
Abstract
A novel DNA methylation class of tumor within the central nervous system, the "neuroepithelial tumor (NET), PATZ1 fusion-positive” has recently been identified in the literature, characterized by EWSR1- and MN1-PATZ1 fusions. The cellular origin of this tumor type remains unknown, wavering between glioneuronal or mesenchymal (as round cell sarcomas with EWSR1-PATZ1 of the soft tissue). Because of the low number of reported cases, this tumor type will not be added to the 2021 World Health Organization Classification of Tumors of the Central Nervous System (CNS). Herein, we report one case of a CNS tumor classified by DNA methylation analysis as NET-PATZ1 but harboring a novel LARGE1-AFF2 fusion which has until now never been described in soft tissue or the CNS. We compare its clinical, histopathological, immunophenotypical, and genetic features with those previously described in NET-PATZ1. Interestingly, the current case presented histopathological (astroblastoma-like features, glioneuronal phenotype), clinical (with a favorable course), genetic (1p loss), and epigenetic (DNA-methylation profiling) similarities to previously reported cases of NET-PATZ1. Our results added data suggesting that different histomolecular tumor subtypes seem to be included within the methylation class “NET, PATZ1 fusion-positive”, including non PATZ1 fusions, and that further cases are needed to better characterize them.
Similar content being viewed by others


Introduction
Neuroepithelial tumors (NET) with PATZ1 fusions (NET-PATZ1) have been isolated by a distinct DNA methylation profile and are characterized by recurrent fusions of PATZ1 in association with EWSR1 or MN1 genes [1]. These tumors present a wide variety of morphologies and immunophenotypes, having been initially classified as glioneuronal tumors, astroblastomas, ependymomas, glioblastomas, pleomorphic xanthoastrocytomas, primary neuroepithelial tumors, and round cell sarcomas, with different histopathological grades [2,3,4,5,6,7,8,9,10,11]. Because extra-CNS sarcomas may also harbor an EWSR1-PATZ1 fusion and because of the uncertainty of the cellular origin of NET-PATZ1, this tumor type will not be added to the upcoming edition of the World Health Organization Classification of CNS Tumors [12]. Here we report a temporal tumor with a NET-PATZ1 DNA methylation class (MC) but harboring a LARGE1-AFF2 fusion. We compare its clinical, histopathological, immunophenotypical, genetic and epigenetic features with those previously described in NET-PATZ1.
Case presentation
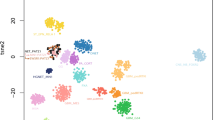
A 3-year-old girl began experiencing seizures. Cerebral magnetic resonance imaging (MRI) showed a left temporal mass with a hyperintense signal on T1-weighted images, a hypointense signal on T2-weighted-images and with a homogeneous and intense contrast enhancement after gadolinium injection (Fig. 1A–D). The mass was cortical, well-circumscribed, solid with a small cyst, having slight perilesional edema and no leptomeningeal attachment. Neither hemorrhagic nor necrotic modification was observed and no calcifications were present on computerized tomodensitometry. Diffusion was not restricted (Fig. 1E). Gross total resection was achieved. Microscopically, the tumor was well-delineated from the brain parenchyma (Fig. 1F), and heterogeneous, presenting a sclerous stroma including isolated cells and nodules of glial cells with microcystic changes (Fig. 1G-I). No ependymal nor astroblastic pseudorosettes, or rhabdoid component were evidenced. The cytoplasm of the tumor cells was abundant and eosinophilic. Some tumor cells were plurinucleated. No mitotic figures, nor necrosis or microvascular proliferation were observed and the MIB-1 labeling index was low (around 5%) (Fig. 1J). Very few perivascular inflammatory infiltrates were present but no eosinophilic granular bodies or ganglion cells were observed. The tumor cells expressed glial markers (GFAP and Olig2 for a subset of cells; Fig. 1K), MAP2 (Fig. 1L) and NeuN (without expression of synaptophysin and chromogranin A), and CD34 (Fig. 1M). There was no immunopositivity for CKAE1/AE3, CK18, CD99, BCOR, SOX10, IDH1R132H or Lin28A. The tumor cells focally expressed desmin but there was no immunoreactivity for smooth muscle actin or myogenin (Fig. 1N). The expression of ATRX, BRG1, INI1 and H3K27me3 was retained. Because of the fibrous stroma, a diagnosis of astroblastoma was initially suggested but a FISH analysis of the MN1 gene failed to reveal a rearrangement. RNA sequencing (including EWSR1, MN1 and PATZ1 genes) evidenced a LARGE1-AFF2 gene fusion (Fig. 2A) and a DNA methylation analysis was conducted. The tumor was classified as NET-PATZ1 (with a strong calibrated score of 0.99) based on the DNA methylation profiling using a random forest machine learning classification algorithm as previously described (v12.3; Fig. 2B) [13]. Twelve months later, the patient is well without adjuvant treatment and no residual tumor on MRI.

