
Abstract
Primary spinal cord tumors contribute to ≤ 10% of central nervous system tumors in individuals of pediatric or adolescent age. Among intramedullary tumors, spinal ependymomas make up ~ 30% of this rare tumor population. A twelve-year-old male presented with an intradural, extramedullary mass occupying the dorsal spinal canal from C6 through T2. Gross total resection and histopathology revealed a World Health Organization (WHO) grade 2 ependymoma. He recurred eleven months later with extension from C2 through T1-T2. Subtotal resection was achieved followed by focal proton beam irradiation and chemotherapy. Histopathology was consistent with WHO grade 3 ependymoma. Molecular profiling of the primary and recurrent tumors revealed a novel amplification of the MYC (8q24) gene, which was confirmed by fluorescence in situ hybridization studies. Although MYC amplification in spinal ependymoma is exceedingly rare, a newly described classification of spinal ependymoma harboring MYCN (2p24) amplification (SP-MYCN) has been defined by DNA methylation-array based profiling. These individuals typically present with a malignant progression and dismal outcomes, contrary to the universally excellent survival outcomes seen in other spinal ependymomas. DNA methylation array-based classification confidently classified this tumor as SP-MYCN ependymoma. Notably, among the cohort of 52 tumors comprising the SP-MYCN methylation class, none harbor MYC amplification, highlighting the rarity of this genomic amplification in spinal ependymoma. A literature review comparing our individual to reported SP-MYCN tumors (n = 26) revealed similarities in clinical, histopathologic, and molecular features. Thus, we provide evidence from a single case to support the inclusion of MYC amplified spinal ependymoma within the molecular subgroup of SP-MYCN.
Similar content being viewed by others

Introduction
Primary spinal cord tumors are rare in children and adolescents, contributing to ≤ 10% of all pediatric central nervous system (CNS) neoplasms [52]. Intramedullary spinal ependymomas (EP) make up ~ 30% of pediatric spinal cord tumors, second to astrocytomas [52]. Histologically, spinal cord ependymomas are categorized into subependymoma (World Health Organization (WHO) grade 1), myxopapillary EP (WHO grade 2), and classic or anaplastic EP (WHO grade 2/3) [34]. Furthermore, molecular classification using DNA methylation array-based profiling distinguishes three distinct molecular subgroups of spinal EP: subependymoma (SP-SE), myxopapillary EP (SP-MP), and anaplastic EP (SP-EP) [38]. Despite these divergent molecular subgroups, spinal cord EP are typically slow growing with universally excellent overall survival rates, especially when gross total resection of the tumor can be achieved [4, 7, 33, 36, 46]. Radiation therapy is usually advocated for subtotally resected grade 2 tumors [3, 18, 32, 46] and all patients with grade 3 tumors [3, 12, 46].
Recently, a novel molecular subgroup of spinal EP with focal high-level MYCN (2p24) amplification was defined and found to be associated with dismal outcomes and malignant progression, despite aggressive management [20, 41, 43, 49]. This molecular pathology will be newly recognized as a distinct subgroup of spinal cord EP (SP-MYCN) in the fifth edition of the WHO Classification of Tumors of the Central Nervous System [16, 35]. Similar to SP-MP, SP-MYCN were found to develop in extramedullary spaces in sharp contrast to the intramedullary growth seen in SP-EP [20, 41, 49]. Herein, we report on an adolescent male with aggressive classic spinal EP harboring a novel focal amplification of the MYC oncogene located on chromosome 8q24.
