
Ling H, Kara E, Revesz T, Lees AJ, Plant GT, Martino D et al (2014) Concomitant progressive supranuclear palsy and chronic traumatic encephalopathy in a boxer. Acta Neuropathol Commun 2:24
Mez J, Alosco ML, Daneshvar DH, Saltiel N, Baucom Z, Abdolmohammadi B et al (2021) Validity of the 2014 traumatic encephalopathy syndrome criteria for CTE pathology. Alzheimers Dement 17(10):1709–1724
Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE et al (2017) Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord 32(6):853–864
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J et al (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134(Pt 9):2456–2477
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF et al (2011) Classification of primary progressive aphasia and its variants. Neurology 76(11):1006–1014
Neumann M, Lee EB, Mackenzie IR (2021) Frontotemporal lobar degeneration TDP-43-immunoreactive pathological subtypes: clinical and mechanistic significance. Adv Exp Med Biol 1281:201–217
Forrest SL, Wagner S, Kim A, Kovacs GG (2022) Association of glial tau pathology and LATE-NC in the ageing brain. Neurobiol Aging 119:77–88
Nolan A, De Paula Franca Resende E, Petersen C, Neylan K, Spina S, Huang E et al (2019) Astrocytic Tau deposition is frequent in typical and atypical Alzheimer disease presentations. J Neuropathol Exp Neurol 78(12):1112–1123
Stern RA, Adler CH, Chen K, Navitsky M, Luo J, Dodick DW et al (2019) Tau positron-emission tomography in former national football league players. N Engl J Med 380(18):1716–1725
Alosco ML, Su Y, Stein TD, Protas H, Cherry JD, Adler CH, et al (2022) Associations between near end-of-life flortaucipir PET and postmortem CTE-related tau neuropathology in six former American football players. Eur J Nucl Med Mol Imaging
Alosco ML, Tripodis Y, Baucom ZH, Adler CH, Balcer LJ, Bernick C, et al (2022) White matter hyperintensities in former American football players. Alzheimers Dement
Gardner RC, Hess CP, Brus-Ramer M, Possin KL, Cohn-Sheehy BI, Kramer JH et al (2016) Cavum septum pellucidum in retired American pro-football players. J Neurotrauma 33(1):157–161
Koerte IK, Hufschmidt J, Muehlmann M, Tripodis Y, Stamm JM, Pasternak O et al (2016) Cavum septi pellucidi in symptomatic former professional football players. J Neurotrauma 33(4):346–353
McAllister D, Akers C, Boldt B, Mitchell LA, Tranvinh E, Douglas D et al (2021) Neuroradiologic evaluation of MRI in high-contact sports. Front Neurol 12:701948
Born CM, Meisenzahl EM, Frodl T, Pfluger T, Reiser M, Möller HJ et al (2004) The septum pellucidum and its variants: an MRI study. Eur Arch Psychiatry Clin Neurosci 254(5):295–302
Stanwell P, Iverson GL, Van Patten R, Castellani RJ, McCrory P, Gardner AJ (2022) Examining for cavum septum pellucidum and ventricular enlargement in retired elite-level rugby league players. Front Neurol 13:817709
Asken BM, Tanner JA, VandeVrede L, Mantyh WG, Casaletto KB, Staffaroni AM et al (2022) Plasma P-tau181 and P-tau217 in patients with traumatic encephalopathy syndrome with and without evidence of alzheimer disease pathology. Neurology 99(6):e594-604
Acknowledgements
Research of Alzheimer Center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. Furthermore, we would like to acknowledge M. Bugiani for the help with the brain autopsy, M. Jacobs, and T.H.J Morrema for their help with pathology preparation and staining, C. Korth for developing and providing the monoclonal antibody IC16 (anti-Aβ/APP), E.R. Bludjea for assisting with the blood assays, and S.J. van der Lee for the help with the genetic analyses. Finally, we would like to gratefully thank the patient and family for their participation and all contributions to this research.
Funding
The NEwTON study received funding from Stichting Dioraphte to make this research possible.