Radiological and histopathological features. A Axial T1-weighted magnetic resonance imaging image showing a left hyperintense temporal lesion. B Axial T1-weighted magnetic resonance imaging image after contrast injection showing an intense homogeneous enhancement of the lesion which is mainly solid with a small cyst (arrow). C Axial T2-weighted magnetic resonance imaging image showing an hypointensity of the lesion and a perilesional edema. D T2-FLAIR-weighted image showing the vasogenic perilesional edema. E Diffusion was not restricted. F The well delimitation of the tumor (HPS, magnification × 50). G The alternance of fibrous stroma containing few tumor cells and highest cellular areas (HPS, magnification × 200). H The collagenous stroma with few spindle cells (HPS, magnification × 400). I The cellular areas monomorphous cells with round to oval nuclei and abundant eosinophilic cytoplasm with microcystic changes (HPS, magnification × 400). J Low MIB-1 labeling index (magnification × 400). K GFAP immunoexpression by many tumor cells (magnification × 400). L MAP2 immunoexpression by a subset of tumor cells (magnification × 400). M Diffuse extravascular CD34 immunoexpression with CD34-positive ramified processes (magnification × 400). N Desmin immunoexpression by some tumor cells (magnification × 400). Black scale bars represent 500 µm for figure F, 250 µm for figure G, and 50 µm for figure H-N. HPS: Hematoxylin Phloxin Saffron.

Illustration of the fusion and t-SNE analysis A RNAseq analysis highlights a fusion between LARGE1 (red) and AFF2 (blue) genes, respectively located on chr22q12.3 and chrX.q28 with a breakpoint in exon 6 for LARGE1 and exon 8 for AFF2. B t-distributed stochastic neighbor embedding (t-SNE) analysis of the DNA methylation profile of the investigated tumor alongside 361 selected reference samples
Discussion and conclusions
Here, we report an intracerebral tumor harboring a novel LARGE1-AFF2 fusion, with clinical, radiological, histopathological, and epigenetic similarities to NET-PATZ1. Like most NET-PATZ1, our observation concerned a supratentorial tumor in a child (Table S1) [1,2,3,4,5,6,7,8, 11]. Whereas neuroradiological data of this recently described tumor type is scarce, our case presented as a solid and cystic lesion with T2-weighted hypointensity suggesting fibrotic content, well-circumscribed from the brain parenchyma, as previously reported [4, 8, 11]. NET-PATZ1 encompassed a wide variety of morphologies in the literature, including glial, glioneuronal, embryonal tumors and sarcomas (Table S1). Based on the literature review and our case, the presence of a collagenous stroma and microcysts seem to be frequent in NET-PATZ1 (Table S1) [1, 4, 8, 11]. Because of this pattern and because some of them present pseudorosettes (not seen in our case), pathologists tend to consider them a differential diagnosis for astroblastoma, MN1-fused [1, 2, 8]. However, NET-PATZ1, as with our case, exhibit frequent glioneuronal immunoprofiles and an extravascular expression of CD34 may be found, which is rare in astroblastomas (Table S1) [1, 11, 14]. The histopathological and epigenetic distinction between sarcomas with EWSR1-PATZ1 fusion and NET, PATZ1-fusion positive is still not clear. Indeed, CNS and extra-CNS tumors with PATZ1 fusion share some histopathological features (microcysts, collagenous stroma and pseudorosettes, and a mesenchymal component described in a part of NET-PATZ1) and a polyphenotypic immunoprofile (expression of glial, neuronal and CD34 in both) [1, 11, 15,16,17]. The DNA methylation analysis (v12.3) classified our case as a NET, PATZ1, although the tumor did not harbor a PATZ1 fusion. Similarly, the case clustered with PATZ1-fused sarcomas, located outside the CNS on the whole RNA sequencing analysis. Our case presented a LARGE1-AFF2 fusion which has not been previously reported in CNS or in soft tissue tumors. Whereas no LARGE1 fusion has been described in tumors, several tumor types have been reported with AFF2 fusions in association with different partners (DEK