
Abstract
The type 1 ryanodine receptor (RyR1) is an intracellular calcium (Ca2+) release channel on the sarcoplasmic/endoplasmic reticulum that is required for skeletal muscle contraction. RyR1 channel activity is modulated by ligands, including the activators Ca2+ and ATP. Patients with inherited mutations in RyR1 may exhibit muscle weakness as part of a heterogeneous, complex disorder known as RYR1-related myopathy (RYR1-RM) or more recently termed RYR1-related disorders (RYR1-RD). Guided by high-resolution structures of skeletal muscle RyR1, obtained using cryogenic electron microscopy, we introduced mutations into putative Ca2+ and ATP binding sites and studied the function of the resulting mutant channels. These mutations confirmed the functional significance of the Ca2+ and ATP binding sites identified by structural studies based on the effects on channel regulation. Under normal conditions, Ca2+ activates RyR1 at low concentrations (µM) and inhibits it at high concentrations (mM). Mutations in the Ca2+-binding site impaired both activating and inhibitory regulation of the channel, suggesting a single site for both high and low affinity Ca2+-dependent regulation of RyR1 function. Mutation of residues that interact with the adenine ring of ATP abrogated ATP binding to the channel, whereas mutating residues that interact with the triphosphate tail only affected the degree of activation. In addition, patients with mutations at the Ca2+ or ATP binding sites suffer from muscle weakness, therefore impaired RyR1 channel regulation by either Ca2+ or ATP may contribute to the pathophysiology of RYR1-RM in some patients.
Similar content being viewed by others


Significance
The ryanodine receptor/calcium release channel (RyR1) is required for skeletal muscle excitation–contraction (EC) coupling. Mutations in RyR1 that render the channel leaky (unable to close properly) to calcium (Ca2+) cause an inherited form of muscle weakness known as RyR1-related disorders (RyR1-RD). Using the high-resolution RyR1 structure solved in our laboratory we identified binding sites for the channel activators Ca2+ and ATP. Mutagenesis of these sites combined with functional studies confirmed that they are indeed the key ligand binding sites. Both the ATP and Ca2+ sites are involved in disease-causing mutations that alter the response of the channel to these physiological activators and likely contribute to the pathophysiology of RyR1-RD.
Introduction
Calcium is a vital second messenger [6, 38] that regulates numerous cellular signaling pathways, including muscle contraction [38], hormone secretion [57], and synaptic transmission [64]. Ryanodine receptors (RyRs) are located on the sarcoplasmic/endoplasmic reticulum (SR/ER) and mediate the release of Ca2+ from intracellular stores [56]. The three mammalian isoforms, RyR1, RyR2, and RyR3, share approximately 70% sequence identity. RyR1 and RyR2 are widely expressed and are the major SR Ca2+ release channels in skeletal and cardiac muscles, respectively [54, 77]. RyR3 was originally found in the brain, but it is also expressed in other tissues [58].
RyR1 is required for excitation–contraction (EC) coupling in skeletal muscle. RyR1 is a homotetramer comprised of four 565 kDa protomers and as such is the largest known ion channel. In addition, regulatory and targeting proteins for enzymes including protein kinase A (PKA) and CaM kinase II (CAMKII), are associated with the channel and regulate its function [41, 45].