Case presentation
A 12-year-old male with no significant past medical history presented to the emergency room with a three-month history of back pain and acute onset of weakness in the left lower extremity. Neurological examination was significant for left lower extremity weakness and ataxia. Magnetic resonance imaging (MRI) of the brain and spine revealed a localized avidly enhancing intradural, extramedullary mass occupying the dorsal spinal canal from C6 through T2. The tumor resulted in severe cord compression and mild edema (Fig. 1a, b). He underwent a gross total resection of the tumor followed by observation with serial imaging. Eleven months later, he re-presented with acute onset of lower extremity paresthesia and left-handed weakness. Spine MRI revealed tumor recurrence with further extension anteriorly and posterior to the cord from C2 through T1-T2 with resultant severe cord compression, again demonstrating avid enhancement (Fig. 1c, d). Management included subtotal resection of the recurrent mass, focal proton beam irradiation (50.4 Gy), followed by four cycles of chemotherapy with vincristine, etoposide, cyclophosphamide, and cisplatin, as per ACNS0831 [45], then oral vorinostat for seven months. The patient experienced further progression with distal metastases in the thoraco-lumbar region distal to his primary tumor, 16 months after first relapse (Fig. 1e, f), and completed intensity-modulated radiation therapy (IMRT) of 36 Gy to the entire thecal sac. On his post-radiation MRI, there was evidence of progression of the cervical region tumor and the patient is therefore now undergoing focal re-irradiation of the cervical region tumor. A clinical summary is presented in Table 1.

MRI images at presentation: sagittal a T1-weighted and b T2-weighted showing avidly enhancing intradural mass, occupying the dorsal spinal canal from C6 through T1-T2. MRI images at first relapse: sagittal c T1-weighted and d T2-weighted showing avidly enhancing tumor, now extending from C2 through T1-T2. MRI images at second relapse: sagittal e T1-weighted and f T2-weighted showing new noncontiguous separate nodules scattered along the surface of the cord from C7 through L1
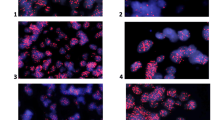
Histopathologic examination of the initial resection specimen revealed a compact lobulated glial neoplasm with variable morphology. The majority of the tumor showed classic ependymoma features, including mildly pleomorphic tumor cells with round to oval nuclei arranged in perivascular pseudo-rosette (nuclear-free zone) formations, often surrounding capillaries showing microvascular proliferation (Fig. 2a). Other tumor areas showed clear cell change (Fig. 2b) or tanycytic morphology with elongate spindle cells. Densely cellular nests of mitotically active cells displaying conspicuous nucleoli were also present (Fig. 2c). Mitoses were counted at 3–4 per 10 high power fields (HPF). Squash preparations showed a branching angiocentric pattern of tumor cells (Additional File 1: Fig. S1a). Immunohistochemical stains demonstrated prominent perivascular glial fibrillary acidic protein (GFAP) staining (Fig. 2d) while Olig-2 nuclear staining was rare (Fig. 2e). Epithelial membrane antigen (EMA) showed perinuclear dot-like immunoreactivity (Fig. 2f), while ring-like staining was much less frequent. The Ki-67 proliferation index was focally up to 20% (Additional File 1: Fig. S1b). A diagnosis of ependymoma, WHO grade 2, with histopathologic features bordering between WHO grade 2 and WHO grade 3. The subsequent recurrent/residual tumor specimen showed similar histologic features, with even more extensive regions of hypercellular tumor and higher mitotic index (up to 8 per 10/HPF) diagnostic of anaplastic ependymoma, WHO grade 3 based on the 2016 WHO Classification.

Histologic features of the primary tumor assessed by routine Hematoxylin and Eosin stain demonstrating perivascular pseudo-rosettes surrounding capillaries showing a microvascular proliferation (20× magnification), with b focal clear cell ependymoma (20× magnification). c Mitotically active cells are denoted by the asterisk (*) (40× magnification). The tumor cells demonstrated d strong perivascular GFAP (20× magnification), e sparse Olig-2 nuclear staining (20× magnification), and f dot-like EMA reactivity (40× magnification). The histopathologic findings were similar in the primary and recurrent tumors
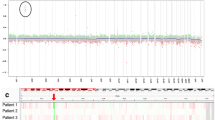