in squamous carcinomas, RET in lung cancer, STAG2 in T-cell lymphoma) [18,19,20,21]. The fusion may drive the oncogenesis by deregulation of transcription as AFF2 encodes a RNA-binding protein through the C-terminal domain that can activate transcription [22]. Moreover, the chimeric protein is predicted to contain the major functional domains of both LARGE1 and AFF2 proteins. Interestingly, LARGE1 (22q12.3) and PATZ1 (22q12.2) genes are found in close proximity on chromosome 22, and PATZ1 was highly expressed at the RNA level (whereas LARGE1 and AFF2 are not) as observed in sarcomas with PATZ1 fusion. Because DNA methylation profiles are thought to represent a combination of both somatically acquired DNA methylation changes and a signature reflecting the cell of origin [23], it is reasonable to assume that our case represents a subtype of NET-PATZ1. Because of the limited follow-up data and the heterogeneity of treatments applied in NET-PATZ1 cases from the literature, no precise prognosis has been defined [1]. And despite histopathological signs of aggressivity, a probable intermediate grade has been suggested considering the better outcome associated with NET-PATZ1 compared to high-grade tumors [1]. Our case is in line with these data, showing no recurrence one year after gross total resection without adjuvant treatment.
In conclusion, we expanded the defined MC NET-PATZ1 genetic spectrum with one novel fusion that does not involve the PATZ1 gene. This case illustrates that further studies are needed to characterize in detail this rare type of tumor in terms of cellular origin, histopathology, genetic features and outcome.
References
Alhalabi KT, Stichel D, Sievers P, Peterziel H, Sommerkamp AC, Sturm D et al (2021) PATZ1 fusions define a novel molecularly distinct neuroepithelial tumor entity with a broad histological spectrum. Acta Neuropathol (Berl) 142:841–857
Zschernack V, Jünger ST, Mynarek M, Rutkowski S, Garre ML, Ebinger M et al (2021) Supratentorial ependymoma in childhood: more than just RELA or YAP. Acta Neuropathol (Berl) 141:455–466
Siegfried A, Rousseau A, Maurage C-A, Pericart S, Nicaise Y, Escudie F et al (2019) EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol Zurich Switz 29:53–62
Rossi S, Barresi S, Giovannoni I, Alesi V, Ciolfi A, Colafati GS, et al. (2020) Expanding the spectrum of EWSR1-PATZ1 rearranged CNS tumors: an infantile case with leptomeningeal dissemination. Brain Pathol Zurich Switz., e12934.
Bridge JA, Sumegi J, Druta M, Bui MM, Henderson-Jackson E, Linos K, et al. (2019) Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod Pathol Off J U S Can Acad Pathol Inc., 32:1593–604
Pei J, Zhao X, Patchefsky AS, Flieder DB, Talarchek JN, Testa JR, et al. (2019) Clinical application of RNA sequencing in sarcoma diagnosis: an institutional experience. Med (Baltimore), 98:e16031
Lopez-Nunez O, Cafferata B, Santi M, Ranganathan S, Pearce TM, Kulich SM et al (2021) The spectrum of rare central nervous system (CNS) tumors with EWSR1-non-ETS fusions: experience from three pediatric institutions with review of the literature. Brain Pathol Zurich Switz 31:70–83
Chadda KR, Holland K, Scoffings D, Dean A, Pickles JC, Behjati S, et al. (2021) A rare case of paediatric astroblastoma with concomitant MN1-GTSE1 and EWSR1-PATZ1 gene fusions altering management. Neuropathol Appl Neurobiol
Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD et al (2016) Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol (Berl) 131:833–845
Johnson A, Severson E, Gay L, Vergilio J, Elvin J, Suh J et al (2017) Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist 22:1478–1490
Burel-Vandenbos F, Pierron G, Thomas C, Reynaud S, Gregoire V, Duhil de Benaze G, et al. (2020) A polyphenotypic malignant paediatric brain tumour presenting a MN1-PATZ1 fusion, no epigenetic similarities with CNS High-Grade Neuroepithelial Tumour with MN1 Alteration (CNS HGNET-MN1) and related to PATZ1-fused sarcomas. Neuropathol Appl Neurobiol, 46: 506–509
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncol., noab106.