Author information
Authors and Affiliations
Contributions
SA: contributed to the design of the study protocol, performed data collection, and drafted the manuscript. SK, KKMK, YALP, PS, CET, FB, RO, AJMR, RAS: contributed intellectually and critically appraised the manuscript. KKMK: has performed immunostaining and post-mortem analysis. PS: has been involved as a neurologist during the whole clinical course of the patient’s disease. CET: has been responsible for blood/CSF assays and interpretation. FB: has been responsible for MRI reports as a radiologist. AJMR: has been responsible for the autopsy, post-mortem evaluation, and interpretation. JJMH: has been responsible for the pathological interpretation, has overseen data collection and supervised the manuscript draft. EGBV: PI of the NEwTON study, has been involved as a neurologist of the patient, has overseen data collection, and supervised the manuscript draft. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The patient has provided written consent to use his clinical data and biomaterial for future research purposes, as part of the Amsterdam Dementia Cohort, which was approved by the Medical Ethics Committee of the VU medical center under the reference number 2016/061. Before death, the patient’s next of kin gave written informed consent for brain donation to the NEwTON cohort of the Netherlands Brain Bank (NBB), according to the ethical guidelines of brain banking. The NBB has been approved by the Medical Ethics Committee of the VU medical center under the reference number 2009/148. After death, the brain autopsy has eventually been performed by the Amsterdam Medical Centers Department of Pathology instead of the NBB due to logistical reasons, after consent of the family.
Consent for publication
Before submission, all closest next of kin gave written informed consent to submit and publish this case report. They understand that all materials are published without a name attached but that anonymity is not guaranteed.
Competing interests
PS has received consultancy fees (paid to the institutions) from AC Immune, Alkermes, Alnylam, Alzheon, Anavex, Axoltis, Brainstorm Cell, Cortexyme, Denali, EIP, ImmunoBrain Checkpoint, GemVax, Genentech, Green Valley, Novartis, Novo Nordisk, PeopleBio, Renew LLC, and Roche. He received payment or honoraria from Nutricia. He is a PI of studies with AC Immune, CogRx, FUJI-film/Toyama, IONIS, UCB, and Vivoryon. He is a part-time employee of Life Sciences Partners Amsterdam, and he serves on the board of New Amsterdam Pharma. CET is a member of the Innogenetics International Advisory Boards of Fujirebio/Innogenetics and Roche. She has received research reagents from ADxNeurosciences and Euroimmun and has a collaboration contract with ADx Neurosciences and with Quanterix. She has performed contract research or received grants from AC Immune, AxonNeurosciences, Biogen, Boehringer, Brainstorm Therapeutics, Celgene, CogRx, EIP Pharma, Esai, Fujirebio, Janssen prevention center, PeopleBio, Probiodrug, Roche, Toyama, and Vivoryon. Research from CET is supported by the European Commission (Marie Curie International Training Network, JPND), Health Holland, the Dutch Research Council (ZonMW), The Weston Brain Institute, Alzheimer Netherlands, and Alzheimer Association. FB is supported by the NIHR biomedical research center at UCLH. RAS has been a paid consultant to Biogen (Cambridge, MA, USA) and Lundbeck (Copenhagen, Denmark). He is a member of the Board of Directors of King-Devick Technologies, Inc. (Chicago, IL, USA), and he receives royalties for published neuropsychological tests from Psychological Assessment Resources, Inc. (Lutz, FL, USA). He was a member of the Medical Science Committee for the National Collegiate Athletic Association Student-Athlete Concussion Injury Litigation. EGBV has received consultancy fees (paid to the institution) from Biogen, Brainstorm Therapeutics, ImmunoBrain Checkpoint, New Amsterdam Pharma, ReMynd and Treeway, Vivoryon, and Vigil Neuroscience. He is the PI of studies with ACImmune, CogRx, Green Valley, IONIS, Janssen, Roche, Rodin Therapeutics, Sanofi, UCB, and Vivoryon. JJMH is the Editor-in-Chief of Acta Neuropathologica Communications.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Supplementary Materials.
This file includes a detailed description of the immunostaining methods and the search queries for the literature search.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
van Amerongen, S., Kamps, S., Kaijser, K.K.M. et al. Severe CTE and TDP-43 pathology in a former professional soccer player with dementia: a clinicopathological case report and review of the literature. acta neuropathol commun 11, 77 (2023). https://doi.org/10.1186/s40478-023-01572-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-023-01572-3