To further characterize the tumor, the patient was consented on an Institutional Review Board-approved translational research protocol and underwent comprehensive molecular profiling, including paired tumor/normal enhanced exome sequencing (eES) and RNA-sequencing (Additional File 1: Materials and Methods). This analysis included an evaluation of small single nucleotide variants (SNV), small insertion-deletions, copy number alterations (CNA), gene fusions, and aberrant gene expression. We sequenced disease-involved tissue from both the primary spinal cord tumor and localized tumor recurrence occurring 11 months after the primary tumor. We did not identify any cancer- or disease-associated SNVs or CNAs from the germline comparator peripheral blood. In addition, we did not identify any clearly medically meaningful somatic SNVs, small indels, or gene fusions. In both analyzed timepoints, the CNA profile was notable for a focal amplification of the MYC gene on 8q24 (Fig. 3a) and biallelic loss of 17p, including TP53 and likely consistent with an isochromosome 17q (Fig. 3b, Additional File 1: Table S1). Other CNA described from eES included segmental biallelic losses of 8q, 10q (including PTEN), and 19q (Additional File 1: Fig. S2, Additional File 1: Table S1). The University of California Santa Cruz (UCSC) Treehouse Initiative (https://treehousegenomics.ucsc.edu/explore-our-data/) is a collaborative data sharing initiative whereby RNA-sequencing data from a breadth of tumor types are publicly available. We utilized a cohort of pediatric and adolescent/young adult central and peripheral nervous system tumors (n = 563) from the UCSC Treehouse Initiative to compare MYC gene expression. Consistent with the identified gene amplification, MYC was found to be overexpressed in both primary (log2 fold change: 3.95, P = 0.0007) and recurrent (log2 fold change: 4.57, P = 9.57 × 10–5) tumors. Visualization of MYC expression for our described patient case relative to ependymoma (n = 41), glioma (n = 300), medulloblastoma (n = 129), and neuroblastoma (n = 199) patients from the UCSC Treehouse Initiative and our internal cohort confirmed this overexpression (Fig. 3c).

Somatic copy number alterations (CNA) on chromosome 8 (a) and chromosome 17 (b) are shown as derived from enhanced exome sequencing data. The blue points represent log2 values based on sequence depth in 100 bp windows. The red line indicates segmented CNA calls. The MYC (8q24) amplification is highlighted by the arrow. c Distribution of MYC gene expression in ependymomas (n = 41), gliomas (n = 300), medulloblastomas (n = 129), and neuroblastoma (n = 199) amid the UCSC Treehouse cohort and patients enrolled on our translational cancer protocol with the red points indicating our described patient case. The shape indicates timepoint (circle = primary tumor, triangle = recurrent tumor). d Fluorescence in situ hybridization (FISH) of the MYC locus (red) demonstrates gene amplification (> 20 signals compared to chromosome 8 centromere in green) with signal pattern most consistent with double minute formation
Fluorescence in situ hybridization (FISH) of the MYC (8q24) and MYCN (2p24) loci was performed from formalin-fixed paraffin-embedded tissue to confirm the presence of MYC amplification. Greater than 20 copies of MYC were detected relative to a 2-copy state for the centromere of chromosome 8 with the amplification signal pattern most consistent with double minute formation (primary tumor: nuc ish(MYC amp)[74/100]; recurrent tumor: nuc ish(MYC amp)[92/100]) (Fig. 3d). In comparison, the MYCN locus was present at a 2-copy state with two signals detected for both MYCN and the centromere of chromosome 2 (primary tumor: nuc ish(MYCNx2)[89/100]), recurrent tumor: nuc ish(MYC amp)[87/100]) (Additional File 1: Fig. S3). These results provide orthogonal confirmation of next generation sequencing data supporting the identification of a spinal ependymoma harboring a novel MYC amplification.
DNA methylation-based molecular classification was performed to assign the described patient case to one of the ten established EPN groups (SP-MP, SP-EP, SP-SE, SP-MYCN, ST-SE (supratentorial subependymoma), ST-YAP1 (supratentorial ependymoma YAP1-fused), ST-ZFTA (supratentorial ependymoma ZFTA-fused), PF-SE (posterior fossa subependymoma), PFA (posterior fossa group A), PFB (posterior fossa group B)). Unsupervised clustering with a reference cohort of 501 methylation profiles spanning all 10 established molecular EPN groups clearly assigned the tumor from our described patient case to the SP-MYCN group (Fig. 4a) [20, 38]. This result was confirmed in a repeated clustering restricted to reference cases of the four molecular spinal EPN groups (n = 66, Fig. 4b). Analyses of CNA plots of 52 spinal tumors predicted as SP-MYCN revealed a focal amplification of MYCN in 50/52 tumors, but no additional case with MYC amplification (Fig. 4c). Furthermore, when clustered with a cohort of ~ 80,000 DNA methylation profiles covering the entire spectrum of existing molecular CNS tumor classes, the described patient case classified with the SP-MYCN EPN subgroup. SP-MYCN will be included as a new reference group in the upcoming version (v12.3) of the Heidelberg Brain Tumor Methylation Classifier. This most recent version (v12.3) of the classifier assigned the described patient case to the SP-MYCN group with a calibrated score of 0.99, which is above the cut-off for confident class prediction (0.9) [10], and thus confirms the assignment of the case to the molecular group of SP-MYCN.

