
Abstract
Myasthenia gravis (MG) is an autoantibody-mediated autoimmune disorder of the neuromuscular junction. A small subset of patients (<10%) with MG, have autoantibodies targeting muscle-specific tyrosine kinase (MuSK). MuSK MG patients respond well to CD20-mediated B cell depletion therapy (BCDT); most achieve complete stable remission. However, relapse often occurs. To further understand the immunomechanisms underlying relapse, we studied autoantibody-producing B cells over the course of BCDT. We developed a fluorescently labeled antigen to enrich for MuSK-specific B cells, which was validated with a novel Nalm6 cell line engineered to express a human MuSK-specific B cell receptor. B cells (≅ 2.6 million) from 12 different samples collected from nine MuSK MG patients were screened for MuSK specificity. We successfully isolated two MuSK-specific IgG4 subclass-expressing plasmablasts from two of these patients, who were experiencing a relapse after a BCDT-induced remission. Human recombinant MuSK mAbs were then generated to validate binding specificity and characterize their molecular properties. Both mAbs were strong MuSK binders, they recognized the Ig1-like domain of MuSK, and showed pathogenic capacity when tested in an acetylcholine receptor (AChR) clustering assay. The presence of persistent clonal relatives of these MuSK-specific B cell clones was investigated through B cell receptor repertoire tracing of 63,977 unique clones derived from longitudinal samples collected from these two patients. Clonal variants were detected at multiple timepoints spanning more than five years and reemerged after BCDT-mediated remission, predating disease relapse by several months. These findings demonstrate that a reservoir of rare pathogenic MuSK autoantibody-expressing B cell clones survive BCDT and reemerge into circulation prior to manifestation of clinical relapse. Overall, this study provides both a mechanistic understanding of MuSK MG relapse and a valuable candidate biomarker for relapse prediction.
Similar content being viewed by others


Introduction
Autoimmune myasthenia gravis (MG) is a prototypical autoantibody-mediated disease. Pathogenic autoantibodies in MG target proteins within the neuromuscular junction (NMJ), which interrupts neuromuscular transmission [16, 74]. The largest subset (≈ 85%) of MG patients have autoantibodies that target the acetylcholine receptor (AChR), but a small subset of patients, who are negative for AChR autoantibodies, harbor autoantibodies that bind to muscle-specific tyrosine kinase (MuSK) [26], low-density lipoprotein receptor-related protein 4 (LRP4) [25, 50, 83] or agrin [83].
The MG subtypes that are defined by the autoantibody specificity appear clinically similar, but the underlying immunopathology is remarkably distinct [13]. This is well highlighted by comparing the AChR and MuSK subtypes. While AChR MG is driven by the IgG1 and IgG3 subclass [56], MuSK MG is largely governed by the IgG4 subclass that mediates pathology by inhibiting the interaction between MuSK and LRP4 [30], which is essential for MuSK phosphorylation and subsequent effective AChR clustering and signaling. The phenotype of the B cells that produce pathogenic autoantibodies in AChR and MuSK MG also appear divergent. It is currently thought that short-lived plasmablasts are key autoantibody producers in MuSK MG [63, 64], while plasma cells mostly account for the production of AChR autoantibodies [59, 77, 81]. These differences are highlighted by the outcomes reached through the use of biological therapeutics in clinical trials. Specifically, this is evident by the poor response to CD20-mediated (rituximab) B cell depletion therapy (BCDT) in AChR MG [47], in contrast to the significant response in MuSK MG [10, 33]. In clinical practice, most MuSK MG patients achieve complete stable remission following BCDT [10, 44, 48], which often includes reduction of autoantibody titer to undetectable levels. This fits well with an emerging pattern of BCDT efficacy in patients with other diseases mediated by pathogenic IgG4 autoantibodies, such as pemphigus with autoantibodies directed against the desmoglein adhesion molecules [32] and chronic inflammatory demyelinating polyneuropathy with paranodal protein-specific autoantibodies [52]. The clinically proven efficacy of BCDT notwithstanding, after an initial remission some MuSK MG patients experience relapse [4, 22, 48, 55], which can associate with an increased frequency of plasmablasts and memory B cells, including populations that express MuSK-specific autoantibodies [64, 67]. Not all of these B cells develop de novo after BCDT, as a proportion of B cell clones persist through treatment [31]. However, it is not clear how pathogenic MuSK autoantibody-expressing B cell clones behave during a given clinical course, as it includes phases of relapse and periods of remission induced by repeated BCDT treatment cycles.
To that end, we sought to isolate MuSK-specific autoantibody-producing B cells, validate their specificity, and determine whether these B cells are present in BCDT-treated patients over a period of time. We developed a monomeric fluorescently labeled MuSK antigen to enrich for MuSK-specific B cells and authenticated this antigen bait using a novel B cell line (Nalm6 cells) that expressed a human MuSK-specific B cell receptor (BCR). We found that MuSK-specific B cells are exceptionally rare in the circulation, yet we were able to isolate two distinct clones from two different patients, the specificity of which was validated through testing of human recombinant monoclonal autoantibodies (mAbs). We collected upwards of 149,000 B cell receptor sequences from serial samples of these two patients to search for clonal variants. We found that MuSK-specific B cell clones persisted through repeated rounds of BCDT and reemerged prior to clinical relapse in association with increased autoantibody titers.
Material and methods
Human specimens
This study was approved by the Human Investigation Committee at the Yale School of Medicine (clinicaltrials.gov || NCT03792659). Informed written consent was obtained from all patients. All MuSK MG patients met definitive diagnostic criteria for MG, including positive serology for MuSK autoantibodies.
Fluorescently labeled MuSK, Nalm6 cells, and cell sorting
Recombinant human MuSK and the negative control protein recombinant human growth hormone (hGH) (BioLegend; 778006) were fluorescently labeled using the Alexa Fluor™ 647 Microscale Protein Labeling Kit (Invitrogen; A30009). Nalm6 cells (CRL-3273™) and Nalm6 cells containing the MuSK-specific MuSK3-28 B cell receptor (Nalm6_3-28) were cultured in RPMI 1640 media containing 10% FBS, 1% P/S and 1% HEPES. On the day of the CBA for the validation of the MuSK reagent, the fluorescently labeled MuSK was added at a final concentration of 10, 1, 0.1 or 0.01 μg/ml. For sorting MuSK-reactive B cells, B cells were enriched from cryopreserved PBMCs using negative selection beads (Stemcell Technologies; 19554). They were incubated with live/dead stain, then stained for 30 minutes on ice with 1 μg/ml of fluorescently labeled MuSK antigen together with fluorescently labeled antibodies against CD3 (BD Biosciences, V500; UCTH1), CD14 (Invitrogen, Pacific orange; TUK4), CD19 (BioLegend, PE Cy7; SJ25C1), CD27 (BD Biosciences, PE; M-T271), IgD (BD Biosciences, FITC; IA6-2), IgM (BioLegend, PerCP/Cyanine5.5; MHM-88) and CD38 (BioLegend, BV421; HB-7) using manufacturer’s recommended dilutions. The cells were sorted on an FACSAria (BD Biosciences) instrument. For general B cell immunophenotyping, B cells were defined as live CD3–CD14–CD19+ cells, memory B cells as live CD3–CD14–CD19+CD27+CD38– cells, and antibody-secreting cells (plasmablast phenotype) as CD3–CD14–CD19+CD27+CD38hi. We sorted for CD3-CD14-CD19+IgD-CD27+IgM-MuSK+ cells for the single cell B cell culture experiments.
B cell culture, molecular cloning, and IgG subclass determination
MS40Llo cells (kindly provided by Drs. Garnett Kelsoe and Dongmei Liao of Duke University; [40]) were maintained in IMDM media (Invitrogen, 12440-053) supplemented with 10% FCS (Thermo Scientific, SH30070.03), 1% Pen/Strep (Invitrogen, 15140-122) and 55 µM 2-ME (Invitrogen, 21985). For B cell culture the cells were suspended in B cell media (RPMI, 10% FBS, 1% Pen/Strep, 1% HEPES, 1% Sodium pyruvate, 1% MEM NEAA and 55 µM of 2-ME) and plated at a concentration of 3 × 103 cells per well into 96 well plates. The B cell media was changed the following day to contain additionally 50 ng/ml IL-2 (Peprotech 100-02), 10 ng/ml IL-4 (Peprotech 200-04), 10 ng/ml IL-21 (Peprotech 200-21) and 10 ng/ml BAFF (Peprotech 310-13) and single B cells were sorted into each well. The culture was maintained for 20-30 days, then the supernatant was harvested and screened for MuSK-reactivity using a MuSK-specific cell-based assay (CBA). The RNA of cells in MuSK antibody positive wells was purified using the RNeasy Plus Mini Kit (Qiagen), followed by cDNA synthesis, single-cell PCR, IgG subclass determination and molecular cloning into the corresponding heavy, kappa or lambda vectors as previously described [67, 75].