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474
Tauziède-Espariat A, Pagès M, Roux A, Siegfried A, Uro-Coste E, Nicaise Y et al (2019) Pediatric methylation class HGNET-MN1: unresolved issues with terminology and grading. Acta Neuropathol Commun 7:176
Park KW, Cai Y, Benjamin T, Qorbani A, George J (2020) Round cell sarcoma with EWSR1-PATZ1 gene fusion in the neck: case report and review of the literature. Laryngoscope 130:E833–E836
Chougule A, Taylor M, Nardi V, Chebib I, Cote GM, Choy E et al (2019) Spindle and round cell sarcoma with EWSR1-PATZ1 gene fusion: a sarcoma with polyphenotypic differentiation. Am J Surg Pathol 43:220–228
Michal M, Rubin BP, Agaimy A, Kosemehmetoglu K, Rudzinski ER, Linos K, et al. (2021) EWSR1-PATZ1-rearranged sarcoma: a report of nine cases of spindle and round cell neoplasms with predilection for thoracoabdominal soft tissues and frequent expression of neural and skeletal muscle markers. Mod Pathol Off J U S Can Acad Pathol Inc., 34:770–785
Kuo Y-J, Lewis JS, Zhai C, Chen Y-A, Chernock RD, Hsieh M-S, et al. (2021) DEK-AFF2 fusion-associated papillary squamous cell carcinoma of the sinonasal tract: clinicopathologic characterization of seven cases with deceptively bland morphology. Mod Pathol Off J U S Can Acad Pathol Inc., 34: 1820–1830
Rooper LM, Agaimy A, Dickson BC, Dueber JC, Eberhart CG, Gagan J, et al. (2021) DEK-AFF2 carcinoma of the sinonasal region and skull base: detailed clinicopathologic characterization of a distinctive entity. Am J Surg Pathol.
Li B, Qu H, Zhang J, Pan W, Liu M, Yan X et al (2021) Genomic characterization and outcome evaluation of kinome fusions in lung cancer revealed novel druggable fusions. NPJ Precis Oncol 5:81
Ohki K, Kiyokawa N, Watanabe S, Iwafuchi H, Nakazawa A, Ishiwata K et al (2021) Characteristics of genetic alterations of peripheral T-cell lymphoma in childhood including identification of novel fusion genes: the Japan Children’s Cancer Group (JCCG). Br J Haematol 194:718–729
Bensaid M, Melko M, Bechara EG, Davidovic L, Berretta A, Catania MV et al (2009) FRAXE-associated mental retardation protein (FMR2) is an RNA-binding protein with high affinity for G-quartet RNA forming structure. Nucleic Acids Res 37:1269–1279
Hovestadt V, Jones DTW, Picelli S, Wang W, Kool M, Northcott PA et al (2014) Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510:537–541
Acknowledgements
We would like to thank the laboratory technicians at the GHU Paris Neuro Sainte-Anne for their assistance, as well as the Integragen platform for their technical assistance with DNA-methylation analyses. We thank P. Sievers from the Department of Neuropathology of the University Hospital Heidelberg for performing an unsupervised t-SNE analysis of the DNA methylation data together with a reference cohort.
Funding
The authors declare that they have not received any funding.
Author information
Authors and Affiliations
Consortia
Contributions
ATE, GC, VDR, NB, CI, and EG compiled the MRI and clinical records; ATE, GC, AM, FC and PV conducted the neuropathological examinations; FLL, RA and JB conducted the molecular studies; ATE, LH, and PV drafted the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the GHU Paris Psychiatrie Neurosciences, Sainte-Anne Hospital’s local ethic committee.
Consent for publication
The patient signed informed consent forms before treatment was started.
Competing interests
The authors declare that they have no conflicts of interest directly related to the topic of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Table S1. Summary of clinical, histopathological and molecular data of CNS PATZ1-fused tumors reported in the literature
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tauziède-Espariat, A., Chotard, G., le Loarer, F. et al. A novel LARGE1-AFF2 fusion expanding the molecular alterations associated with the methylation class of neuroepithelial tumors with PATZ1 fusions. acta neuropathol commun 10, 15 (2022). https://doi.org/10.1186/s40478-022-01317-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-022-01317-8