a t-SNE showing DNA methylation array-based clustering of the described patient case (enlarged black dot) with a reference cohort of n = 501 tumors spanning across ten established molecular EPN groups [20, 38] and b a subset restricted to the four established spinal EPN groups. In both analyses, the MYC-amplified case (enlarged black dot) clustered with the SP-MYCN group. SP-MP (spinal myxopapillary ependymoma), SP-EP (spinal anaplastic ependymoma), SP-SE (spinal subependymoma), SP-MYCN (spinal ependymoma, MYCN-amplified), ST-SE (supratentorial subependymoma), ST-YAP1 (supratentorial ependymoma YAP1-fused), ST-ZFTA (supratentorial ependymoma ZFTA-fused), PF-SE (posterior fossa subependymoma), PFA (posterior fossa group A), PFB (posterior fossa group B). c Copy number plot showing a prominent amplification at the MYC locus (chr8), but no alteration at the MYCN locus (chr2)
Discussion and conclusions
This report describes a novel case of an aggressive recurrent progressive spinal cord ependymoma with histologic features of an anaplastic ependymoma harboring focal MYC amplification. Interestingly, DNA methylation-based classification assigned this case to the molecular group of SP-MYCN. MYC (8q24) encodes the c-MYC protein, a transcription factor that interacts with other proteins to regulate gene expression, including those that promote cell growth and proliferation [25, 48]. Deregulation of MYC has been shown to stimulate and maintain tumorigenesis in ex vivo models [28, 48]. MYC alterations are recurrently described amongst many different types of neoplasms, including pediatric brain tumors [6, 25], with amplification being most frequently reported [25, 28]. Glial and non-glial brain tumors harboring MYC amplification demonstrated a significantly worse prognosis [8, 22, 27, 30, 37, 42, 54].
Despite the high frequency of MYC gene alteration in human cancers, it has been rarely reported in ependymoma. A single individual was reported with a recurrent anaplastic ependymoma harboring an abnormal karyotype 46,XX,der(8)t(8;11)(q24;p11), − 11,add(?)t(?;11)(?;q13)) and MYC overexpression [13]. Notably, this tumor was not located in the spine but rather in the supratentorial region of the brain. Despite MYC overexpression, no evidence of MYC gene rearrangements nor amplification were identified [13]. Given the paucity of literature describing MYC alterations in ependymoma, we performed a literature review describing the clinical, histologic, and molecular features of the 26 reported spinal ependymomas with MYCN (2p24) amplification (Table 1 and Additional File 1: Table S1) [20, 41, 43, 49]. Similar to our described patient case, the SP-MYCN tumors had distinct growth patterns, and typically arose intradurally and extramedullary with invasion of the spinal cord. Most of the SP-MYCN tumors were located in the cervical or thoracic spine and were commonly associated with nodular metastatic spread and diffuse leptomeningeal involvement [20, 41, 43, 49]. Compared to other spinal ependymomas, SP-MYCN tumors were associated with aggressive behavior and unfavorable outcomes, despite intensive multi-modal therapies [20, 41]. In the described patient case, the histopathologic features including perivascular pseudo-rosettes, microvascular proliferation, densely cellular nests of mitotically active cells, prominent perivascular GFAP staining, sparse Olig-2 nuclear staining, a dot-like staining pattern with EMA, as well as increased Ki-67 proliferation indices were also described in prior reported examples of the molecular group SP-MYCN [20, 49]. Furthermore, loss of chromosome 10 may be a recurrent finding among this tumor subgroup, seen in 8/19 (42%) individuals with available copy number data (Additional File 1: Table S1). Our described patient case with MYC amplification also demonstrated segmental losses across chromosome 10q, including the tumor suppressor PTEN. Amplification of MYC in combination with disruption of PTEN has been shown in prostate cancer to contribute to aggressive disease and poor outcomes [23, 29]. Larger studies to assess the association of MYC or MYCN amplification with chromosome 10 loss and patient outcomes may be warranted.