Recombinant expression of MuSK and human mAbs and Fabs
The extracellular domain of MuSK (AA 22-452) was produced in stably transfected Drosophila S2 cells kindly provided by Dr. Patrick Waters (University of Oxford) as previously described [67]. The 2E6 and 6C6 mAbs and were expressed as IgG1 subclass whole antibodies as previously described [67]. The negative control mAb D12 was generated from patient MuSK MG-3. It was derived from single cell sorting and subsequent single cell PCR. The sort enriched for IgG4-specific memory B cells/plasmablasts (CD3neg, CD14neg, CD19+, IgDneg, CD27+, IgG4+ (biotinylated IgG4-specific clone MH164-1, with APC streptavidin (Biolegend #405207) used for detection). The antibody subclass usage of this clone was verified as IgG4 through subclass PCR. The D12 mAb was expressed as an IgG4 subclass whole antibody, and then used as a negative control as no binding to MuSK via CBA was observed. The antigen binding fragments (Fabs) of 2E6 and 6C6 were expressed in a human Fab expression vector (see below for heavy chain plasmid description) using the same expression system as described for the mAbs. Protein G Sepharose® 4 Fast Flow beads were used for mAb purification and the 6XHis-tagged Fabs and MuSK were affinity purified using HisPurTM Cobalt Resin according to manufacturer’s protocol.
Immunofluorescence of mouse muscle sections
The binding of the mAb 2E6 and 6C6 to mouse MuSK was performed as previously described [67]. Briefly, positive control AChR-specific mAb 637 [18], 6C6 and 2E6 mAbs were added at a concentration of 4 μg/mL each combined with Alexa Fluor 647–conjugated α-bungarotoxin (1:300, B35450, Thermo Fischer Scientific). Human Fc-γ–specific Alexa Fluor 488–conjugated goat F(ab′)2 (3 μg/mL, 109-546-170, Jackson ImmunoResearch) was subsequently added. Endplates were identified using the immunofluorescence of the α-bungarotoxin staining. Triple-fluorescent photomicrographs of the endplate regions were acquired using μManager software ver2.0 [69] on an Olympus BX51WI spinning-disk confocal fluorescence microscope with a Hamamatsu EM-CCD C9100 digital camera. Endplates were analyzed using ImageJ software (NIH) as described [17, 69].
Autoantibody binding cell-based assay
HEK293T cells were transfected with either full-length MuSK-GFP (kindly provided by Drs. David Beeson, Angela Vincent and Patrick Waters, Neurosciences Group at the Weatherall Institute of Molecular Medicine, University of Oxford) or different ectodomain variants of MuSK-GFP (previously described in [67]). The cell-based assay (CBA) was performed as previously decribed [14]. The autoantibody titer of human sera was measured using 10 two-fold dilutions ranging from 1:20 to 1:10240. The binding of each mAb was detected with Alexa Fluor®-conjugated AffiniPure Rabbit Anti-Human IgG, Fcγ (309-605-008, Jackson Immunoresearch) on a BD LSRFortessa® (BD Biosciences). FlowJo software (FlowJo, LLC) was used for analysis.
Human Fab expression vector construction
A human Fab expression vector was engineered from our human IgG1 heavy chain expression vector [49], following a human Fab VH vector design demonstrated to work with mammalian antibody expression [72]. The region coding for the IgG1 constant region was modified to terminate near the junction of the CH1 region and the upper hinge (TKVDKKV - EPKSC). At this region a linker sequence (GS) was added followed by a 6XHis-tag, then a stop codon (TKVDKKV – EPKSC – GS – HHHHHH - stop). A synthetic DNA fragment (gBlock™, Integrated DNA Technologies) coding for the modified constant region was amplified by PCR (GoTaq® DNA Polymerase; Promega (M3001)) and cloned into the original human IgG1 heavy chain expression vector at the Apa I site—located at the beginning of the CH1—and a Bam HI site, which is located at the end of CH3 immediately downstream of the stop codon. The sequence integrity of this new human Fab heavy chain expression vector was confirmed by both Sanger sequencing of the insert and sequencing of the entire plasmid with the Oxford Nanopore platform (Plasmidsaurus). The variable regions coding for the heavy chains of the mAbs 2E6, 6C6, MuSK1A and IgG4 D12 were then subcloned into the Fab expression vector at the Afe I and Apa I sites following standard procedures. The plasmids were transformed into NEB® 5-alpha Competent E. coli (New England BioLabs, Inc.). Plasmid DNA was then isolated with the QIAprep Spin Miniprep Kit (Qiagen) and sequenced by Sanger sequencing to confirm the presence of each specific variable region.
Polyreactivity, HEp-2 ELISA and AChR clustering assay
Recombinant human mAbs were tested for polyreactivity on microplates coated with 20 µg/ml double-stranded DNA (dsDNA), 10 µg/ml lipopolysaccharide (LPS), or 15 µg/ml recombinant human insulin (all purchased from SIGMA-Aldrich) using a previously described approach [75]. Highly polyreactive antibody ED38 was used as positive control [62]. Purified recombinant IgGs were tested for autoreactivity on a commercially available human epithelial type 2 (HEp-2) cell lysate ELISA kit (INOVA) according to the manufacturer’s instructions with minor modifications that have been described previously [84]. The ELISA plates were read using a PowerWave XS (BIO-TEK). The C2C12 AChR clustering assay was performed as previously reported [14, 67].
Bulk library preparation
RNA was isolated from frozen PBMCs using the RNeasy Mini Kit (Qiagen, 74104) according to manufacturer’s instructions. BCR libraries were either generated using the NEBNext Immune Sequencing Kit (NEB) as previously published [31] or the SMARTer® Human BCR IgG IgM H/K/L Profiling Kit (Takara Bio USA, Inc.) using the primers targeting IgA, IgG and IgG-subclasses. Four libraries were pooled in equimolar amounts and prepared for sequencing on the Pacific Biosciences (PacBio) Sequel II machine by B Cell Receptor Repertoire SMRTbell® Library (PacBio) preparation.
Single-cell library preparation
B cells were enriched from cryopreserved PBMCs using negative selection beads (Stemcell Technologies; 19554). They were incubated with live/dead stain, then stained for 30 minutes on ice with fluorescently labeled antibodies against CD3 (BD Biosciences, V500; UCTH1), CD14 (Invitrogen, Pacific orange; TUK4), CD19 (BioLegend, PE Cy7; SJ25C1), CD27 (BD Biosciences, PE; M-T271), IgD (BD Biosciences, FITC; IA6-2), IgM (BioLegend, PerCP/Cyanine5.5; MHM-88) and CD38 (BioLegend, BV421; HB-7) using manufacturer’s recommended dilutions. The cells were sorted on an FACSAria (BD Biosciences) instrument and the population of CD3-CD14-CD19+IgD-CD27+IgM-CD38+ was collected for subsequent analysis by 10x Genomics. Sorted B cells were then loaded into the Chromium Controller (10 × Genomics). Single-cell gene expression libraries were prepared using the Chromium Single-cell 5′ Reagent Kit (10x Genomics; V 2.0) according to manufacturer’s instructions. Samples were sequenced on the NovaSeq 6000 Sequencing System (Illumina) with HiSeq paired-end, 150bp reads for 10 × Single cell BCR (BCR libraries) and 10 × Single cell 5 Prime (gene expression).
Single B cell RNA-seq analysis
Single cell RNA-seq gene expression information from all subjects was processed using Seurat v4.1.1 [23] in R v4.1.0. To remove apoptotic or lysing cells, cells with a ≥10% of RNA transcripts from mitochondrial genes were excluded. To exclude poor quality cells, only cells with reads from > 400 features were retained. Read counts were log-normalized using a scaling factor of 104. To account for variability in gene expression, log-normalized read counts were then scaled and centered for each feature. The top 2000 variable genes were then identified using Seurat’s “vst” method. V, D, and J genes from the IGH, IGL, and IGK loci were removed so that the properties of the BCR expressed by the cell would remain independent of the cluster to which it was assigned. Seurat’s IntegrateData function was then used to combine data from both sequencing runs. Integration was performed using the previously identified top variable genes of each run, and the first 20 dimensions. Following integration, variable gene expression values were re-scaled and centered. This data was then reduced to the first 20 principal components. To annotate B cell subtypes, cells were clustered by Seurat’s shared nearest neighbor clustering algorithm with a resolution of 0.5. The B cell subtype of each cluster was then determined by gene expression correlations to cell types in the immunoStates database [5]. This resulted in 3 clusters identified as plasmablasts, 6 identified as memory B cells, and 2 identified as naïve B cells. These cell type annotations were verified using known marker genes for plasmablasts (PRDM1, XBP1), memory B cells (CD24, TNFRSF13B) and naïve B cells (IGHD, IL4R, TCL1A). One cluster of plasmablasts was further identified as “proliferating plasmablasts” due to high expression of MKI67.
