
Abstract
Huntington disease is characterized by progressive neurodegeneration, especially of the striatum, and the presence of polyglutamine huntingtin (HTT) inclusions. Although HTT inclusions are most abundant in the neocortex, their neocortical distribution and density in relation to the extent of CAG repeat expansion in the HTT gene and striatal pathologic grade have yet to be formally established. We immunohistochemically studied 65 brains with a pathologic diagnosis of Huntington disease to investigate the cortical distributions and densities of HTT inclusions within the calcarine (BA17), precuneus (BA7), motor (BA4) and prefrontal (BA9) cortices; in 39 of these brains, a p62 immunostain was used for comparison. HTT inclusions predominate in the infragranular cortical layers (layers V-VI) and layer III, however, the densities of HTT inclusions across the human cerebral cortex are not uniform but are instead regionally contingent. The density of HTT and p62 inclusions (intranuclear and extranuclear) in layers V-VI increases caudally to rostrally (BA17 < BA7 < BA4 < BA9) with the median burden of HTT inclusions being 38-fold greater in the prefrontal cortex (BA9) than in the calcarine cortex (BA17). Conversely, intranuclear HTT inclusions prevail in the calcarine cortex irrespective of HTT CAG length. Neocortical HTT inclusion density correlates with CAG repeat expansion, but not with the neuropathologic grade of striatal degeneration (Vonsattel grade) or with the duration of clinical disease since motor onset. Extrapolation of these findings suggest that HTT inclusions are at a regionally-contingent, CAG-dependent, density during the advanced stages of HD. The distribution and density of HTT inclusions in HD therefore does not provide a measure of pathologic disease stage but rather infers the degree of pathogenic HTT expansion.
Similar content being viewed by others


Introduction
Hallmarks of many neurodegenerative diseases are the relative selective vulnerability of specific cell types to injury and the accretion of pathologic protein inclusions in the central nervous system (CNS). In Huntington disease (HD), the cardinal pathologic features are the worsening, topographic, degeneration of the neostriatum (caudate nucleus, putamen, and nucleus accumbens), in which the medium spiny neuron is the most vulnerable cell type, and the appearance of huntingtin inclusions due to abnormal polyglutamine (polyQ) expansion of the huntingtin protein (HTT) [13, 63, 64]. Although the brunt of neurodegeneration in HD is concentrated in the neostriatum, morphometric studies of extra-striatal brain regions, such as the frontal neocortex, white matter, thalamus and brainstem, collectively emphasize widespread degeneration in the HD brain [10, 22, 23, 38, 49,50,51,52]. Yet despite the well-described neurodegenerative changes that occur in the HD brain, data on the neuroanatomic distributions of pathologic inclusions are lacking. Furthermore, how these proteins relate to striatal neurodegeneration remain uncertain [49,50,51,52]. If aggregated HTT and its oligomers are toxic, then the distribution of these inclusions may be clinically significant and their removal would be therapeutically relevant [2, 3, 13, 16, 18, 41, 59, 66].
Wild-type huntingtin has a myriad of functions throughout life (reviewed in [54]) and is expressed in neurons of the central nervous system, predominating in the cytoplasm with a small proportion also present within the nucleus [14, 17, 60]. Antibodies directed against the N-terminal region of HTT have revealed visible pathologic HTT aggregates (inclusions) within the HD brain and in murine model systems of HTT expansion [13, 18, 25, 31, 37, 63]. In the HD brain, these inclusions may be found within neuronal nuclei (neuronal intranuclear inclusions) and outside the nucleus (extranuclear inclusions) as seen within the perikaryal cytoplasm or the cytoplasm of axons and dendritic spines as dystrophic neurites. Human postmortem studies have shown that HTT inclusions are most abundant in the neocortex, prevailing in layers III, V and VI, which correspond to the same layers that are most vulnerable to neuronal loss [13, 21, 58]. Brains with juvenile-onset HD show a greater number of intranuclear inclusions, whereas brains with adult-onset HD have mostly extranuclear inclusions [3, 9, 13, 18, 42]. But beyond this, immunohistochemical studies that have investigated the distribution of HTT aggregation in the cortex vary considerably, likely due to small sample sizes and variation of antibodies utilized. Herndon et al. applied the 1C2 antibody, which targets the polyQ stretch of the HTT protein, to a series of 19 HD brains and found widespread inclusions in the brains without noticeable differences of abundance between neocortices [25]. Conversely, Van Roon-Mom et al. utilized the EM48 antibody and found a higher density of inclusions in the prefrontal cortex (BA9) than in the posterior frontal cortex (BA4/6) in 7 HD brains [61]. The EM48 antibody was raised against a.a. 1–256 of the HTT protein with the epitope mapping to the C-terminus of HTT exon 1 [65]. Becher et al. found step-wise increases in intranuclear HTT inclusion density with higher CAG repeat expansions in the cortex of 20 HD brains using a variety of HTT-specific antibodies and demonstrated intercortical variability of intranuclear inclusion density [1]. Finally, Gutekunst et al. found that EM48-labeled HTT inclusions are present in the cortex before onset of clinical signs and are larger in grade 4 (n = 2) than in grade 1 brains (n = 2) [18]. Cortical differences in HTT densities were noted within individual HD brains but a defined distribution in 12 HD brains was not established.