In vitro and in vivo MYC inhibition has demonstrated tumor regression, across numerous tumor types [1, 11, 47]; nevertheless, direct inhibition of MYC is challenging. Thus, efforts have shifted towards targeting MYC transcriptional targets and regulatory domains [11, 25, 51, 53], including the study of BET inhibitors [2, 21], CDK inhibitors [5], mTOR inhibitors [26], Aurora A-kinase inhibitors [9, 17] and CHK1 inhibitors [40]. Additionally, histone deacetylase (HDAC) inhibitors have been shown to impede MYC-amplified Group 3 medulloblastoma tumor growth in vitro [14, 15, 39, 44]. Due to the focal MYC amplification, our patient received single-agent vorinostat, an oral HDAC inhibitor that is well tolerated in children with relapsed CNS tumors [19, 24, 31, 50], following his first relapse. However, he presented with progressive disease after seven months.
In summary, we report a unique case of an adolescent male with an aggressive spinal ependymal tumor harboring focal MYC amplification. DNA array-based methylation profiling confidently classified this tumor as SP-MYCN, a recently described subgroup of spinal ependymoma. Our described patient case demonstrates clinical, histologic, and molecular overlap with the newly described SP-MYCN subgroup. Thereby, we provide evidence to support the inclusion of MYC amplified spinal ependymoma within the molecular subgroup of SP-MYCN. Testing for MYC or MYCN gene amplification may be warranted in newly diagnosed spinal tumors to aid in tumor characterization. Future strategies should focus on investigating the efficacy of indirect MYC-targeting strategies, introducing new possibilities for improving the prognosis in patients with SP-MYCN.
Availability of data and material
The datasets generated and/or analyzed during the current study are available in the dbGaP repository (https://www.ncbi.nlm.nih.gov/gap/) accession phs001820.v1.p1., under submitter: Institute for Genomic Medicine (IGM) Clinical Laboratory, Nationwide Children's Hospital. Details are provided in the Additional File 1: Materials and Methods.
References
Arvanitis C, Felsher DW (2006) Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol 16:313–317. https://doi.org/10.1016/j.semcancer.2006.07.012
Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S et al (2014) BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res 20:912–925. https://doi.org/10.1158/1078-0432.CCR-13-2281
Benesch M, Frappaz D, Massimino M (2012) Spinal cord ependymomas in children and adolescents. Childs Nerv Syst 28:2017–2028. https://doi.org/10.1007/s00381-012-1908-4
Benesch M, Weber-Mzell D, Gerber NU, von Hoff K, Deinlein F, Krauss J, Warmuth-Metz M, Kortmann RD, Pietsch T, Driever PH et al (2010) Ependymoma of the spinal cord in children and adolescents: a retrospective series from the HIT database. J Neurosurg Pediatr 6:137–144. https://doi.org/10.3171/2010.5.Peds09553
Bolin S, Borgenvik A, Persson CU, Sundstrom A, Qi J, Bradner JE, Weiss WA, Cho YJ, Weishaupt H, Swartling FJ (2018) Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 37:2850–2862. https://doi.org/10.1038/s41388-018-0135-1
Borgenvik A, Cancer M, Hutter S, Swartling FJ (2020) Targeting MYCN in molecularly defined malignant brain tumors. Front Oncol 10:626751. https://doi.org/10.3389/fonc.2020.626751
Bostrom A, von Lehe M, Hartmann W, Pietsch T, Feuss M, Bostrom JP, Schramm J, Simon M (2011) Surgery for spinal cord ependymomas: outcome and prognostic factors. Neurosurgery 68:302–308. https://doi.org/10.1227/NEU.0b013e3182004c1e (discussion 309)
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. https://doi.org/10.1016/j.cell.2013.09.034
Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, Jamin Y, Thway K, Robinson SP, Roels F et al (2016) Small molecule inhibitors of aurora-a induce proteasomal degradation of N-Myc in childhood neuroblastoma. Cancer Cell 30:357–358. https://doi.org/10.1016/j.ccell.2016.07.002