B cell receptor sequence processing
Bulk and single cell B cell receptor sequences were obtained from four different data sources: 10X Genomics single cell RNAseq + BCR sequencing, bulk heavy-chain only BCR from New England Biolabs NEBNext sequencing kits, Takaras SMARTer® Human BCR IgG IgM H/K/L Profiling Kit, and previously published, processed bulk BCR sequences from the same patients, which we previously reported [43]. All BCR repertoire sequence data were analyzed using the Immcantation (www.immcantation.org) framework. Heavy and light chain BCR sequence data from 10X Genomics scRNA-seq + BCR sequencing began with the filtered V(D)J contigs from Cell Ranger. To obtain V and J gene assignments, these contigs were aligned to the IMGT GENE-DB [19] (v3.1.23, obtained August 3, 2019) human germline reference database using IgBlast v1.13.0 [80]. Preprocessing of NEBNext BCR sequences was performed using pRESTO v0.6.2 [71]. Quality control was first performed by removing all reads with a Phred quality score < 20. Reads which did not match to a constant region primer (maximum error rate 0.2) or template switch sequence (maximum error rate 0.5) were discarded. A unique molecular identifier (UMI) was assigned to each read using the first 17 nucleotides following the template switch site. Sequences within each UMI group were then collapsed into consensus sequences. Clusters with error rates exceeding 0.1 or majority isotype found in less than 60% of sequences were discarded. Positions containing more than 50% gap sequences were removed from the consensus. Mate-pairs were assembled into sequences with a minimum overlap of 8 nucleotides and a maximum error of 0.3. Isotypes were then assigned by local alignment of the 3’ end of the assembled sequences to known isotype-constant region sequences with a maximum error rate of 0.3. Duplicate sequences were collapsed except if assigned to different isotypes. Sequences represented by a single reconstructed mate-pair were discarded. To obtain V and J gene assignments, remaining sequences were aligned to the IMGT GENE-DB human reference database (v3.1.23, obtained August 3, 2019) using IgBlast v1.13.0.
Pacific Biosciences B cell receptor sequence preprocessing
HiFi reads generated from single molecule, real-time (SMRT) sequencing data were first demultiplexed using the Lima tool (https://github.com/PacificBiosciences/pbbioconda) based on the Illumina indices integrated during library construction. Sample-level demultiplexed reads were then further processed using pRESTO [71], following a similar workflow to that used for the NEBNext-generated libraries. Briefly, reads with Phred quality scores < 20 were removed, followed by the identification of the universal primer on the 5’ end of each read; reads with primer annotation error rates exceeding 0.3 were discarded. Sequences with the same UMIs were then clustered and aligned to generate collapsed consensus sequences representing each unique UMI. Consensus reads with < 2 representative sequences from the dataset were removed. The remaining reads for each sample were assigned to respective IGHV, IGHD, and IGHJ genes using IgBLAST with the IMGT database as the reference (downloaded February 21, 2022).
B cell clonal analysis
BCR sequences from all data sources were pooled together and grouped by subject. Nonproductive heavy chains were removed. Light chain sequences were excluded from clonal clustering analysis. To limit low-coverage sequences, all sequences with fewer than 300 unambiguous nucleotide characters (ATCG) were discarded. Novel IGHV alleles and subject-specific IGHV genotypes were inferred using TIgGER v1.0.0 [15]. To identify B cell clones, sequences were first partitioned based on common V and J gene annotations, as well as junction length. Within these groups, sequences differing from one another by a Hamming distance threshold of 0.15 within the junction region were clustered into clones using single linkage hierarchical clustering [20, 21] implemented in Change-O v1.2.0 [21]. This Hamming distance threshold was determined by manual inspection of distance to the nearest sequence neighbor plot using shazam v1.1.0 [79]. Three clones containing both high-throughput BCR sequences and monoclonal antibody sequences were identified. Unmutated germline V and J gene sequences were then reconstructed for each of these clones using the createGermlines function within dowser v1.0.0 [27]. To infer lineage trees, tree topologies, branch lengths, and substitution model parameters were estimated first under the GY94 model [46] and then under the HLP19 model [28]. All lineage tree analysis used IgPhyML v1.1.4 [28] and dowser v1.0.0 [27]. Trees were visualized using dowser v1.0.0, ggtree v3.0.4 [82], and ggplot v3.3.6 [76]. All B cell clonal analyses were performed using R v4.1.0. Scripts for performing B cell receptor sequence processing and analyses are available at https://bitbucket.org/kleinstein/projects.
Statistics
Statistical significance was assessed with Prism Software (GraphPad; version 8.0) by multiple comparison ANOVA with Dunnett’s correction for AChR clustering in the C2C12 assay.
Data Availability
Anonymized data will be shared on request from qualified investigators and completion of materials transfer agreements. The anonymized BCR sequencing data reported in this paper have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus Archive, https://www.ncbi.nlm.nih.gov/geo (BioProject ID: PRJNA886711; GEO accession: GSE215237). Code used in this study is available in a publicly accessible repository at https://bitbucket.org/ kleinstein/projects.
Results
Validation of fluorescently labeled MuSK
We generated a fluorescently labeled human MuSK ectodomain to specifically identify and isolate MuSK-specific B cells from patient samples (Additional file 1 : Supplement Fig. 1). We verified the utility of the labeled MuSK by testing it with a human B cell precursor line (Nalm6), which was modified to express a MuSK-specific human recombinant mAb (MuSK3-28) in the form of a BCR [11, 64, 67]. Fluorescently labeled recombinant human growth hormone (hGH) was used as a negative control antigen. The fluorescently labeled MuSK was tested over a broad range of concentrations (0.01, 0.1, 1.0 and 10 µg/mL), and showed strong binding to the Nalm6 cells, which expressed the MuSK3-28 BCR (Additional file 1: Supplement Fig. 1a, b). No binding of either antigen was observed with the unmodified Nalm6 cells (Additional file 1: Supplement Fig. 1c) or with the hGH tested on the Nalm6 cells, expressing the MuSK3-28 BCR.
Generation of MuSK-specific human mAbs from circulating B cells
We developed a flow cytometry panel to isolate MuSK-specific B cells (CD3-CD14-CD19+IgD-CD27+IgM-MuSK+) using the fluorescently labeled MuSK antigen (Additional file 1: Supplement Fig. 2). We started with 107 PBMCs from each of twelve samples, derived from nine unique MuSK-MG patients (Additional file 1: Supplement Table 1), then after B cell enrichment, sorted single MuSK positive B cells. These single B cells (N=672) were individually cultured using a well-established system that induces differentiation to antibody-secreting cells (ASC) such that the supernatant can be screened for antibody specificity [66]. Screening identified two different MuSK autoantibody-expressing cells isolated from patients MuSK MG-1 and MuSK MG-4 (Fig. 1a; Table 1). Index sorting showed that both originated from plasmablasts (Table 1), subclass PCRs showed that both expressed IgG4, and they had acquired somatic mutations including those coding for variable region glycosylation sites. Next, mAbs (referred to as 2E6 and 6E6) were generated from the BCRs of these two cells and their MuSK binding specificity was tested with three independent techniques. The mAbs bound MuSK over a broad range of concentrations (ranging from ≈0.03-10µg/mL) in a live cell-based assay (Fig. 1b; Additional file 1: Supplement Fig. 3). This binding specificity was also verified by a clinical radioimmunoassay (Additional file 1: Supplement Table 2) tested with both MuSK and AChR; both mAbs bound only to the former. Additionally, binding to MuSK expressed on muscle tissue was tested by immunofluorescent staining of murine neuromuscular junctions. Both mAbs bound to MuSK, closely associated with the AChR at the neuromuscular junction (Additional file 1: Supplement Fig. 4).

MuSK autoantibody binding. A live cell-based assay (CBA) was used to screen B cell culture media for MuSK IgG and to validate the binding of the human MuSK specific mAbs 2E6 and 6C6. a To generate MuSK mAbs, patient-derived B cells were sorted for single cell culture, after which the secreted IgG was tested for MuSK-specificity using a CBA. The contour plots from this screening show that MuSK-specific IgG are present in the supernatant of two culture wells from which the mAbs 2E6 and 6C6 were subsequently derived. The x-axis represents GFP fluorescence intensity and, consequently, the fraction of HEK cells transfected with MuSK. The y-axis represents Alexa Fluor 647 fluorescence intensity, which corresponds to secondary anti–human IgG Fc antibody binding and, consequently, primary antibody binding to MuSK. Hence, transfected cells are located in the right quadrants and cells with MuSK antibody binding in the upper quadrants. b Binding to MuSK by mAbs 2E6 and 6C6 was tested over a series of ten two-fold dilutions ranging from 10-0.02 µg/ml. The ∆MFI was calculated by subtracting the signal acquired from non-transfected cells from the signal of transfected cells. The MuSK-specific human mAb MuSK1A was used as a positive control and the AChR-specific human mAb 637 used as a negative control. Each data point represents the mean value from three independent experiments. Bars or symbols represent means and error bars SDs. Values greater than the mean + 4SD of the negative control mAb at 1.25 µg/ml (indicated by the horizontal dotted line) were considered positive
Characterization of MuSK mAbs 2E6 and 6C6
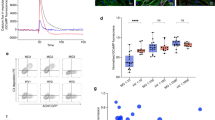
The extracellular domain of MuSK is comprised of three Ig-like domains and a frizzled-like domain (Fig. 2a). Most pathogenic MuSK autoantibodies recognize the Ig1-like domain [29, 30] while a smaller subset bind to the Ig2-like domain [67]. We found that both 2E6 and 6C6 bound to the Ig1-like domain of MuSK and showed no reactivity towards other domains (Fig 2b, c). Valency plays an important role in the pathogenic capacity of MuSK autoantibodies. Monovalent antibodies or Fabs—emulating fab-arm exchanged IgG4—are more potent in disrupting the MuSK-LRP4 interaction which is necessary for the clustering and functionality of the AChR [14, 29, 73]. Therefore, we evaluated the pathogenic capacity of 2E6 and 6C6 as both divalent mAbs and monovalent Fabs by using an established in vitro AChR clustering assay [67]. This assay specifically evaluates the capability of autoantibodies to interfere with AChR-clustering, which is dependent upon the MuSK-LRP4 pathway. The number of AChR clusters that formed in response to agrin alone was assigned a value of 100%, and the number that formed in the presence of the mAb or monovalent Fab (tested at equimolar concentrations) was expressed relative to this value. The mAb 2E6 reduced the number of clusters by 53% and mAb 6C6 by 71%. The monovalent Fab of 2E6 reduced the AChR clusters by 61%, while the monovalent Fab of 6C6 reduced the clusters by 96%, similar to the Fab of the positive control MuSK-specific human mAb MuSK1A (Fig 2d).