Attempts to address any differential neurodegeneration in the HD neocortex have provided mixed results. Three morphometric studies that examined the macroscopic thickness of the cortex of coronal sections of the cerebral hemispheres demonstrated diffuse, even, and symmetric cortical thinning, except for relative sparing of the medial temporal lobe by Halliday et al. [10, 20, 61]. In contrast, one of the first volumetric studies on human HD tissue claimed that the occipital neocortex is the most severely afflicted in terms of neuronal loss and that the pattern of progression within the neocortex may be caudal to rostral [48]. Caudo-rostral progression of cortical thinning has also been proposed by Rosas et al. in addition to hemispherical asymmetry, with the left cerebral hemisphere being more involved than the right by structural MRI [47, 48]. However, the small series of stereological studies in HD by Rüb and others show similar degrees of neuronal loss (32–33% loss compared with control brains) between the calcarine cortex and other cortical regions arguing against differential neuronal loss in the neocortex [24, 51].
The entire neocortex sends corticostriatal glutamatergic projections to the neostriatum in a topographic manner, a subset of which synapse onto the medium striatal spiny neurons [32, 33]. Given these topographic projections to the neostriatum and the progression of caudal to rostral, medial to lateral, and dorsal to ventral degeneration in HD, if HTT is neurotoxic to cortical neurons, one might predict lower abundances of inclusions in the frontal neocortices that project to the dorsomedial head of the caudate nucleus and the sensorimotor regions of the putamen and a greater burden in the occipital and parietal neocortices that project to the more caudal aspects of the caudate nucleus in early stage HD brains [8, 19, 32, 57]. This assumption would also be dependent on the premise of uniform neuronal loss across the cortex. Alternatively, if neuronal degeneration is predominant within caudal neocortices, such as the calcarine cortex, then the density of HTT inclusions may be least in the occipital cortex in contrast with more rostral cortices at postmortem provided there is clearance of HTT following neuronal death.
With this in mind, we sought to characterize the neocortical distribution of pathologic inclusions in 65 HD brains. These were selected to encompass the spectrum of severity in HD, including juvenile- and adult-onset individuals with CAG repeat expansions ranging from 38- 71 repeats and ages at death between 24 and 89 years-of-age. HTT inclusions and their relationship to CAG repeat expansion, metrics of disease burden (Vonsattel grade, CAG age product (CAP) score, length of time from motor onset to death) were investigated. The selected distribution of HTT inclusions in the brain, correlation with CAG repeat expansion and relationship to grade and symptom duration are discussed.
Materials and methods
Ethical approval
All brain donors were consented for autopsy and brain donation. All procedures performed were in accordance with the ethical standards of the institution and with the 1964 Helsinki declaration and its later amendments.
Study design
The 65 brains of this study had a pathologic diagnosis of HD and known HTT CAG repeat size. All brains were processed using the standardized protocol in place at the New York Brain Bank as outlined previously [28, 62]. The half brain or whole brain that was assigned for neuropathologic diagnosis, was fixed by immersion in 10% neutral-buffered formalin solution for approximately two weeks and then cut and blocked as per the standardized New York Brain Bank protocol as previously described [62]. Both cerebral hemispheres were sliced in the coronal plane at 0.3 cm intervals. The Brodmann map was utilized for blocking cortical regions to allow for consistent comparisons of neocortical regions across multiple brains [7]. Formalin-fixed tissue blocks of BA17 (calcarine), BA7 (precuneus), BA4 (motor cortex/ leg region), and BA9 (prefrontal cortex/ superior frontal gyrus) were then processed, embedded in paraffin wax, sectioned perpendicular to the cortical surface at 7 μm thickness and collected on positively-charged slides. These four neocortical areas were chosen because they are aligned parasagittally enabling for caudal to rostral assessment without significant lateral deviation and are away from watershed territories so as to be less influenced by peri-mortem hypotension.
Immunohistochemical staining
Paraffin sections were immunostained on an automated immunostaining platform using a DAB with or without alkaline phosphatase dual staining system on a Roche Ventana staining platform (Table 1). Hematoxylin counterstains were performed. We used the 2B4 antibody which targets amino acids 1–82 of the N-terminal end of HTT to study HTT inclusions; this antibody has been validated in human HD brains previously [11, 12, 25, 28]. To corroborate the distribution of HTT, we also examined sections stained with an antibody directed against p62 in a subset of cases (n = 39). P62 is an autophagy adaptor protein that binds to ubiquitinated HTT for subsequent degradation by macroautophagy [4, 35].
Assessment of inclusion density
Immunohistochemically stained slides were blinded by demographics, HTT CAG repeat length, clinical disease duration, and neuropathologic grade before examination and analysis. All slides were reviewed by one board-certified neuropathologist (RAH). HTT and p62 inclusions were defined as a protein aggregate visible by light microscopy that were immunolabeled by their respective antibody (anti-HTT or anti-p62). Intranuclear inclusions were defined as inclusions localized to within the hematoxylin-counterstained nucleus; extranuclear inclusions were defined as any inclusion outside of the nucleus, i.e. within the visible perikaryal cytoplasm of a cell or as inclusions that appear within the neuropil. Since HTT inclusions predominate in the infragranular layers, quantification of inclusions was performed in cortical layers V–VI. For HTT quantification, inclusions labeled with brown chromogen with the huntingtin/ p62 double stain were analyzed. For p62 quantification, inclusions labeled with brown chromogen with the p62 single stain were analyzed. For both HTT and p62 quantification, three photomicrographs per cortical region (BA17, BA7, BA4, BA9) of layers V–VI were obtained at an original magnification of 200x. Then, using ImageJ 1.52P and the Cell counter plugin, intranuclear and extranuclear inclusions were manually counted per image field (RAH) [56]. The total inclusion count of three representative fields was calculated and then divided by the total field area to determine the inclusion density (number of inclusions per mm2).
Metrics of disease burden
Several measures have been proposed to assess disease severity. One reliable and widely used measure of neurodegeneration within the neostriatum is the Vonsattel grading system; all brains studied were diagnosed and graded either by JPV or RAH [64]. Another measure of disease burden is the CAP score (age × (CAG repeat length–33.66)/432.3326) [40, 67]. Age-at-death provides a measure of the length and severity of exposure to mutant HTT at the end of life. For individuals with a known age-of-onset of HD motor signs (n = 22), the duration of clinical disease was calculated by subtracting the age of clinical onset of motor signs from the age at death.