RYR1-related myopathies (RYR1-RM), or as recently proposed RYR1-related disorders (RyR1-RD) [31], are rare, inherited disorders, the prevalence of which have likely been underestimated at 1:90,000 individuals [1]. Indeed, RYR1-RD is the most common form of non-dystrophic muscle disease and includes individuals with malignant hyperthermia susceptibility that affects ~ 1:3000–1:8500 and possibly as many as 1 in 400 [31]. RYR1-RD exhibits both autosomal dominant and recessive inheritance as well as de novo occurrences. RYR1-RD is characterized by pleotropic clinical presentations ranging from mild to severe muscle weakness, and moderate to severe respiratory insufficiency, which is more often apparent in recessive cases. Some mutations in RYR1 (19q13.2) result in leaky channels that promote muscle weakness and damage in RyR1-RD patients [27]. Although there are currently no approved treatments, a clinical trial using a novel Rycal drug that fixes the leak in RyR1 channels is currently underway at the NIH (NCT04141670).
The RyR1 macromolecular complex includes calstabin [43, 45, 69, 74], PKA [52], CaMKIIδ [26, 70], the phosphatases PP1 and PP2A [15, 42], the phosphodiesterase PDE4D3 [32, 57], sorcin [19], calmodulin [49, 51, 53, 66, 78], triadin [55], junction [33], and calsequestrin [4]. RyR channels are regulated by posttranslational modifications including phosphorylation [27, 44, 45], oxidation [2, 59, 60], and nitrosylation [5]. RyR channels exhibit a bell-shaped response to cytosolic Ca2+, with activation at micromolar levels and inhibition at millimolar concentrations [7]. ATP is a potent activator of RyR [48] and millimolar ATP concentrations enhance Ca2+-dependent activation of RyR1, manifested as increased open probability (Po) [8, 17, 30, 50, 63, 65]. In disease states, RyR channels may exhibit a stressed-induced leak that contributes to the pathophysiology of heart failure [22, 39, 40, 45], cardiac arrhythmias [69], diabetes [57], muscular dystrophy [3], age-dependent loss of muscle function [2], cancer-associated muscle weakness [68], post-traumatic stress disorder [36], Alzheimer’s Disease [9, 28], and Huntington’s Disease [16].
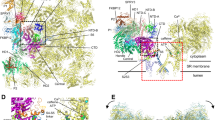
In 2015, three cryogenic electron microscopy (cryo-EM) studies, including our own, described the high-resolution architecture of the closed state of RyR1 [18, 73, 76], revealing that RyR1 belongs to the six transmembrane (6TM) cation channel family. As opposed to most members of the 6TM family, RyR is not voltage gated; however, it contains an evolutionarily conserved pseudo-voltage-sensor domain (pVSD) [76] which lacks the positively charged residues present in voltage-gated channels. We also solved the structure of the open state of RyR1, activated by Ca2+, ATP, and caffeine, revealing the structural basis of channel gating and ligand-dependent activation of RyR1 [14]. The cytosolic shell of RyR is composed of alpha-solenoid repeats, including two N-terminal beta-trefoil domains (NTD-A and NTD-B) [67], three SPRY domains (SPRY1-SPRY3) [29], two pairs of RYR repeats (RY1&2 and RY3&4) [61, 75], and a pair of EF-hands (EF1&2) [71] inserted in the core solenoid [76]. The activation domain contains a thumb and forefinger motif (TaF), which clamps the zinc finger-containing C-terminal domain (CTD) and provides allosteric coupling between the movement of the cytosolic shell and dilation of the pore aperture. The core solenoid (C-Sol), which is part of the activation domain, links the pore domain to the shell. Binding sites for Ca2+, caffeine, and ATP were identified at interdomain interfaces of the C-terminal domain and the transmembrane domain [14] where they likely stabilize interdomain interactions and amplify the effects of Ca2+ binding on the gating of the channel pore (Fig. 1).