Epitope specificity and pathogenic capacity of MuSK-specific mAbs 2E6 and 6C6. The mAbs 2E6 and 6C6 were tested for domain binding with a CBA expressing MuSK-GFP domain variants. a Illustration of the full-length MuSK domains. b, c The ectodomain of MuSK consists of several different Ig-like domains and a frizzled-like domain. Different mutations of the MuSK protein either consisting of a domain deletion or specific domain-only construct were tested for binding by the mAbs. MuSK-specific human mAbs MuSK1A and MuSK3B were used as positive controls (MuSK1A for the Ig2-like domain and MuSK3B for frizzled-like domain) and the AChR-specific human mAb 637 as the negative control. The mAbs were added at a concentration of 10 µg/ml. Results for each mAb are shown. The ∆MFI was calculated by subtracting the signal acquired from non-transfected cells from the signal of transfected cells. Each data point represents a separate replicate within the same experiment, which was measured in triplicate. Bars represent means and error bars SDs. Values greater than the mean + 4SD of the negative control mAb 637, indicated by horizontal dotted lines, were considered positive. d AChR-clustering assay in C2C12 mouse myotubes demonstrates pathogenic capacity of MuSK mAbs. The presence of agrin in C2C12 myotube cultures leads to dense clustering of AChRs that can be readily visualized with fluorescent α-bungarotoxin and then quantified. Pathogenic MuSK autoantibodies disrupt this clustering. The mAbs 2E6 and 6C6 were tested for their ability to disrupt the AChR clustering. They were tested as divalent mAbs (1µg/mL) and monovalent Fabs (0.3µg/mL). Clonal variant, CVA, of mAb 2E6 was tested with either the mature (mutated) or an unmutated common ancestor (UCA) of the light chain from mAb 2E6, given that the clonal variants were identified with heavy chain-only sequencing. Quantitative measurements of the C2C12 clustering were normalized to the agrin-only effect of each individual experiment. Each data point represents the mean value from 2-8 individual values from a total of 4-10 independent experiments. Bars represent the mean of means and error bars SDs. Multiple comparisons ANOVA (against the pooled results for the three human non- MuSK-specific mAbs), Dunnett’s test; * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, only shown when significant
Unmutated common ancestors (UCA) approximate germline encoded versions of mature antibodies. UCA versions of MuSK mAbs can bind to the cognate self-antigen with high affinity [14]. Therefore, we investigated whether the UCAs of 2E6 and 6C6 can bind to MuSK. We found that the UCA of 6C6 was able to bind to MuSK over a broad range of concentrations (10 - 0.3 µg/ml), while the UCA of 2E6 only showed binding at the highest concentration tested (10 µg/ml) (Additional file 1: Supplement Fig. 5). Next, we tested whether the binding of the mAbs and their CA counterparts is a consequence of polyspecificity (Additional file 1: Supplement Fig. 6). We used a well-established approach to test binding to insulin, dsDNA, LPS and a HEp-2 cell lysate [75]. We found that 6C6, the UCA of 6C6 and 2E6 were neither polyreactive nor specific for the HEp-2 lysate, while the UCA of 2E6 was both polyreactive and specific for the HEp-2 lysate (Additional file 1: Supplement Fig. 6a, b).
Phenotype of plasmablasts in MuSK MG at time of relapse after BCDT
The 2E6 and 6C6 mAbs were both isolated from IgG4-expressing plasmablasts during post-BCDT relapse, as were several human MuSK mAbs that we previously produced [64, 67]. Given the increased frequency of plasmablasts in MuSK MG patients during relapse and that these cells can express pathogenic autoantibodies, we sought to explore whether their gene expression profile displayed any unique characteristics, such as expression of pro-survival genes that have been associated with BCDT resistance [9].
To that end, we used single cell gene expression profiling to investigate the phenotype of the plasmablasts at the time of relapse. Single-cell BCR sequencing was performed at the same time to search for clones related to 2E6 and 6C6, and also to explore the MuSK specificity of the IgG4-expressing plasmablast compartment. We used the specific samples from which 2E6 and 6C6 were obtained; MG-1 timepoint 70 months and MG-4 timepoint 25 months respectively (Additional file 1: Supplement Tables 1 and 3). B cells were sorted to enrich for plasmablasts (CD3-CD14-CD19+IgD-CD27+IgM-CD38+), but with gating that did not entirely exclude other B cell phenotypes. By clustering cells based on gene expression information, we identified 11 distinct clusters, annotated as naïve, memory and plasmablast populations (Fig. 3a, b; Additional file 1: Supplement Fig. 7). We found IgG4 enriched among specific populations in the plasmablast and memory B cell clusters (Additional file 1: Supplement Figs. 7b, 8c). Plasmablasts were defined by expression of XBP1, IRF4, PRDM1 and high levels of somatic hypermutations (Cluster 2,8 and 10; Fig. 3c, Additional file 1: Supplement Fig. 7). One plasmablast cluster (number 10) expressed high levels of MKI67, which is associated with proliferation (Fig. 3a, b, c). We found that CD20 (MS4A1) was expressed at low levels in 25% of the cells within the plasmablast subpopulations, while low level expression of CD19 was found in 25-50% of the population (Fig. 3b). The expression of high levels of TACI (TNFRSF13B) and BCMA (TNFRSF17) ranged between 25-100% in all three plasmablast subpopulations, while the expression levels of BAFF-R (TNFRSF13C) were low (0–10%) (Fig. 3b). Using the single-cell BCR sequencing data, we did not identify clonal relatives of 2E6 or 6C6 indicating that MuSK-specific clones comprise only a small fraction of circulating plasmablasts in these patients.

Single cell RNAseq characterization of MuSK MG patient B cells at the time of relapse. Single cell transcriptional profiling was used to characterize B cell samples from patients MG-1 and MG-4 taken at timepoint 70 and 25 months respectively, when MuSK-specific mAbs 2E6 and 6C6 were isolated. Both patients were experiencing a relapse at the time of sample collection. a, b Uniform Manifold Approximation and Projection (UMAP) of B cells showing 10 populations. Each point represents a single cell. c Dot plots showing expression of selected B cell marker genes for naive, memory, and plasmablast B cell subsets for the clusters identified in (a, b)
MuSK-specific B cell clones persist through BCDT and reemerge during relapses
We next investigated whether we could detect historic clonal variants of the pathogenic mAbs 2E6 and 6C6. We had collected serial samples from patient MG-1 (from whom mAb 2E6 was cloned) that made it possible to follow the clinical and treatment course for 79 months, and from patient MG-4 (from whom mAb 6C6 was cloned) for 25 months (Additional file 1: Supplement Table 3). We produced BCR repertoire libraries using bulk heavy chain only RNA sequencing and single-cell BCR sequencing to collectively obtain 50,948 unique clones for patient MG-1 and 13,029 clones for patient MG-4 (Additional file 1: Supplement Table 4). We identified B cell clones–cells that descend from a common V(D)J rearrangement–by clustering BCR heavy chain sequences by sequence similarity (Methods, Additional file 1: Supplement Fig. 9). No historic clones or clonal variants (CV) of mAb 6C6 were found. However, we identified three clonal variants of 2E6, two of which (CVA and CVB) were identified in samples collected prior to that which harbored the plasmablast that produced mAb 2E6, and one (CVC) was identified in the sample that produced mAb 2E6 (Fig. 4a; Additional file 1: Supplement Table 3).