Statistical analysis
Statistical analyses were performed on GraphPad Prism version 9.1.2. Normality tests were performed on all numerical datasets using the D’Agostino Pearson test. For data that was not normally distributed, non-parametric tests were utilized. Average values are presented as means ± standard deviation (SD) if normally distributed, or as median ± interquartile range (IQR) if non-normal. Multiple comparisons of median inclusion densities between brain region were initially assessed by pairwise comparison using the Freidman test, followed by post-hoc Dunn’s multiple comparison test for regional differences. Correlational analyses between CAG repeat length and inclusion density were performed using Spearman’s rank correlational coefficient. Regression analyses assume linearity and significant differences in slopes of regression lines were assessed between BA9 and BA17 using Prism’s built-in function that is equivalent to the ‘analysis of covariance’ (ANCOVA) analysis [1]. A P value < 0.05 was deemed significant (*) and a P value < 0.01 was considered highly significant (**); P values < 0.001 and < 0.0001 were also designated on graphs as *** and ****, respectively.
Results
Cohort
The mean age at death of the 65 brain donors was 56.3 years ± 14.1 years (SD) and 31 (47.7%) individuals were men. The Vonsattel grade of HD neuropathologic changes ranged from 1–4 with a mean grade of 3 ± 0.83 (SD). The median CAG repeat expansion was 45 (IQR: 4).
Inclusion patterns and intracellular localization
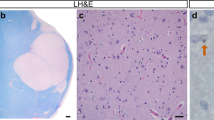
As previously described, both intranuclear and extranuclear HTT and p62 inclusions predominated within neocortical layers V-VI and III, with some inclusions present in the immediate subcortical white matter [13, 18]. In the adult-onset HD cases, extranuclear inclusions vastly outnumbered the intranuclear inclusions overall (Fig. 1a, b). In instances where the perikaryal cytoplasm could be discerned around its respective nucleus, inclusions were either found within the cytoplasm or within the nucleus, but never concomitantly in both compartments. Intranuclear inclusions were nearly always singular, whereas perikaryal inclusions could be singular or multiple (Fig. 1a). Occasionally, groups of closely spaced cortical neurons contained intranuclear HTT or p62 inclusions (Fig. 1b). Using dual immunoperoxidase labeling against HTT and p62, a subset of HTT inclusions were enwrapped by p62, a protein that is important in autophagic removal of aggregated proteins (Fig. 1c).

Intracellular distribution of HTT and p62 inclusions within HD. a Numerous HTT or p62 cytoplasmic inclusions can be found within some neurons, such as in this Betz cell of a 48-year-old woman with 45 CAG repeats. b Intranuclear HTT inclusions are occasionally found in nuclear clusters, particularly in individuals with > 50 CAG repeats. This example was from the calcarine cortex (BA17) of a 30-year-old man with 61 CAG repeats. c Dual staining for HTT (brown)/p62 (red) highlights p62 colocalization with HTT and wrapping of p62 around HTT. Inset (c′) shows a fortuitous dystrophic neurite with a long extension of p62 that emanates from and enwraps an extranuclear HTT inclusion. d The proportion of intranuclear HTT inclusions relative to total HTT inclusions varies in the neocortex and prevails in caudal neocortices, especially the calcarine cortex (BA17). Vertical lines indicate the median value for the respective neocortical region. Note that in the instance of BA17 where the absolute number of inclusions is least of all four cortical regions, only 54 individuals could have calculated ratios because 11 brains had no inclusions in BA17. Brown arrows indicate HTT labeling; red arrows indicate p62 labeling. Brown arrows with a red edge indicate p62 colocalizing with HTT. Scale bars: a–c: 50 µm, c′: 10 µm
The ratio of intranuclear inclusions/total inclusions increases in more caudal cortices (P < 0.0001, Kruskall Wallis test; BA17 vs. BA9, P = 0.007; BA7 vs. BA9, P = 0.013, post-hoc Dunn test, Fig. 1d). The median ratio of intranuclear inclusions/ total of the calcarine cortex was 0.25 ± 0.52 (IQR), whereas that of the prefrontal cortex was 0.038 ± 0.057 (IQR). Thirteen of fifty-four individuals had predominantly intranuclear HTT inclusions (i.e., a ratio > 0.5) in the calcarine cortex, and only one of these individuals had juvenile-onset HD. Therefore, although intranuclear HTT inclusions are generally considered less prominent than extranuclear HTT inclusions in adult-onset HD brains, there are regional differences in the localization of HTT inclusions in the HD cortex [13, 18].
Neocortical variation in HTT and p62 inclusions
We next quantified total HTT inclusion density (i.e. both intra- and extranuclear HTT inclusions) in the infragranular layers (layers V/VI) of neocortical areas BA17, BA7, BA4, and BA9 of the 65 brains. We found that the median density of HTT inclusions was least in the calcarine cortex (BA17) and that this density sequentially increased in a caudal to rostral direction with a nearly 38-fold difference in median inclusion density between BA17 and BA9 (Figs. 2e-h, 3a). Similarly, an increasing caudal to rostral gradient was found with total p62 inclusion density; there was a 14-fold difference in median p62 density between BA17 and BA9 (Figs. 2i–L, 3b, N = 39). Significant correlations between the HTT and p62 inclusion densities were found in BA17, BA7 and BA9 (r = 0.54, P = 0.0004, r = 0.49, P = 0.0017, r = 0.40, P = 0.012, respectively, n = 39).