Our previous work examined the pathophysiological mechanisms underlying RyR1-RD [27]. The present study extends these observations at the atomic level by focusing on structure/function studies of the ATP and Ca2+ binding sites and disease causing mutations that affect these sites in patients with RyR1-myopathy. In the present study, we used site-directed mutagenesis to assess the functional importance of the ATP and Ca2+ binding sites that we previously identified using cryo-EM. Moreover, RyR1 channel mutations found at the Ca2+ and ATP binding sites of patients with RYR1-RD resulted in defective regulation by Ca2+ and ATP that may contribute to muscle weakness in RYR1-RD patients.
Results
Architecture and function of the RyR1 ATP binding site
The ATP binding site of RyR1 is located at the junction of the cytoplasmic extension of S6 (S6c) tranmembrane helix and the CTD [14]. Based on the structure, T4979 of the CTD contributes to the adenine ring binding site, and the positively charged K4211, K4214, and R4215 residues of the TaF interact with the triphosphate tail of ATP (Fig. 2A). Based on this model, we hypothesized that mutation of T4979 would reduce ATP binding to RyR1, whereas mutation of K4211, K4214, and R4215 would reduce ATP-dependent activation of RyR1, since ADP and AMP are less effective activators of RyR1 [10, 30].

Structure of the ATP binding site. A The adenine ring binding site (T4979) and triphosphate tail interacting residues (K4211, K4214, and R4215) are labeled and depicted in stick representation. B [α-32P]-ATP/[3H]-ryanodine binding to ER microsomes of HEK293 cells expressing WT RyR1, T4979F, and K4211S/K4214S/R4215S. C, [3H]-ryanodine binding to ER microsomes of HEK293 cells expressing WT RyR1 and ATP binding site mutants in response to ATP. Data are presented as the mean ± S.E.M. N = 3 for each group. **P < 0.01
To assess the functional effects of the ATP binding site. T4979 was mutated to phenylalanine (T4979F), near the adenine ring of ATP, and a triple mutant of K4211/K4214/R4215, each to serine, near the triphosphate tail of ATP was generated. These mutants were made in recombinant rabbit RyR1 and expressed in HEK293 cells. To assess the effects of these mutations, [α-32P]-ATP binding, [3H]-ryanodine binding, which is a surrogate measure of channel activity, and single channel recordings in planar lipid bilayers were performed. The RyR1 T4979F mutant channel exhibited reduced [α-32P]-ATP binding, with a Bmax of 2.5 ± 0.25 pmol ATP/pmol RyR1 compared to a Bmax of 8.0 ± 0.77 pmol ATP/pmol RyR1 for WT RyR1. These data confirm that T4979 is a critical residue for ATP binding to RyR1. Furthermore, the Bmax of 8 suggests that there is likely a second ATP binding site in RyR1 with two molecules of ATP are binding per protomer of the homotetrameric channel. In contrast, the K4211S/K4214S/R4215S mutant channel exhibited normal [α-32P]-ATP binding to RyR1 (Fig. 2B), indicating that the complete triphosphate tail is not required for ATP binding to RyR1.
We then used [3H]-ryanodine binding to further evaluate activation of RyR1 by ATP binding. Ryanodine binds to pore of the RyR1 channel in the open state [12, 14] and therefore binding can be used as an indicator of channel activity. In the absence of ATP, the levels of [3H]-ryanodine binding to T4979F RyR1 and K4211S/K4214S/R4215S RyR1 were similar to that of the WT RyR1 (Fig. 2C). In contrast, in the presence of 1 mM ATP, WT RyR1 exhibited significantly increased [3H]-ryanodine binding indicating channel activation, whereas T4979F and K4211S/K4214S/R4215S mutant channels did not (Fig. 2C). Thus, the ATP binding site identified by cryo-EM [14] is a functional site that regulates activity of RyR1.
Previously, we reported that in the presence of 30 µM [Ca2+]cyt, the open probability of WT RyR1 channels was ~ 20%, while in the presence of Ca2+/ATP/caffeine (30 µM, 1 mM, 2 mM), the open probability was ~ 90% [14]. This finding is consistent with many previous reports from multiple laboratories [47]. Single channel recordings were used as an additional assessment of the activation of RyR1 by ATP. WT RyR1 channels exhibited an open probability (Po) of 20%, mean open time (To) of 2.1 ms and mean closed time (Tc) of 30.1 ms at 10 µM [Ca2+]cyt. Mutant RyR1 channels T4979F and K4211S/K4214S/R4215S exhibited similar single channel properties compared to WT RyR1 under this condition, suggesting normal Ca2+ dependent activation. Addition of 1 mM ATP dramatically increased WT RyR1 Po (Fig. 3A, B), with increased To and reduced Tc of single RyR1 channels (Fig. 3A, C, D). However, 1 mM ATP had no effect on the Po of T4979F or K4211S/K4214S/R4215S mutant channels (Fig. 3A–D). These data further indicate that these mutations eliminate ATP-dependent activation of RyR1 and confirm the functional importance of the ATP binding site idenbtified using cryo-EM (Fig. 2C).

Effects of ATP binding site mutations on the gating of RyR1 channels. A Representative single channel traces of WT RyR1 (top), T4979F (middle), and K4211S/K4214S/R4215S (bottom) under 10 µM Ca2+ only (left) or 10 µM Ca2+ and 1 mM ATP (right). B Po, C To, and D Tc of single WT RyR1, T4979F, and K4211S/K4214S/R4215S mutants. Data are presented as mean ± S.E.M from 6 single channels for each group. *P < 0.05
Architecture and function of the RyR1 Ca2+ binding site
Comparison of difference maps calculated from RyR1 preparations with or without 30 μM Ca2+ revealed a Ca2+ binding site located at the interface of the CTD and the C-Sol [14]. This Ca2+ binding site is primarily comprised of five amino acids (Fig. 4A) which are conserved between RyR and the homologous inositol trisphosphate receptor (IP3R) channels [14, 21]. E3893 and E3967 from the C-Sol and T5001 from the CTD directly interact with Ca2+, and H3895 and Q3970 from the C-Sol indirectly interact with Ca2+ (Fig. 4A). In order to assess Ca2+-dependent ryanodine binding, we mutated E3893 and E3967 to either alanine or aspartic acid, expressed each mutant in HEK293 cells, and then determined [3H]-ryanodine binding to isolated ER vesicles. As shown in Fig. 4B, WT RyR1 exhibits a bell-shaped Ca2+ response with peak activation at 100 μM Ca2+. In contrast, E3893A, E3893D, E3967A, and E3967D mutant RyR1 channels exhibited both impaired activation at low [Ca2+] and impaired deactivation at high [Ca2+] (Fig. 4B).