Characteristics of a persistent MuSK-specific B cell clone. Clonal variants of the MuSK-specific mAb 2E6 were identified in longitudinally collected samples from patient MuSK MG-1. a Clonal lineage containing BCR sequences from bulk RNA sequencing of serial samples collected from patient MuSK MG-1 from whom mAb 2E6 was cloned. A maximum likelihood tree of the 2E6 clone, with clonal variants CVA, CVB, and CVC is shown. Edge lengths represent the expected number of intervening somatic mutations between nodes (see scale bar). Colors correspond to the collection time point (months) at which each clone (sequence) was identified in relation to the first available collection time point. b Binding to MuSK by mAb 2E6 and clonal variants was tested in a CBA over a series of ten two-fold dilutions of each mAb ranging from 10-0.02 µg/ml. Clonal variants, CVA, CVB and CVC were each tested with either the mature (mutated) or an unmutated common ancestor (UCA) of the light chain from mAb 2E6, given that the clonal variants were identified with heavy chain-only sequencing. The MuSK-specific human mAb MuSK1A was used as the positive control and AChR-specific human mAb 637 as the negative control. The ∆MFI was calculated by subtracting the signal acquired with the non-transfected cells from the signal of transfected cells. Each data point represents the mean value from three independent experiments. Bars or symbols represent means and error bars SDs. Values greater than the mean + 4SD of the negative control mAb at 1.25 µg/ml (indicated by the horizontal dotted line) were considered positive. c The x-axis depicts the time in months representing the clinical course of patient MuSK MG-1 from whom the mAb 2E6 was isolated. The bold timepoints indicate longitudinal sample collection and clinical assessment; the italicized timepoints indicate BCDT administration. The left y-axis indicates the MuSK autoantibody titer at each timepoint. The autoantibody titer was measured by CBA using 10 two-fold dilutions ranging from 1:20 to 1:10240. The right y-axis shows the MGC score at each timepoint. Dotted vertical lines indicate the timepoints where the patient received rituximab mediated BCDT. Colored boxes indicate the timepoints at which both clonal variants and the original 2E6 mAb were identified. The blue bars show the MGC score, while the green bars show the autoantibody titer
We next tested whether the other members of the 2E6 clone had pathogenic capacity. Given that bulk-RNA sequencing provided only the heavy chain of each clonal variant, we confirmed the specificity of the variants for MuSK by pairing the heavy chain with either the mature or UCA light chain of 2E6 to approximate the range of light chain sequence variation within the clone. We found that all clonal variants of 2E6 bound to MuSK over a wide range of concentrations when paired with either the mature or UCA light chain of 2E6 (10-0.02 µg/ml) (Fig. 4b). The mAb of 2E6 originated from a polyspecific germline-encoded UCA antibody that gained specificity towards MuSK over time through affinity maturation (Additional file 1: Supplement Fig. 6). Thus, we tested whether the earliest-sampled clonal variant of 2E6 (2E6 CVA) showed pathogenic capacity and specific binding to MuSK. We found that 2E6 CVA showed pathogenic capacity in vitro (Fig. 2d). 2E6 CVA paired with the mature light chain reduced the AChR clusters by 43% and 2E6 CVA paired with the UCA light chain by 76% (Fig. 2d). Neither recombinant variants of 2E6 CVA (VL mature and VL UCA) were polyreactive, nor did they react to HEp-2 (Additional file 1: Supplement Fig. 6a, b). Both variants bound to MuSK over a wide range of concentrations (10-0.02 µg/ml) (Fig. 4b). The MuSK MG patient harboring this clone had received rituximab-mediated BCDT 28 months before the patient presented with relapse at the first collection timepoint (Additional file 1: Supplement Table 3). The patient received BCDT six additional times over the course of 79 months (6.6 years) and received two cycles of BCDT between the identification of the first clonal variant 2E6 CVA and 2E6 CVB which was 31 months apart (Fig. 4c; Additional file 1: Supplement Table 3). Thus, 2E6 persisted through BCDT and reemerged over time.
MuSK autoantibody titer may be a biomarker for detecting relapse after BCDT-induced remission [68]. In agreement with this finding, we observed that the MuSK autoantibody titer started to increase several months prior to relapse (range: 7-9 months), while the patient was still free of symptoms (Fig. 4c; Additional file 1: Supplement Table 3). The titer remained at the same level during the subsequent relapse and decreased after BCDT (Fig. 4c; Additional file 1: Supplement Table 3). The clonal variants 2E6 CVA and 2E6 CVB were identified at timepoints that preceded relapse by two months (Fig. 4c). In summary, we found three unique clonal variants of 2E6, which persisted through BCDT. Two of these clonal variants were found during the time at which MuSK autoantibody titer increased and importantly, two months prior to relapse. Together, these results demonstrate the existence of pathogenic B cell clones that survive BCDT and emerge before clinically-detected relapse in MuSK MG.
Discussion
Most MuSK MG patients respond very well to rituximab-mediated BCDT with a rate of remission approaching 100% [10, 33]. However, these patients often experience relapse after approximately 1-3.5 years depending on the rituximab treatment regime [7], and a minority of patients do not respond to BCDT therapy [44]. Plasmablast and memory B cell populations are increased at the time of relapse [64, 67] and a subpopulation of these B cells produce MuSK-reactive antibodies [64]. These B cells can, in part, be traced back to clones that existed before BCDT [31].
The isolation of the two mAbs, 6C6 and 2E6, was achieved through enriching PBMCs for IgG-expressing memory B cells and plasmablasts that bound to a soluble MuSK antigen. This process required screening of 2.6 x 106 B cells from 12 patient samples; 672 of which were MuSK positive and sorted into plates. Of these 672 sorted cells, two clones were validated and 10 showed high MuSK reactivity, as measured by flow cytometry during the sorting procedure. Although the technical approach is not without limitations, it appears that B cells producing MuSK autoantibodies are exceptionally rare, and that their frequency varies through the course of disease. These findings are consistent with other studies; our own and from other groups who have isolated MuSK mAbs [29, 64, 67]. Furthermore, the number of unique clones in an individual patient appears to be low. In this study we isolated only one clone from each patient and in our other studies we isolated as few as one or two clones from single patients. Although the plasmablast compartment frequency—relative to the B cell population—increases at disease exacerbation, it appears that MuSK-specific plasmablasts make up a very minor population of this expanded compartment. This observation contrasts with findings from studies of acute responses to environmental antigens, where expanded plasmablast clones producing antibodies to COVID or influenza antigens are predominantly antigen-specific and include a diverse clonal repertoire [70, 78]. MuSK autoimmunity may differ in this regard in that only few clones—or even a single clone—could contribute to the production of circulating pathogenic autoantibody, and the expanded plasmablast compartment appears to include many specificities, few of which are MuSK-specific.
The characterization of the MuSK mAbs 2E6 and 6C6 showed that MuSK autoantibodies are heterogenous. Although both MuSK mAbs are not polyreactive, their pathogenic development is quite different. Previously, we had found that the UCA of MuSK mAbs recognize their cognate self-antigen without being polyreactive and that these antibodies develop exceptionally high affinities through the process of affinity maturation necessary to reach their pathogenic potential [14]. These self-reactive antibodies within the naïve B cell repertoire are most likely the consequence of impaired elimination of self-/poly-reactive clones during central and peripheral tolerance checkpoints during B cell development [37, 45, 75]. The approximated naïve precursor (UCA) of 6C6 does recognize MuSK and it was not polyreactive—consistent with our previous findings [14]. However, 2E6 developed from a polyreactive precursor gaining specificity for MuSK through affinity maturation. Furthermore, 2E6 and 6C6 differ in terms of how valency effects their pathogenic capacity. The pathogenic capacity of 6C6 increases as a monovalent Fab, while 2E6 shows similar pathogenic capacity when expressed as a monovalent Fab and divalent mAb. Although it was shown that monovalency increases the pathogenic effect of MuSK antibodies [14, 29, 35, 73], it was also found that an autoantibody requires high affinity towards MuSK to be pathogenic [14]. Therefore, possible explanations for the differing results might be different affinities and binding kinetics of mAbs 2E6 and 6C6, or that heteroligation may be relevant for mAb 2E6.
We demonstrated that the pathogenic 2E6 clone persists through BCDT. The long-term survival of this clone could be the consequence of tissue-based homing, as rituximab is not highly effective in depleting B cells localized in tissues as it is at targeting those in the circulation [2, 36, 42, 54]. Although plasmablasts can express CD20 [31, 51, 64], we found that 25% of plasmablasts in these patients express low levels of CD20 at the time of relapse. Therefore, clone 2E6 might originate from a plasmablast with low expression of CD20 affording it survival through BCDT. Thus, it might be beneficial to use therapies targeting other surface molecules to improve targeting of disease relevant B cell subsets. Recently, new therapies have been developed that deplete B cells, including therapies targeting CD19 [1, 6, 8, 60]. Inebilizumab (anti-CD19) has been approved for the treatment of neuromyelitis optica spectrum disorder (NMOSD) [8], and a clinical trial on the efficacy of inebilizumab in MG (MINT; ClinicalTrials.gov Identifier: NCT04524273) is currently ongoing. We additionally detected high levels of the receptors TACI and BCMA within the plasmablast subpopulations, but low levels of BAFF-R. TACI and BCMA are part of the BAFF/APRIL-system that regulates the survival of B cells [41, 57]. BAFF-R is the third receptor of the system and the detection of low levels of BAFF-R fits well with previous studies, and is indicative of poor response to treatment with rituximab [3, 31, 58]. The low expression of BAFF-R negatively correlates with BAFF levels [61] and high levels of BAFF are associated with autoimmune diseases, including MG [41] [34, 39]. Hence, the BAFF/APRIL axis has been considered as a valuable therapeutic target in the context of autoimmunity and B cell malignancies [38, 65]. Belimumab (anti-BAFF) was already investigated as an add-on therapy in generalized MG patients and showed a subtle positive effect [24]. More recently, chimeric antigen receptor (CAR)-T cells targeting BCMA and/or TACI and anti-BCMA mAbs show promising effects in the treatment of multiple myeloma [38, 53], and autoimmune disease [12], including an ongoing clinical trial in generalized MG (ClinicalTrials.gov Identifier: NCT04146051).