HTT and p62 inclusion density follows an increasing caudal to rostral gradient in parasagittal neocortical regions of HD. a Macroscopic photographs of the formalin fixed right half brain of a 56-year-old man with adult-onset HD (CAG: 47/21) demonstrating the lateral and medial aspects. The brain (fresh weight: 981.6 g) appeared diffusely small. Vertical lines indicate the plane of section to obtain the coronal slices of images (b)–(d). b–d Select coronal slices of this brain from rostral to caudal with rectangles showing the Brodmann areas of interest, BA9 (superior frontal cortex, b), BA4 (motor cortex, c) and BA7/ BA17 (precuneal cortex and calcarine/visual cortex, respectively, d). e–l Immunohistochemical stains against HTT (e–h) and against p62 (i–l) demonstrating gradually increasing inclusion densities from BA17, BA7, BA4 to BA9. Brown arrows indicate a subset of the inclusions seen in these micrographs. Original magnifications: e–l: 400×, scale bars of e–l: 50 µm

Violin plots of total HTT inclusion densities (a) and p62 inclusion densities (b) across neocortical regions showing successive increases from caudal to rostral cortices (BA17 → BA9). Statistical tables for these respective proteins are provided beneath each graph. a, b Dots indicate individual cortical regions; horizontal dashed lines indicate median values while horizontal dotted lines indicate the first and third quartile values
Neocortical HTT and p62 inclusion densities correlate with CAG expansion
Total HTT inclusion density correlated with CAG repeat length of the 65 patients in all four brain regions studied (BA17: r = 0.49, P < 0.0001; BA7: r = 0.60, P < 0.0001; BA4: r = 0.44, P = 0.0003; BA9: r = 0.53, P < 0.0001, n = 65, Spearman’s rank correlation coefficient, Fig. 4a). The increase in HTT inclusion density per CAG repeat rose in a caudal to rostral direction (BA9 vs. BA17, F = 23.1, P < 0.0001, see methods for statistical analysis). Extrapolation of the regression lines demonstrated an incremental decrease in the x-intercept of the regression lines from caudal to rostral cortices (BA17, BA7, BA4, BA9, Fig. 4b). Although the confidence intervals are broad and assuming linearity in the regression lines, the extrapolation raises the possibility that HTT inclusions might be present in the prefrontal cortex in individuals with CAG expansions between CAG22-35 and in the precuneus at CAG34-39 with the x-intercepts of these cortices corresponding to the intermediate expansion and reduced penetrance ranges, respectively.

Bivariate regression plots showing correlations between CAG repeat expansion and neocortical HTT/ p62 inclusion density. a HTT inclusion density correlates with CAG repeat expansion and increasing gradient of HTT burden along a caudal to rostral direction. b The x-intercepts of the extrapolated linear regression lines between HTT and CAG imply that inclusions may be formed in individuals with intermediate CAG expansion. c p62 inclusion density significantly correlates with CAG repeat expansion in BA17, BA7 and BA9
We next examined the relationship between p62 and CAG repeat expansion in a subset of cases (n = 39). Significant correlations were present between p62 inclusion density and CAG repeat expansion in BA17, BA7, and BA9 but not with BA4 (BA17: r = 0.36, P = 0.026, BA7: r = 0.39, P = 0.014; BA4: r = 0.23, P = 0.16; BA9: r = 0.40, P = 0.011, n = 39; Spearman’s rank correlation coefficient, Fig. 4c). Similar to the relationship between HTT density and CAG repeat expansion, the slopes of the p62 lines of BA17, BA7, and BA9 were progressively steeper from caudal to rostral (BA9 vs. BA17, F = 9.9, P = 0.0024, see methods for statistical analysis).
HTT neocortical inclusion density accords with CAG repeat expansion regardless of disease grade
The Vonsattel grading system is a five-point neuropathologic scale used to measure the extent of degeneration within the striatum from symptomatic HD patients [64]. HD brains without any evidence of striatal degeneration, when assessed with conventional clinical neuropathologic methods, are assigned 0/4, while those with severe neuronal loss and widespread gliosis in the striatum are designated 4/4. Therefore, if the striatum degenerates from grades 0 to 4 over the clinical disease course, this system could also potentially assess whether neocortical HTT inclusion density changes with disease progression.
There were no significant differences in neocortical HTT inclusion density across grades 1–4 in cortical regions assessed (Fig. 5e-l, n = 65), except for the precuneal cortex (BA7) between grades 2 and 4 (P = 0.0028, n = 65, Dunn’s multiple comparison test, Fig. 5k). However, there were no differences of HTT inclusion density between grades 1 and 4, which would be counterintuitive if HTT inclusions accumulate with progression of disease.

a–d: Selected examples of brains that ranged from grade 1 to grade 4. a–d High power image of the dorsal head of the caudate nucleus. Arrows indicate neurons and demonstrate increasing neuronal loss with higher grades and worsening gliosis in higher grades. e–h HTT/p62 immunostains demonstrate similar inclusion densities between the prefrontal neocortices (BA9) despite increasing grade. i–l Violin plots showing that HTT inclusion density is stable across neocortical regions, except between grades 2 and 4 in BA7. Horizontal dashed lines indicate the median. m–n Bivariate regression plots demonstrate correlations between CAP score and HTT inclusion density in BA17, BA7 and BA9 (m, n = 65), however, no correlations were found between the clinical disease duration and HTT inclusion densities (n) across BA17, BA7, BA4 and BA9 (n = 22). Scale bar: 50 µm
Significant correlations were found between the CAP score and total HTT inclusion densities in BA9, BA7, and BA17 (BA9: r = 0.42, P = 0.0006, BA4: r = 0.19, P = 0.14, BA7: r = 0.47, P < 0.0001, BA17: r = 0.43, P = 0.0003, n = 65, Spearman’s rank correlation, Fig. 5m). However, no correlation was found between clinical disease duration, i.e. time from motor onset to death, and HTT inclusion density in any of these four cortices (Fig. 5n, n = 22). Overall, these findings raise the possibility that there is no significant accumulation of HTT inclusions with disease progression but rather that the inclusion density is at a steady state and driven by the extent of CAG repeat expansion. Consistent with this, correlations between inclusion density and CAG expansion size were repeatedly stronger than with CAP score across all 65 individuals with known CAG repeat lengths (BA17: r = 0.50 vs. 0.43; BA7: r = 0.62 vs. r = 0.47; n = 65, BA4: r = 0.46 vs. r = 0.19; BA9: r = 0.48 vs. r = 0.42, n = 65).