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Chen H, Liu H, Qing G (2018) Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther 3:5. https://doi.org/10.1038/s41392-018-0008-7
Constantini S, Miller DC, Allen JC, Rorke LB, Freed D, Epstein FJ (2000) Radical excision of intramedullary spinal cord tumors: surgical morbidity and long-term follow-up evaluation in 164 children and young adults. J Neurosurg 93:183–193. https://doi.org/10.3171/spi.2000.93.2.0183
Debiec-Rychter M, Lasota J, Alwasiak J, Liberski PP (1995) Recurrent anaplastic ependymoma with an abnormal karyotype and c-myc proto-oncogene overexpression. Acta Neuropathol 89:270–274. https://doi.org/10.1007/BF00309343
Ecker J, Oehme I, Mazitschek R, Korshunov A, Kool M, Hielscher T, Kiss J, Selt F, Konrad C, Lodrini M et al (2015) Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol Commun 3:22. https://doi.org/10.1186/s40478-015-0201-7
Ecker J, Thatikonda V, Sigismondo G, Selt F, Valinciute G, Oehme I, Muller C, Buhl JL, Ridinger J, Usta D et al (2021) Reduced chromatin binding of MYC is a key effect of HDAC inhibition in MYC amplified medulloblastoma. Neuro Oncol 23:226–239. https://doi.org/10.1093/neuonc/noaa191
Ellison DW, Aldape KD, Capper D, Fouladi M, Gilbert MR, Gilbertson RJ, Hawkins C, Merchant TE, Pajtler K, Venneti S et al (2020) cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol 30:863–866. https://doi.org/10.1111/bpa.12866
Faisal A, Vaughan L, Bavetsias V, Sun C, Atrash B, Avery S, Jamin Y, Robinson SP, Workman P, Blagg J et al (2011) The aurora kinase inhibitor CCT137690 downregulates MYCN and sensitizes MYCN-amplified neuroblastoma in vivo. Mol Cancer Ther 10:2115–2123. https://doi.org/10.1158/1535-7163.MCT-11-0333
Feldman WB, Clark AJ, Safaee M, Ames CP, Parsa AT (2013) Tumor control after surgery for spinal myxopapillary ependymomas: distinct outcomes in adults versus children: a systematic review. J Neurosurg Spine 19:471–476. https://doi.org/10.3171/2013.6.SPINE12927
Fouladi M, Park JR, Stewart CF, Gilbertson RJ, Schaiquevich P, Sun J, Reid JM, Ames MM, Speights R, Ingle AM et al (2010) Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children’s Oncology Group phase I consortium report. J Clin Oncol 28:3623–3629. https://doi.org/10.1200/JCO.2009.25.9119
Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PW, Scheie D, Kruse A, Harter PN, Kastelan M et al (2019) MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol 138:1075–1089. https://doi.org/10.1007/s00401-019-02056-2
Henssen A, Thor T, Odersky A, Heukamp L, El-Hindy N, Beckers A, Speleman F, Althoff K, Schafers S, Schramm A et al (2013) BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 4:2080–2095. https://doi.org/10.18632/oncotarget.1534
Ho B, Johann PD, Grabovska Y, De Dieu Andrianteranagna MJ, Yao F, Fruhwald M, Hasselblatt M, Bourdeaut F, Williamson D, Huang A et al (2020) Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol 22:613–624. https://doi.org/10.1093/neuonc/noz235
Hubbard GK, Mutton LN, Khalili M, McMullin RP, Hicks JL, Bianchi-Frias D, Horn LA, Kulac I, Moubarek MS, Nelson PS et al (2016) Combined MYC activation and Pten loss are sufficient to create genomic instability and lethal metastatic prostate cancer. Cancer Res 76:283–292. https://doi.org/10.1158/0008-5472.CAN-14-3280
Hummel TR, Wagner L, Ahern C, Fouladi M, Reid JM, McGovern RM, Ames MM, Gilbertson RJ, Horton T, Ingle AM et al (2013) A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: a Children’s Oncology Group phase 1 consortium study. Pediatr Blood Cancer 60:1452–1457. https://doi.org/10.1002/pbc.24541
Hutter S, Bolin S, Weishaupt H, Swartling FJ (2017) Modeling and targeting MYC genes in childhood brain tumors. Genes 8:107. https://doi.org/10.3390/genes8040107