In summary, we generated two new MuSK mAbs that bind to the Ig1-like domain of MuSK and show pathogenic capacity in vitro. These autoantibodies were isolated from a sample collected at the time of relapse after BCDT and originated from plasmablasts. The phenotype of the expanded plasmablast population at time of relapse showed variable expression levels of CD20 and CD19, identifying these cells as potential candidates for BCDT, but highlighting that a subpopulation may escape deletion. Clonal variants of a pathogenic, MuSK-specific B cell were identified prior to relapse together with increased MuSK autoantibody levels, both of which may serve as valuable prognostic biomarkers for predicting post-BCDT relapse [68].
References
Agius MA, Klodowska-Duda G, Maciejowski M, Potemkowski A, Li J, Patra K, Wesley J, Madani S, Barron G, Katz E et al (2019) Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Multiple Scler J 25:235–245. https://doi.org/10.1177/1352458517740641
Anolik JH, Barnard J, Owen T, Zheng B, Kemshetti S, Looney RJ, Sanz I (2007) Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum 56:3044–3056. https://doi.org/10.1002/art.22810
Becerra E, De La Torre I, Leandro MJ, Cambridge G (2017) B cell phenotypes in patients with rheumatoid arthritis relapsing after rituximab: expression of B cell-activating factor-binding receptors on B cell subsets. Clin Exp Immunol 190:372–383. https://doi.org/10.1111/cei.13024
Blum S, Gillis D, Brown H, Boyle R, Henderson R, Heyworth-Smith D, Hogan P, Kubler P, Lander C, Limberg N et al (2011) Use and monitoring of low dose rituximab in myasthenia gravis. J Neurol, Neurosurg Psychiat 82:659–663. https://doi.org/10.1136/jnnp.2010.220475
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnol 36:411–420. https://doi.org/10.1038/nbt.4096
Chen D, Blazek M, Ireland S, Ortega S, Kong X, Meeuwissen A, Stowe A, Carter L, Wang Y, Herbst R et al (2014) Single dose of Glycoengineered Anti-CD19 antibody (MEDI551) disrupts experimental autoimmune encephalomyelitis by inhibiting pathogenic adaptive immune responses in the bone marrow and spinal cord while preserving peripheral regulatory mechanisms. J Immunol 193:4823–4832. https://doi.org/10.4049/jimmunol.1401478
Cortés-Vicente E, Rojas-Garcia R, Díaz-Manera J, Querol L, Casasnovas C, Guerrero-Sola A, Muñoz-Blanco JL, Bárcena-Llona JE, Márquez-Infante C, Pardo J et al (2018) The impact of rituximab infusion protocol on the long-term outcome in anti-MuSK myasthenia gravis. Ann Clin Trans Neurol 5:710–716. https://doi.org/10.1002/acn3.564
Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, Fujihara K, Paul F, Cutter GR, Marignier R et al (2019) Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. https://doi.org/10.1016/S0140-6736(19)31817-3
Crickx E, Chappert P, Sokal A, Weller S, Azzaoui I, Vandenberghe A, Bonnard G, Rossi G, Fadeev T, Storck S et al (2021) Rituximab-resistant splenic memory B cells and newly engaged naive B cells fuel relapses in patients with immune thrombocytopenia. Sci Transl Med. https://doi.org/10.1126/scitranslmed.abc3961
Diaz-Manera J, Martinez-Hernandez E, Querol L, Klooster R, Rojas-Garcia R, Suarez-Calvet X, Munoz-Blanco JL, Mazia C, Straasheijm KR, Gallardo E et al (2012) Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 78:189–193. https://doi.org/10.1212/WNL.0b013e3182407982
Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G et al (2016) Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353:179–184. https://doi.org/10.1126/science.aaf6756
Feng J, Xu H, Cinquina A, Wu Z, Chen Q, Zhang P, Wang X, Shan H, Xu L, Zhang Q et al (2021) Treatment of Aggressive T Cell Lymphoblastic Lymphoma/leukemia Using Anti-CD5 CAR T Cells. Stem Cell Rev Rep 17:652–661. https://doi.org/10.1007/s12015-020-10092-9
Fichtner ML, Jiang R, Bourke A, Nowak RJ, O’Connor KC (2020) Autoimmune pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology. Front Immunol 11:776. https://doi.org/10.3389/fimmu.2020.00776
Fichtner ML, Vieni C, Redler RL, Kolich L, Jiang R, Takata K, Stathopoulos P, Suarez PA, Nowak RJ, Burden SJ et al (2020) Affinity maturation is required for pathogenic monovalent IgG4 autoantibody development in myasthenia gravis. J Exp Med. https://doi.org/10.1084/jem.20200513
Gadala-Maria D, Yaari G, Uduman M, Kleinstein SH (2015) Automated analysis of high-throughput B-cell sequencing data reveals a high frequency of novel immunoglobulin V gene segment alleles. Proc Natl Acad Sci U S A: https://doi.org/10.1073/pnas.1417683112
Gilhus NE (2016) Myasthenia gravis. N Engl J Med 375:2570–2581. https://doi.org/10.1056/NEJMra1602678
Gomez AM, Stevens JA, Mane-Damas M, Molenaar P, Duimel H, Verheyen F, Cossins J, Beeson D, De Baets MH, Losen M et al (2016) Silencing of Dok-7 in adult rat muscle increases susceptibility to passive transfer myasthenia gravis. Am J Pathol 186:2559–2568. https://doi.org/10.1016/j.ajpath.2016.05.025
Graus YF, de Baets MH, Parren PW, Berrih-Aknin S, Wokke J, van Breda Vriesman PJ, Burton DR (1997) Human anti-nicotinic acetylcholine receptor recombinant Fab fragments isolated from thymus-derived phage display libraries from myasthenia gravis patients reflect predominant specificities in serum and block the action of pathogenic serum antibodies. J Immunol 158:1919–1929
Guidicelli VCD, Bodmer J, MÅller W, Busin C, Mash S, Bontrop R, Marc L, Malik A, Lefranc MP (1997) IMGT, The International ImMunoGeneTics database. 25: 206-211
Gupta NT, Adams KD, Briggs AW, Timberlake SC, Vigneault F, Kleinstein SH (2017) Hierarchical clustering can identify B cell clones with high confidence in Ig repertoire sequencing data. J Immunol 198:2489–2499. https://doi.org/10.4049/jimmunol.1601850
Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH (2015) Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31:3356–3358. https://doi.org/10.1093/bioinformatics/btv359
Guptill JT, Sanders DB, Evoli A (2011) Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 44:36–40. https://doi.org/10.1002/mus.22006
Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M et al (2021) Integrated analysis of multimodal single-cell data. Cell 184:3573-3587.e3529. https://doi.org/10.1016/j.cell.2021.04.048
Hewett K, Sanders DB, Grove RA, Broderick CL, Rudo TJ, Bassiri A, Zvartau-Hind M, Bril V (2018) Randomized study of adjunctive belimumab in participants with generalized myasthenia gravis. Neurology 90:e1425–e1434. https://doi.org/10.1212/wnl.0000000000005323
Higuchi O, Hamuro J, Motomura M, Yamanashi Y (2011) Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 69:418–422. https://doi.org/10.1002/ana.22312
Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A (2001) Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 7:365–368
Hoehn KB, Pybus OG, Kleinstein SH (2022) Phylogenetic analysis of migration, differentiation, and class switching in B cells. PLoS Comput Biol 18:e1009885. https://doi.org/10.1371/journal.pcbi.1009885
Hoehn KB, Vander Heiden JA, Zhou JQ, Lunter G, Pybus OG, Kleinstein SH (2019) Repertoire-wide phylogenetic models of B cell molecular evolution reveal evolutionary signatures of aging and vaccination. Proc Natl Acad Sci U S A 116:22664–22672. https://doi.org/10.1073/pnas.1906020116
Huijbers MG, Vergoossen DL, Fillie-Grijpma YE, van Es IE, Koning MT, Slot LM, Veelken H, Plomp JJ, van der Maarel SM, Verschuuren JJ (2019) MuSK myasthenia gravis monoclonal antibodies: Valency dictates pathogenicity. Neurol Neuroimmunol Neuroinflamm 6:e547. https://doi.org/10.1212/NXI.0000000000000547
Huijbers MG, Zhang W, Klooster R, Niks EH, Friese MB, Straasheijm KR, Thijssen PE, Vrolijk H, Plomp JJ, Vogels P et al (2013) MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci U S A 110:20783–20788. https://doi.org/10.1073/pnas.1313944110
Jiang R, Fichtner ML, Hoehn KB, Pham MC, Stathopoulos P, Nowak RJ, Kleinstein SH, O’Connor KC (2020) Single-cell repertoire tracing identifies rituximab-resistant B cells during myasthenia gravis relapses. JCI Insight: https://doi.org/10.1172/jci.insight.136471
Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, Caillot F, Golinski ML, Labeille B, Picard-Dahan C et al (2017) First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet (London, England) 389:2031–2040. https://doi.org/10.1016/s0140-6736(17)30070-3