Discussion
By utilizing 65 well-characterized HD brains, we found a wide variation of the distribution and density of HTT inclusions across the human HD neocortex. Importantly, HTT inclusion density follows an increasing caudal to rostral gradient (BA17 < BA7 < BA4 < BA9) that is irrespective of cortical cytoarchitecture (heterotypical vs. homotypical). HTT inclusion density correlates with CAG repeat expansion and rostral cortices have greater increases in inclusion density per CAG repeat than caudal cortices. Although intranuclear inclusions are overall more frequent in the juvenile HD cortex with higher CAG repeat expansions than in adult-onset HD, we found that the ratio of intranuclear HTT inclusion density / total HTT inclusion density increases in more caudal cortices in HD brains regardless of the size of CAG repeat expansion and can predominate in the calcarine cortex in adult-onset HD brains [13, 18]. Therefore, the subcellular localization of HTT inclusions in the cortex is regionally dependent. Furthermore, there is an apparent mutual exclusion of the cellular localization of the inclusions, which are either within the nucleus or cytoplasm.
Pathogenic trinucleotide repeat expansion of the HTT gene increases the length of the polyglutamine tract of the HTT protein and results in aberrant splicing of the HTT transcript in a CAG repeat length dependent manner [53]. This produces the small polyadenylated HTT1a transcript that encodes the highly aggregation-prone and pathogenic exon 1 HTT protein and may initiate aggregation within certain compartments of the cell, depending on its subcellular concentration [45, 46]. In no instance were inclusions found concomitantly in both the nucleus or cytoplasm of a neuron. This mutual exclusion may be because inclusion formation in one subcellular compartment may recruit exon 1 HTT from another compartment [36]. In this model, the formation of intranuclear inclusions would depend on whether exon 1 HTT could cross the nuclear membrane, which would be contingent on its size and governed by the length of the polyQ tract. Genome wide association studies have indicated that somatic CAG repeat expansion drives the onset and progression of HD and somatic CAG expansion would result in increased levels of exon 1 HTT with longer polyQ tracts [39, 43]. The relatively higher density of intranuclear inclusions compared with extranuclear inclusions in BA17 than other regions of the cortex, may therefore reflect differences in somatic CAG repeat expansion between these cortical regions.
The burden of HTT inclusions predominate in the infragranular layers of the cortex (layers V/VI), which was our rationale to focus our study on these layers [13, 18]. Studies have demonstrated a relationship between the size of the CAG repeat expansion and the extent of HTT aggregation [34, 55]. Scherzinger demonstrated that polyglutamine expansion above 30 was sufficient to cause HTT aggregation in vitro and that increasing size of polyglutamine stretches and exon 1 HTT concentration, increased aggregation [55]. An increased propensity for self-propagation by mutant HTT with increased polyQ lengths partly explains the correlation between CAG length and HTT inclusion density. However, the cause for regional differences in inclusion density is not clear. One possible cause of the increasing caudal to rostral gradient of HTT burden is that there is a gradient of somatic CAG repeat expansion across the neocortex that influences HTT inclusion density, consistent with the concept of variable CAG repeat expansions across the cortex [44]. Another possibility is that increased neuronal loss of more caudal cortices earlier in the disease would result in fewer neurons at the end of life and reduced HTT burden relative to rostral cortices at the time of autopsy provided there is clearance of HTT inclusions during life [47, 48].
Pathologic grading of HD is widely used to assess the severity of neurodegeneration for an individual postmortem. The general finding that the HTT inclusion burden does not differ across grade (except for one instance in BA7) suggests that there is a steady burden during the clinical disease. Ubiquitinated and HTT inclusions are indeed present in prodromal HD gene carriers, as well as in non-HD individuals with pathogenic HTT expansion and amyotrophic lateral sclerosis (ALS) [12, 15, 18, 26, 27]. Collectively, this evidence implies that the presence and caudal to rostral distribution of pathologic HTT inclusions are a phenotypic expression of HTT gene CAG expansion that does not necessarily signify clinical disease or pathologic grade. This contrasts with other more common neurodegenerative diseases (e.g., primary age-related tauopathy, Alzheimer disease, Parkinson disease), where pathologic inclusions have a tendency to accumulate over time and involve successive brain regions allowing for reliable disease staging [5, 6, 29].
There are several limitations to this study. Firstly, there was limited clinical annotation to many of these brains, partly because HD is rare and the brain bank relies upon referrals from across the nation to acquire this large collection often with limited clinical information. Secondly, only one HTT immunostain was employed in this study and may not have bound all HTT species. However, the p62 immunostain confirmed the presence of an increasing caudal to rostral gradient of inclusions and essentially supported the data gained from the HTT immunostains. Thirdly, like other autopsy studies, this work is correlative and cross-sectional thus preventing the intravitam study of HTT production and clearance, i.e., turnover, within individuals.