Johnsen JI, Segerstrom L, Orrego A, Elfman L, Henriksson M, Kagedal B, Eksborg S, Sveinbjornsson B, Kogner P (2008) Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene 27:2910–2922. https://doi.org/10.1038/sj.onc.1210938
Jung M, Russell AJ, Liu B, George J, Liu PY, Liu T, DeFazio A, Bowtell DD, Oberthuer A, London WB et al (2017) A Myc activity signature predicts poor clinical outcomes in Myc-associated cancers. Cancer Res 77:971–981. https://doi.org/10.1158/0008-5472.CAN-15-2906
Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, Tamachi A, Tu WB, Penn LZ (2017) MYC deregulation in primary human cancers. Genes 8:151. https://doi.org/10.3390/genes8060151
Kim J, Eltoum I-EA, Roh M, Wang J, Abdulkadir SA (2009) Interactions between cells with distinct mutations in c-MYC and pten in prostate cancer. PLoS Genet 5:e1000542. https://doi.org/10.1371/journal.pgen.1000542
Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-van Meeteren A, van Vuurden D et al (2012) Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123:473–484. https://doi.org/10.1007/s00401-012-0958-8
Leary S, Kilburn L, Geyer JR, Olson JM, MacDonald T, Ellison D, Kocak M, Onar A, Ermoian R, Poussaint TY et al (2015) MB-32: feasibility of vorinostat combined with isotretinoin and chemotherapy for young children with newly diagnosed embryonal brain tumors: a pediatric brain tumor consortium study. Neuro Oncol 17:iii27–iii27. https://doi.org/10.1093/neuonc/nov061.108
Lin Y, Smith ZA, Wong AP, Melkonian S, Harris DA, Lam S (2015) Predictors of survival in patients with spinal ependymoma. Neurol Res 37:650–655. https://doi.org/10.1179/1743132815Y.0000000041
Lonjon M, Goh KY, Epstein FJ (1998) Intramedullary spinal cord ependymomas in children: treatment, results and follow-up. Pediatr Neurosurg 29:178–183. https://doi.org/10.1159/000028718
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. https://doi.org/10.1007/s00401-016-1545-1
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 23:1231–1251. https://doi.org/10.1093/neuonc/noab106
Lucchesi KM, Grant R, Kahle KT, Marks AM, DiLuna ML (2016) Primary spinal myxopapillary ependymoma in the pediatric population: a study from the Surveillance, Epidemiology, and End Results (SEER) database. J Neurooncol 130:133–140. https://doi.org/10.1007/s11060-016-2218-6
Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE et al (2012) Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488:49–56. https://doi.org/10.1038/nature11327
Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. https://doi.org/10.1016/j.ccell.2015.04.002
Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, Maier DL, Udaka YT, Murad N, Morrissy S et al (2016) HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell 29:311–323. https://doi.org/10.1016/j.ccell.2016.02.011
Prince EW, Balakrishnan I, Shah M, Mulcahy Levy JM, Griesinger AM, Alimova I, Harris PS, Birks DK, Donson AM, Davidson N et al (2016) Checkpoint kinase 1 expression is an adverse prognostic marker and therapeutic target in MYC-driven medulloblastoma. Oncotarget 7:53881–53894. https://doi.org/10.18632/oncotarget.10692
Raffeld M, Abdullaev Z, Pack SD, Xi L, Nagaraj S, Briceno N, Vera E, Pittaluga S, Lopes Abath Neto O, Quezado M et al (2020) High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun 8:101. https://doi.org/10.1186/s40478-020-00973-y
Roussel MF, Robinson GW (2013) Role of MYC in Medulloblastoma. Cold Spring Harb Perspect Med 3:a014308. https://doi.org/10.1101/cshperspect.a014308
Scheil S, Bruderlein S, Eicker M, Herms J, Herold-Mende C, Steiner HH, Barth TF, Moller P (2001) Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol 11:133–143. https://doi.org/10.1111/j.1750-3639.2001.tb00386.x
Shofuda T, Kanemura Y (2021) HDACs and MYC in medulloblastoma: how do HDAC inhibitors control MYC-amplified tumors? Neuro Oncol 23:173–174. https://doi.org/10.1093/neuonc/noaa292