Keung B, Robeson KR, DiCapua DB, Rosen JB, O’Connor KC, Goldstein JM, Nowak RJ (2013) Long-term benefit of rituximab in MuSK autoantibody myasthenia gravis patients. J Neurol Neurosurg Psychiatry 84:1407–1409. https://doi.org/10.1136/jnnp-2012-303664
Kim JY, Yang Y, Moon JS, Lee EY, So SH, Lee HS, Park KD, Choi YC (2008) Serum BAFF expression in patients with myasthenia gravis. J Neuroimmunol 199:151–154. https://doi.org/10.1016/j.jneuroim.2008.05.010
Koneczny I, Stevens JA, De Rosa A, Huda S, Huijbers MG, Saxena A, Maestri M, Lazaridis K, Zisimopoulou P, Tzartos S et al (2017) IgG4 autoantibodies against muscle-specific kinase undergo Fab-arm exchange in myasthenia gravis patients. J Autoimmun 77:104–115. https://doi.org/10.1016/j.jaut.2016.11.005
Leandro MJ, Cambridge G, Ehrenstein MR, Edwards JCW (2006) Reconstitution of peripheral blood B cells after depletion with rituximab in patients with rheumatoid arthritis. Arthritis & Rheumatism 54:613–620. https://doi.org/10.1002/art.21617
Lee JY, Stathopoulos P, Gupta S, Bannock JM, Barohn RJ, Cotzomi E, Dimachkie MM, Jacobson L, Lee CS, Morbach H et al (2016) Compromised fidelity of B-cell tolerance checkpoints in AChR and MuSK myasthenia gravis. Ann Clin Transl Neurol 3:443–454. https://doi.org/10.1002/acn3.311
Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, Onuoha S, Thomas S, Baldan V, Bughda R et al (2018) An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood 131:746–758. https://doi.org/10.1182/blood-2017-05-781351
Lisak RP, Ragheb S (2012) The role of B cell-activating factor in autoimmune myasthenia gravis. Ann New York Acad Sci 1274:60–67. https://doi.org/10.1111/j.1749-6632.2012.06842.x
Luo XM, Maarschalk E, O’Connell RM, Wang P, Yang L, Baltimore D (2009) Engineering human hematopoietic stem/progenitor cells to produce a broadly neutralizing anti-HIV antibody after in vitro maturation to human B lymphocytes. Blood 113:1422–1431. https://doi.org/10.1182/blood-2008-09-177139
Mackay F, Schneider P (2009) Cracking the BAFF code. Nature Rev Immunol 9:491–502. https://doi.org/10.1038/nri2572
Mamani-Matsuda M, Cosma A, Weller S, Faili A, Staib C, Garçon L, Hermine O, Beyne-Rauzy O, Fieschi C, Pers J-O et al (2008) The human spleen is a major reservoir for long-lived vaccinia virus–specific memory B cells. Blood 111:4653–4659. https://doi.org/10.1182/blood-2007-11-123844
Mandel-Brehm C, Fichtner ML, Jiang R, Winton VJ, Vazquez SE, Pham MC, Hoehn KB, Kelleher NL, Nowak RJ, Kleinstein SH et al (2021) Elevated N-linked glycosylation of IgG V regions in myasthenia gravis disease subtypes. J Immunol. https://doi.org/10.4049/jimmunol.2100225
Marino M, Basile U, Spagni G, Napodano C, Iorio R, Gulli F, Todi L, Provenzano C, Bartoccioni E, Evoli aA, (2020) Long lasting rituximab-induced reduction of specific - but not of total- IgG4 in MuSK-positive myasthenia gravis. Front Immunol. https://doi.org/10.3389/fimmu.2020.00613
Meffre E, O’Connor KC (2019) Impaired B-cell tolerance checkpoints promote the development of autoimmune diseases and pathogenic autoantibodies. Immunol Rev 292:90–101. https://doi.org/10.1111/imr.12821
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936. https://doi.org/10.1093/genetics/148.3.929
Nowak RJ, Coffey CS, Goldstein JM, Dimachkie MM, Benatar M, Kissel JT, Wolfe GI, Burns TM, Freimer ML, Nations S et al (2021) Phase 2 trial of rituximab in acetylcholine receptor antibody-positive generalized myasthenia gravis: The BeatMG Study. Neurology 98: 376-389 doi https://doi.org/10.1212/wnl.0000000000013121
Nowak RJ, DiCapua DB, Zebardast N, Goldstein JM (2011) Response of patients with refractory myasthenia gravis to rituximab: a retrospective study. Therapeutic Adv Neurol Dis 4:259–266. https://doi.org/10.1177/1756285611411503
Owens GP, Bennett JL, Lassmann H, O’Connor KC, Ritchie AM, Shearer A, Lam C, Yu X, Birlea M, Dupree C et al (2009) Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann Neurol 65:639–649. https://doi.org/10.1002/ana.21641
Pevzner A, Schoser B, Peters K, Cosma NC, Karakatsani A, Schalke B, Melms A, Kroger S (2012) Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 259:427–435. https://doi.org/10.1007/s00415-011-6194-7
Quach TD, Rodriguez-Zhurbenko N, Hopkins TJ, Guo X, Hernandez AM, Li W, Rothstein TL (2016) Distinctions among circulating antibody-secreting cell populations, including B-1 cells, in human adult peripheral blood. J Immunol 196:1060–1069. https://doi.org/10.4049/jimmunol.1501843
Querol L, Rojas-García R, Diaz-Manera J, Barcena J, Pardo J, Ortega-Moreno A, Sedano MJ, Seró-Ballesteros L, Carvajal A, Ortiz N et al (2015) Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurology(R) neuroimmunology & neuroinflammation https://doi.org/10.1212/nxi.0000000000000149
Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, Liedtke M, Rosenblatt J, Maus MV, Turka A et al (2019) Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. The New England journal of medicine 380:1726–1737. https://doi.org/10.1056/NEJMoa1817226
Ramwadhdoebe TH, van Baarsen LGM, Boumans MJH, Bruijnen STG, Safy M, Berger FH, Semmelink JF, van der Laken CJ, Gerlag DM, Thurlings RM et al (2019) Effect of rituximab treatment on T and B cell subsets in lymph node biopsies of patients with rheumatoid arthritis. Rheumatology 58:1075–1085. https://doi.org/10.1093/rheumatology/key428
Robeson KR, Kumar A, Keung B, DiCapua DB, Grodinsky E, Patwa HS, Stathopoulos PA, Goldstein JM, O’Connor KC, Nowak RJ (2016) Durability of the Rituximab Response in Acetylcholine Receptor Autoantibody-Positive Myasthenia Gravis. JAMA Neurol: https://doi.org/10.1001/jamaneurol.2016.4190
Rodgaard A, Nielsen FC, Djurup R, Somnier F, Gammeltoft S (1987) Acetylcholine receptor antibody in myasthenia gravis: predominance of IgG subclasses 1 and 3. Clin Exp Immunol 67:82–88
Samy E, Wax S, Huard B, Hess H, Schneider P (2017) Targeting BAFF and APRIL in systemic lupus erythematosus and other antibody-associated diseases. Int Rev Immunol 36:3–19. https://doi.org/10.1080/08830185.2016.1276903
Sarantopoulos S, Stevenson KE, Kim HT, Washel WS, Bhuiya NS, Cutler CS, Alyea EP, Ho VT, Soiffer RJ, Antin JH et al (2011) Recovery of B-cell homeostasis after rituximab in chronic graft-versus-host disease. Blood 117:2275–2283. https://doi.org/10.1182/blood-2010-10-307819
Scadding GK, Vincent A, Newsom-Davis J, Henry K (1981) Acetylcholine receptor antibody synthesis by thymic lymphocytes: correlation with thymic histology. Neurology 31:935–943
Schiopu E, Chatterjee S, Hsu V, Flor A, Cimbora D, Patra K, Yao W, Li J, Streicher K, McKeever K et al (2016) Safety and tolerability of an anti-CD19 monoclonal antibody, MEDI-551, in subjects with systemic sclerosis: a phase I, randomized, placebo-controlled, escalating single-dose study. Arthritis Res Therapy 18:131. https://doi.org/10.1186/s13075-016-1021-2
Sellam J, Miceli-Richard C, Gottenberg JE, Ittah M, Lavie F, Lacabaratz C, Gestermann N, Proust A, Lambotte O, Mariette X (2007) Decreased B cell activating factor receptor expression on peripheral lymphocytes associated with increased disease activity in primary Sjögren’s syndrome and systemic lupus erythematosus. Ann Rheumat Dis 66:790–797. https://doi.org/10.1136/ard.2006.065656
Sng J, Ayoglu B, Chen JW, Schickel J-N, Ferre EMN, Glauzy S, Romberg N, Hoenig M, Cunningham-Rundles C, Utz PJ et al (2019) AIRE expression controls the peripheral selection of autoreactive B cells. Sci Immunol. https://doi.org/10.1126/sciimmunol.aav6778
Stathopoulos P, Kumar A, Heiden JAV, Pascual-Goni E, Nowak RJ, O’Connor KC (2018) Mechanisms underlying B cell immune dysregulation and autoantibody production in MuSK myasthenia gravis. Ann N Y Acad Sci 1412:154–165. https://doi.org/10.1111/nyas.13535
Stathopoulos P, Kumar A, Nowak RJ, O’Connor KC (2017) Autoantibody-producing plasmablasts after B cell depletion identified in muscle-specific kinase myasthenia gravis. JCI Insight 2:e94263–e94275. https://doi.org/10.1172/jci.insight.94263
Stohl W, Scholz JL, Cancro MP (2011) Targeting BLyS in rheumatic disease: the sometimes-bumpy road from bench to bedside. Curr Opin Rheumatol 23:305–310. https://doi.org/10.1097/BOR.0b013e328344c15e