Conclusions
In summary, we have demonstrated a wide variation in HTT inclusion density within the human HD neocortex and a variable propensity for aggregation for different neocortices. The uncertainty remains as to whether these inclusions are toxic to the CNS. A higher burden of HTT inclusions in the neocortex did not unequivocally associate with higher grades of disease. Instead, these data suggest that the presence and distribution of HTT inclusions is a gain-of-function phenotypic expression of HTT gene expansion. The spectrum of pathologic effects of pathogenic HTT gene expansion has now expanded to include not simply the HD neurodegenerative phenotype, but also to include abnormalities during neurodevelopment, including a greater propensity for developmental malformations in the brain, as well as ALS [12, 27, 28, 30]. Further investigation as to the biological mechanism for why some cortical regions have disparate burdens of HTT inclusions and whether this signifies variable resistance to HTT accumulation across cortices may reveal possible therapeutic avenues for patients with pathogenic HTT gene expansion.
Availability of data and materials
Data is available upon reasonable request.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- ANCOVA:
-
Analysis of covariance
- BA:
-
Brodmann area
- BA4:
-
Brodmann area 4 (frontal lobe, paracentral)
- BA7:
-
Brodmann area 7 (parietal lobe, precuneus)
- BA9:
-
Brodmann area 9 (frontal lobe, prefrontal cortex)
- BA17:
-
Brodmann area 17 (occipital lobe, primary visual cortex)
- CAP:
-
CAG age product
- CNS:
-
Central nervous system
- HD:
-
Huntington disease
- HTT:
-
Huntingtin
- IQR:
-
Interquartile range
- polyQ:
-
Polyglutamine
- SD:
-
Standard deviation
References
Comparing slopes and intercepts https://www.graphpad.com/guides/prism/7/curve-fitting/reg_comparingslopesandintercepts.htm
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810. https://doi.org/10.1038/nature02998
Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA (1998) Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis 4:387–397. https://doi.org/10.1006/nbdi.1998.0168
Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–614
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259. https://doi.org/10.1007/bf00308809
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Brodmann K (1909) Vergleichende Lokalisationslehre der Grosshirnrinde in ihren Prinzipien dargestellt auf Grund des Zellenbaues. Barth, City
Caan W, Perrett DI, Rolls ET (1984) Responses of striatal neurons in the behaving monkey. 2. Visual processing in the caudal neostriatum. Brain Res 290:53–65. https://doi.org/10.1016/0006-8993(84)90735-2
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90:537–548. https://doi.org/10.1016/s0092-8674(00)80513-9
de la Monte SM, Vonsattel JP, Richardson EP Jr (1988) Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J Neuropathol Exp Neurol 47:516–525
Dehay B, Weber C, Trottier Y, Bertolotti A (2007) Mapping of the epitope of monoclonal antibody 2B4 to the proline-rich region of human Huntingtin, a region critical for aggregation and toxicity. Biotechnol J 2:559–564. https://doi.org/10.1002/biot.200600249
Dewan R, Chia R, Ding J, Hickman RA, Stein TD, Abramzon Y, Ahmed S, Sabir MS, Portley MK, Tucci A et al (2021) Pathogenic huntingtin repeat expansions in patients with frontotemporal dementia and amyotrophic lateral sclerosis. Neuron 109:448-460.e444. https://doi.org/10.1016/j.neuron.2020.11.005
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science (New York, NY) 277:1990–1993
Ferrante RJ, Gutekunst C-A, Persichetti F, McNeil SM, Kowall NW, Gusella JF, MacDonald ME, Beal MF, Hersch SM (1997) Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J Neurosci 17:3052–3063
Gómez-Tortosa E, MacDonald ME, Friend JC, Taylor SA, Weiler LJ, Cupples LA, Srinidhi J, Gusella JF, Bird ED, Vonsattel JP et al (2001) Quantitative neuropathological changes in presymptomatic Huntington’s disease. Ann Neurol 49:29–34
Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu X-H, Tao J, Yamazaki I, Li S-H, Sun YE (2008) Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci 28:6182–6195
Gutekunst C-A, Levey AI, Heilman CJ, Whaley WL, Yi H, Nash NR, Rees HD, Madden JJ, Hersch SM (1995) Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc Natl Acad Sci 92:8710–8714
Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ (1999) Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci 19:2522–2534. https://doi.org/10.1523/jneurosci.19-07-02522.1999
Haber SN (2016) Corticostriatal circuitry. Dialogues Clin Neurosci 18:7–21
Halliday GM, McRitchie DA, Macdonald V, Double KL, Trent RJ, McCusker E (1998) Regional specificity of brain atrophy in Huntington’s disease. Exp Neurol 154:663–672. https://doi.org/10.1006/exnr.1998.6919
Hedreen JC, Peyser CE, Folstein SE, Ross CA (1991) Neuronal loss in layers V and VI of cerebral cortex in Huntington’s disease. Neurosci Lett 133:257–261. https://doi.org/10.1016/0304-3940(91)90583-f
Heinsen H, Rüb U, Bauer M, Ulmar G, Bethke B, Schüler M, Böcker F, Eisenmenger W, Götz M, Korr H et al (1999) Nerve cell loss in the thalamic mediodorsal nucleus in Huntington’s disease. Acta Neuropathol 97:613–622. https://doi.org/10.1007/s004010051037
Heinsen H, Rüb U, Gangnus D, Jungkunz G, Bauer M, Ulmar G, Bethke B, Schüler M, Böcker F, Eisenmenger W et al (1996) Nerve cell loss in the thalamic centromedian-parafascicular complex in patients with Huntington’s disease. Acta Neuropathol 91:161–168. https://doi.org/10.1007/s004010050408