Smith A, Onar-Thomas A, Ellison D, Owens-Pickle E, Wu S, Leary SES, Fouladi M, Merchant T, Gajjar A, Foreman N (2020) EPEN-54. ACNS0831, phase III randomized trial of post-radiation chemotherapy in patients with newly diagnosed ependymoma ages 1 to 21 years. Neuro Oncol 22:iii318–iii319. https://doi.org/10.1093/neuonc/noaa222.187
Sofuoglu OE, Abdallah A (2018) Pediatric spinal ependymomas. Med Sci Monit 24:7072–7089. https://doi.org/10.12659/MSM.910447
Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI (2008) Modelling Myc inhibition as a cancer therapy. Nature 455:679–683. https://doi.org/10.1038/nature07260
Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV (2015) MYC, metabolism, and cancer. Cancer Discov 5:1024–1039. https://doi.org/10.1158/2159-8290.CD-15-0507
Swanson AA, Raghunathan A, Jenkins RB, Messing-Junger M, Pietsch T, Clarke MJ, Kaufmann TJ, Giannini C (2019) Spinal cord ependymomas with MYCN amplification show aggressive clinical behavior. J Neuropathol Exp Neurol 78:791–797. https://doi.org/10.1093/jnen/nlz064
van Tilburg CM, Milde T, Witt R, Ecker J, Hielscher T, Seitz A, Schenk JP, Buhl JL, Riehl D, Fruhwald MC et al (2019) Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma, or leukemia. Clin Epigenetics 11:188. https://doi.org/10.1186/s13148-019-0775-1
Wang C, Zhang J, Yin J, Gan Y, Xu S, Gu Y, Huang W (2021) Alternative approaches to target Myc for cancer treatment. Signal Transduct Target Ther 6:117. https://doi.org/10.1038/s41392-021-00500-y
Wilson PE, Oleszek JL, Clayton GH (2007) Pediatric spinal cord tumors and masses. J Spinal Cord Med 30(Suppl 1):S15-20. https://doi.org/10.1080/10790268.2007.11753963
Wolf E, Eilers M (2020) Targeting MYC proteins for tumor therapy. Annu Rev Cancer Biol 4:61–75. https://doi.org/10.1146/annurev-cancerbio-030518-055826
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46:444–450. https://doi.org/10.1038/ng.2938
Acknowledgements
We thank the patient and their family for participating in our translational research protocol. We would like to acknowledge the Treehouse Childhood Cancer Initiative at the University of California Santa Cruz Genomics Institute.
Funding
We thank the Nationwide Foundation Pediatric Innovation Fund for generously supporting sequencing, data production, and analysis. This work was supported by fellowships of the Mildred-Scheel doctoral program of the German Cancer Aid and the German Academic Scholarship Foundation (D.R.G.) as well as the CERN Research Fellowship and the Ein Kiwi gegen Krebs-Foundation (to K.W.P.).
Author information
Authors and Affiliations
Contributions
MS, DK, EAS, MSA, JLF, DSO were involved in the clinical care of the patient. KMS, CD, EH, SL, KEM, JF, BK, KL, RP, PW, VM, RKW, ERM, CEC were involved in analysis and/or interpretation of genomic and transcriptomic sequencing and fluorescence in situ hybridization. DLT, CRP, DRB were involved in the histopathological work-up for this patient. JJ, DPR were involved in the radiologic work-up for this patient. DRG and KWP were involved in performing DNA methylation array-based analysis. MS and KMS drafted the initial manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was provided by the patient’s parents and assent was provided from the patient for genomic sequencing. This research is under a protocol approved by the Institutional Review Board at Nationwide Children’s Hospital (IRB17-00206).
Consent for publication
Written consent was provided by the patient’s parents for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Supplementary methods and data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shatara, M., Schieffer, K.M., Klawinski, D. et al. Clinically aggressive pediatric spinal ependymoma with novel MYC amplification demonstrates molecular and histopathologic similarity to newly described MYCN-amplified spinal ependymomas. acta neuropathol commun 9, 192 (2021). https://doi.org/10.1186/s40478-021-01296-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01296-2