Su K-Y, Watanabe A, Yeh C-H, Kelsoe G, Kuraoka M (2016) Efficient culture of human naive and memory B cells for use as APCs. J Immunol 197:4163–4176. https://doi.org/10.4049/jimmunol.1502193
Takata K, Stathopoulos P, Cao M, Mane-Damas M, Fichtner ML, Benotti ES, Jacobson L, Waters P, Irani SR, Martinez-Martinez P et al (2019) Characterization of pathogenic monoclonal autoantibodies derived from muscle-specific kinase myasthenia gravis patients. JCI Insight. https://doi.org/10.1172/jci.insight.127167
Triplett JD, Hardy TA, Riminton DS, Chu SYK, Reddel SW (2019) Association between musk antibody concentrations and the myasthenia gravis composite score in 3 patients: a marker of relapse? Muscle & nerve 60:307–311. https://doi.org/10.1002/mus.26609
Tse N, Morsch M, Ghazanfari N, Cole L, Visvanathan A, Leamey C, Phillips WD (2014) The neuromuscular junction: measuring synapse size, fragmentation and changes in synaptic protein density using confocal fluorescence microscopy. J Vis Exp: https://doi.org/10.3791/52220
Turner JS, O’Halloran JA, Kalaidina E, Kim W, Schmitz AJ, Zhou JQ, Lei T, Thapa M, Chen RE, Case JB et al (2021) SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature 596:109–113. https://doi.org/10.1038/s41586-021-03738-2
Vander Heiden JA, Yaari G, Uduman M, Stern JN, O’Connor KC, Hafler DA, Vigneault F, Kleinstein SH (2014) pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30:1930–1932. https://doi.org/10.1093/bioinformatics/btu138
Vazquez-Lombardi R, Nevoltris D, Luthra A, Schofield P, Zimmermann C, Christ D (2018) Transient expression of human antibodies in mammalian cells. Nature Protocols 13:99–117. https://doi.org/10.1038/nprot.2017.126
Vergoossen DLE, Plomp JJ, Gstottner C, Fillie-Grijpma YE, Augustinus R, Verpalen R, Wuhrer M, Parren P, Dominguez-Vega E, van der Maarel SM et al (2021) Functional monovalency amplifies the pathogenicity of anti-MuSK IgG4 in myasthenia gravis. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.2020635118
Vincent A (2002) Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol 2:797–804
Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC (2003) Predominant autoantibody production by early human B cell precursors. Science 301:1374–1377. https://doi.org/10.1126/science.10869071086907[pii]
Wickham H (2016) ggplot2: Elegant Graphics for Data Analysis Springer-Verlag New York, City
Willcox HN, Newsom-Davis J, Calder LR (1984) Cell types required for anti-acetylcholine receptor antibody synthesis by cultured thymocytes and blood lymphocytes in myasthenia gravis. Clin Exp Immunol 58:97–106
Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C et al (2008) Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453:667–671. https://doi.org/10.1038/nature06890
Yaari G, Vander Heiden JA, Uduman M, Gadala-Maria D, Gupta N, Stern JN, O’Connor KC, Hafler DA, Laserson U, Vigneault F et al (2013) Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front Immunol 4:358. https://doi.org/10.3389/fimmu.2013.00358
Ye J, Ma N, Madden TL, Ostell JM (2013) IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 41:W34-40. https://doi.org/10.1093/nar/gkt382
Yi JS, Guptill JT, Stathopoulos P, Nowak RJ, O’Connor KC (2018) B cells in the pathophysiology of myasthenia gravis. Muscle Nerve 57:172–184. https://doi.org/10.1002/mus.25973
Yu G, Lam TT, Zhu H, Guan Y (2018) Two methods for mapping and visualizing associated data on phylogeny using ggtree. Mol Biol Evol 35:3041–3043. https://doi.org/10.1093/molbev/msy194
Yu Z, Zhang M, Jing H, Chen P, Cao R, Pan J, Luo B, Yu Y, Quarles BM, Xiong W et al (2021) Characterization of LRP4/Agrin antibodies from a patient with myasthenia gravis. Neurology. https://doi.org/10.1212/wnl.0000000000012463
Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, Nussenzweig MC (2005) Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med 201:703–711. https://doi.org/10.1084/jem.20042251
Acknowledgements
We thank Dr. Gianvito Masi for critical reading of the manuscript, Drs. Lesley Devine and Chao Wang in the Yale Flow Cytometry Core for their assistance with cell sorting, and Lindsay Young for administrative support. The Yale Flow Cytometry Core is supported in part by an NCI Cancer Center Support Grant # NIH P30 CA016359. We also thank Yale Center for Genome Analysis and Keck Microarray Shared Resource for providing the necessary Pacific Biosciences sequencing services, which is funded, in part, by the National Institutes of Health instrument grant 1S10OD028669-01.
Funding
MLF is supported through a DFG Research fellowship (FI 2471/1-1). KCO is supported by the National Institute of Allergy and Infectious Diseases of the NIH under award numbers R01-AI114780 and R21-AI142198, and through an award provided through the Rare Diseases Clinical Research Consortia of the NIH and MGNet (award number U54-NS115054). SHK is supported by the National Institute of Allergy and Infectious Diseases of the NIH under award number R01AI104739. PMM and MMD were supported by the the Kootstra Talent Fellowship (Fall 2019, Maastricht University) and the ZonMW/NWO Aspasia Program [grant number 015.011.033] and a NIH grant (R21-AI142198). ASP and SO were supported by sponsored research from Cabaletta Bio. The funders had no role in the decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
This study was originally conceived, initiated, and directed by KCO and MLF. MLF led the laboratory work at Yale, designing, executing, and interpreting experiments associated with the B cell isolation, mAb expression, functional and binding assays, BCR sequencing, and 10x sample preparation. KBH and SHK performed the computational biology for the 10x and BCR sequencing analysis from the NEB data sets and PacBio sets transformed into the Change-O format. EEF, MLS and CTW performed the computational biology for the BCR sequencing analysis from the PacBio data sets. MMD, PMM, and ML performed and interpreted the mAb tissue staining on mouse muscle. SO and ASP engineered and generated the NALM cells expressing MuSK specific BCRs. PW performed the RIA binding studies and interpreted the data. RJN provided the clinical specimens from which the mAbs were derived and provided insight on the clinical and therapeutic relevance of the findings. The manuscript was initially drafted by MLF and KCO. All authors contributed substantially to the intellectual content of the work, and all discussed results, and made key contributions to editing and revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
KCO has received research support from Alexion, now part of AstraZeneca, and Viela Bio, now part of Horizon Therapeutics, and Cabaletta Bio. KCO is a consultant and equity shareholder of Cabaletta Bio. During the last two years, KCO has served as consultant/advisor for Alexion Pharmaceuticals, now part of AstraZeneca, and for Roche, and he has received speaking fees from Alexion, Roche, Genentech, Viela Bio, now part of Horizon Therapeutics, and UCB. MLF received a SPIN award from Grifols outside the submitted work and is a member of the Alexion-Akademie. KBH receives consulting fees from Prellis Biologics. SHK receives consulting fees from Peraton. ASP has received equity, research support, patent licensing and other payments from Cabaletta Bio; patent licensing payments from Novartis; and consultant fees from Janssen. SO has received patent licensing payments from Cabaletta Bio. The research of PMM and MMD at the School of Mental Health and Neuroscience, Faculty of Health, Medicine and Life Sciences was financially supported by Apellis, Argenx, Genmab, Neurotune and Takeda. PMM is a co-inventor of the following patent: Use of effector-function-deficient antibodies for treatment of auto-immune diseases (Patent number: 9181344). These competing interests played no role in the research design, reference collection, decision to publish, or preparation of the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary figures and tables.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fichtner, M.L., Hoehn, K.B., Ford, E.E. et al. Reemergence of pathogenic, autoantibody-producing B cell clones in myasthenia gravis following B cell depletion therapy. acta neuropathol commun 10, 154 (2022). https://doi.org/10.1186/s40478-022-01454-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-022-01454-0