Heinsen H, Strik M, Bauer M, Luther K, Ulmar G, Gangnus D, Jungkunz G, Eisenmenger W, Götz M (1994) Cortical and striatal neurone number in Huntington’s disease. Acta Neuropathol 88:320–333. https://doi.org/10.1007/bf00310376
Herndon ES, Hladik CL, Shang P, Burns DK, Raisanen J, White CL 3rd (2009) Neuroanatomic profile of polyglutamine immunoreactivity in Huntington disease brains. J Neuropathol Exp Neurol 68:250–261. https://doi.org/10.1097/NEN.0b013e318198d320
Hickman R, Dewan R, Cortes E, Traynor B, Vonsattel J-P (2021) Comorbid neurodegenerative pathologies in brains with huntington disease. J Neuropathol Exp Neurol, pp 560–560
Hickman RA, Dewan R, Cortes E, Traynor BJ, Marder K, Vonsattel J-P (2021) Amyotrophic lateral sclerosis is over-represented in two Huntington’s disease brain bank cohorts: further evidence to support genetic pleiotropy of pathogenic HTT gene expansion. Acta Neuropathol, pp 1–4
Hickman RA, Faust PL, Rosenblum MK, Marder K, Mehler MF, Vonsattel JP (2021) Developmental malformations in Huntington disease: neuropathologic evidence of focal neuronal migration defects in a subset of adult brains. Acta Neuropathol 141:399–413. https://doi.org/10.1007/s00401-021-02269-4
Hickman RA, Flowers XE, Wisniewski T (2020) Primary age-related tauopathy (PART): addressing the spectrum of neuronal tauopathic changes in the aging brain. Curr Neurol Neurosci Rep 20:39. https://doi.org/10.1007/s11910-020-01063-1
Hickman RA, O’Shea SA, Mehler MF, Chung WK (2022) Neurogenetic disorders across the lifespan: from aberrant development to degeneration. Nat Rev Neurol, pp 1–8
Hodgson JG, Agopyan N, Gutekunst C-A, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J (1999) A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23:181–192
Kemp JM, Powell TP (1970) The cortico-striate projection in the monkey. Brain 93:525–546. https://doi.org/10.1093/brain/93.3.525
Kemp JM, Powell TP (1971) The termination of fibres from the cerebral cortex and thalamus upon dendritic spines in the caudate nucleus: a study with the Golgi method. Philos Trans R Soc Lond B Biol Sci 262:429–439. https://doi.org/10.1098/rstb.1971.0105
Krobitsch S, Lindquist S (2000) Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A 97:1589–1594. https://doi.org/10.1073/pnas.97.4.1589
Kuusisto E, Kauppinen T, Alafuzoff I (2008) Use of p62/SQSTM1 antibodies for neuropathological diagnosis. Neuropathol Appl Neurobiol 34:169–180. https://doi.org/10.1111/j.1365-2990.2007.00884.x
Landles C, Milton RE, Ali N, Flomen R, Flower M, Schindler F, Gomez-Paredes C, Bondulich MK, Osborne GF, Goodwin D (2020) Subcellular localization and formation of huntingtin aggregates correlates with symptom onset and progression in a Huntington’s disease model. Brain Commun 2: fcaa066
Landles C, Sathasivam K, Weiss A, Woodman B, Moffitt H, Finkbeiner S, Sun B, Gafni J, Ellerby LM, Trottier Y (2010) Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J Biol Chem 285:8808–8823
Lange H, Thorner G, Hopf A, Schroder KF (1976) Morphometric studies of the neuropathological changes in choreatic diseases. J Neurol Sci 28:401–425
Lee J-M, Wheeler VC, Chao MJ, Vonsattel JPG, Pinto RM, Lucente D, Abu-Elneel K, Ramos EM, Mysore JS, Gillis T (2015) Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162:516–526
Lee JK, Conrad A, Epping E, Mathews K, Magnotta V, Dawson JD, Nopoulos P (2018) Effect of trinucleotide repeats in the huntington’s gene on intelligence. EBioMedicine 31:47–53. https://doi.org/10.1016/j.ebiom.2018.03.031
Li H, Li S-H, Yu Z-X, Shelbourne P, Li X-J (2001) Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J Neurosci 21:8473–8481
Maat-Schieman ML, Dorsman JC, Smoor MA, Siesling S, Van Duinen SG, Verschuuren JJ, den Dunnen JT, Van Ommen GJ, Roos RA (1999) Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J Neuropathol Exp Neurol 58:129–137. https://doi.org/10.1097/00005072-199902000-00003
Monckton DG (2021) The contribution of somatic expansion of the CAG repeat to symptomatic development in Huntington’s disease: a historical perspective. J Huntington’s Dis 10:7–33. https://doi.org/10.3233/JHD-200429
Mouro Pinto R, Arning L, Giordano JV, Razghandi P, Andrew MA, Gillis T, Correia K, Mysore JS, Grote Urtubey DM, Parwez CR et al (2020) Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum Mol Genet 29:2551–2567. https://doi.org/10.1093/hmg/ddaa139
Neueder A, Dumas AA, Benjamin AC, Bates GP (2018) Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat Commun 9:1–13
Neueder A, Landles C, Ghosh R, Howland D, Myers RH, Faull RL, Tabrizi SJ, Bates GP (2017) The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci Rep 7:1–10
Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH, van der Kouwe A, Jenkins BG, Dale AM, Fischl B (2002) Regional and progressive thinning of the cortical ribbon in Huntington’s disease. Neurology 58:695–701. https://doi.org/10.1212/wnl.58.5.695
Rosas HD, Salat DH, Lee SY, Zaleta AK, Pappu V, Fischl B, Greve D, Hevelone N, Hersch SM (2008) Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain 131:1057–1068. https://doi.org/10.1093/brain/awn025
Rüb U, Hentschel M, Stratmann K, Brunt E, Heinsen H, Seidel K, Bouzrou M, Auburger G, Paulson H, Vonsattel JP et al (2014) Huntington’s disease (HD): degeneration of select nuclei, widespread occurrence of neuronal nuclear and axonal inclusions in the brainstem. Brain Pathol 24:247–260. https://doi.org/10.1111/bpa.12115
Rüb U, Hoche F, Brunt ER, Heinsen H, Seidel K, Del Turco D, Paulson HL, Bohl J, von Gall C, Vonsattel JP et al (2013) Degeneration of the cerebellum in Huntington’s disease (HD): possible relevance for the clinical picture and potential gateway to pathological mechanisms of the disease process. Brain Pathol 23:165–177. https://doi.org/10.1111/j.1750-3639.2012.00629.x
Rüb U, Seidel K, Vonsattel JP, Lange HW, Eisenmenger W, Gotz M, Del Turco D, Bouzrou M, Korf HW, Heinsen H (2015) Huntington’s disease (HD): neurodegeneration of Brodmann’s primary visual area 17 (BA17). Brain Pathol 25:701–711. https://doi.org/10.1111/bpa.12237
Rüb U, Vonsattel JP, Heinsen H, Korf HW (2015) The Neuropathology of Huntington s disease: classical findings, recent developments and correlation to functional neuroanatomy. Adv Anat Embryol Cell Biol 217:1–146
Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RL, Roos RA, Howland D et al (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A 110:2366–2370. https://doi.org/10.1073/pnas.1221891110
Saudou F, Humbert S (2016) The biology of huntingtin. Neuron 89:910–926
Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for Huntington’s disease pathology. Proc Natl Acad Sci 96:4604. https://doi.org/10.1073/pnas.96.8.4604
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. https://doi.org/10.1038/nmeth.2089
Seger C (2013) The visual corticostriatal loop through the tail of the caudate: circuitry and function. Front Syst Neurosci. https://doi.org/10.3389/fnsys.2013.00104
Sotrel A, Paskevich PA, Kiely DK, Bird ED, Williams RS, Myers RH (1991) Morphometric analysis of the prefrontal cortex in Huntington’s disease. Neurology 41:1117–1123. https://doi.org/10.1212/wnl.41.7.1117
Takeuchi T, Nagai Y (2017) Protein misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci. https://doi.org/10.3390/brainsci7100128
Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L et al (1995) Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 378:403–406. https://doi.org/10.1038/378403a0
van Roon-Mom WM, Hogg VM, Tippett LJ, Faull RL (2006) Aggregate distribution in frontal and motor cortex in Huntington’s disease brain. NeuroReport 17:667–670. https://doi.org/10.1097/00001756-200604240-00022
Vonsattel JP, Del Amaya MP, Keller CE (2008) Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol 115:509–532. https://doi.org/10.1007/s00401-007-0311-9
Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57:369–384
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr (1985) Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 44:559–577
Wang C-E, Zhou H, McGuire JR, Cerullo V, Lee B, Li S-H, Li X-J (2008) Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J Cell Biol 181:803–816
Williams AJ, Paulson HL (2008) Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 31:521–528. https://doi.org/10.1016/j.tins.2008.07.004
Zhang Y, Long JD, Mills JA, Warner JH, Lu W, Paulsen JS (2011) Indexing disease progression at study entry with individuals at-risk for Huntington disease. Am J Med Genet B Neuropsychiatr Genet 156:751–763. https://doi.org/10.1002/ajmg.b.31232
Acknowledgements
We extend our sincere gratitude to all of the patients, families, and caregivers who were critical for accomplishing this study. In particular, we wish to thank the Hereditary Disease Foundation and Nancy Wexler for their ongoing support for HD brain banking and Gregory Suter and Norberta Robertson of the Hereditary Neurological Disease Center in Wichita, Kansas for their continuing efforts in HD brain donations over the past four decades. Gratitude is also extended to Scott Reid, Elizabeth Delaney, Anisha Viswanathan, Matthew Surface, Drs. Roy Alcalay, and Lawrence S. Honig for their coordination of HD brain donations to Columbia University Irving Medical Center. We are grateful to the immunohistochemistry core in the Department of Pathology and Cell Biology at Columbia University Irving Medial Center for performing the immunohistochemical stains. We thank Dr. Wendy Chung of Columbia University Irving Medical Center and Prof. Gill Bates, FRS, of University College London for their critical reviews of this manuscript.
Funding
RAH was supported by grant funding from the Huntington Disease Society of America and Hereditary Disease Foundation and is a Columbia University Irving Medical Center Alzheimer Disease Research Center Research Education Component trainee (P30 AG066462-01 PI: Small, Scott, MD). The New York Brain Bank is supported by the NIH, National Institute on Aging P50 AG008702 (PI Scott Small, MD). KM receives funding from the HDSA Center of Excellence. This work was supported in part by grant funding from the NIH, National Institute of Neurological Disorders and Stroke, R01NS077111 (PI Ai Yamamoto, PhD).
Author information
Authors and Affiliations
Contributions
RAH, AY and JPV designed the study. RAH performed the study, including the statistical analysis. RAH and JPV performed the neuropathological examination of the brains. RAH wrote the manuscript. All authors reviewed, edited and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All brain donors were consented for autopsy and brain donation. All procedures performed were in accordance with the ethical standards of the institution and with the 1964 Helsinki declaration and its later amendments.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hickman, R.A., Faust, P.L., Marder, K. et al. The distribution and density of Huntingtin inclusions across the Huntington disease neocortex: regional correlations with Huntingtin repeat expansion independent of pathologic grade. acta neuropathol commun 10, 55 (2022). https://doi.org/10.1186/s40478-022-01364-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-022-01364-1