Structure of the calcium binding site. A ribbon structure of the Ca2+ binding site showing interacting residues. B [3H]-ryanodine binding to ER microsomes of HEK293 cells expressing WT RyR1 and calcium binding site mutants. Data are presented as mean ± S.E.M from 6 single channels for each group. **P < 0.01
To further assess the role of the Ca2+ binding site in RyR1 channel gating, we examined the single channel properties of the E3893A, E3893D, E3967A, and E3967D RyR1 channel mutants. WT and Ca2+-binding site mutants appropriately displayed low Po at 150 nM cytosolic Ca2+ (Fig. 5); however, at 10 μM [Ca2+]cyt, WT RyR1 channels were activated (Fig. 5A), whereas E3893A, E3893D, E3967A, and E3967D mutant channels were not activated (Fig. 5B–F). Furthermore, 10 mM cytosolic Ca2+ inhibited RyR1 WT channels, but the Ca2+-binding site mutants were not inhibited (Fig. 5B–F). Thus, the Ca2+-binding site mutants were insensitive to both Ca2+-dependent activation at [Ca2+]cyt below 100 μM, and Ca2+-dependent inhibition at [Ca2+]cyt above 1 mM (Fig. 5B–F). Furthermore, these data suggest that a single Ca2+-binding site confers both the high and low affinity [Ca2+]cyt dependence of RyR1 channel activity.

Effects of mutations in the Ca2+ binding site on the gating of RyR1 channels. Representative single channel traces of WT RyR1 (A), E3893A (B), E3893D (C), E3967A (D), and E3967D (E), under 150 nM Ca2+, 10 µM Ca2+, or 10 mM Ca2+. F, Po versus Ca2+ concentration curve. Data are presented as mean ± S.E.M from 6 single channels for each group
RYR1-RD-associated mutations near ATP and Ca2+ binding sites
RYR1 mutations can cause skeletal muscle dysfunction in children and adults, resulting in a wide range of disabilities, and are the most common cause of congenital myopathy [27]. We have established an RYR1-RD database by assembling genetic, structural, biophysical, and clinical information from more than 2200 RYR1-RM affected patients [27]. This database contains an RyR1 mutation at T4980M (rabbit RyR1 T4979M) associated with congenital myopathy [23, 24, 37]. RyR1-T4980 is located in the ATP binding site where it may interact with the adenine ring of ATP (Fig. 2A). The patient in the database with this mutation also had a second mutation, A538T; however, this mutation is located in the NTD of RyR1, which is far from ligand binding sites and myopathy hotspots. To study the effects of these RyR1 myopathic mutations, T4979M, A538T and A538T/T4979M mutant channels were expressed in HEK293 cells. 1 mM ATP did not increase the Po of RyR1 T4979M and A538T/T4979M mutant channels, whereas the mutant RyR1 A538T channel responded normally to ATP (Fig. 6A). A538T channels exhibited similar ATP-dependent activation as WT RyR1 channels (Fig. 6B). RyR1 T4979M and A538T/T4979M mutant channels also displayed significantly decreased [α-32P]-ATP binding compared to WT RyR1 channels, while RyR1 A538T exhibited normal ATP binding (Fig. 6C), Taken together, these results show that mutation of threonine to methionine at 4980 of human RyR1 significantly attenuates ATP binding and activation of the RyR1 channel. Impaired ATP-dependent activation of RyR1 may contribute to muscle weakness in RYR1-RD affected patients.

ATP binding and single channel analyses of the RyR1-RM patient-related ATP binding site mutants. A [α-32P]-ATP/ [3H]-ryanodine binding to ER microsomes of HEK293 cells expressing WT RyR1, T4979M, A538T and A538T/T4979M. B Single channel analysis of WT RyR1, T4979M, A538T and A538T/T4979M (bottom) under 10 µM Ca2+ only (left) or 10 µM Ca2+ and 1 mM ATP (right). C [3H]-ryanodine binding to ER microsomes of HEK293 cells expressing WT RyR1, T4979M, A538T and A538T/T4979M. Data are presented as mean ± S.E.M from 6 for each group
There are also RYR1-RD associated mutations near the Ca2+-binding site, including the human RyR1 mutation Q3969K (rabbit Q3970K) and S4028L. The Q3970K mutation is linked to a form of RYR1-RD formerly referred to as multi-minicore disease [62]. Q3970K mutant channels displayed a right shift in Ca2+ dependent activation measured using single channel determinations and [3H]-ryanodine binding (Fig. 7A–C). Our previous work regarding the S4028L mutation showed that the RyR1 channel from this patient’s muscle biopsy exhibited elevated Po at low, non-activating [Ca2+]cyt, consistent with a leaky RyR1 channel that likely plays a role in the patient’s muscle weakness [27]. To assess the role of posttranslational modifications in determining the leaky behavior of disease-associated mutant RyR1 channels, RyR1-S4028L patient muscle lysates were treated with protein phosphatase 1 (PP1) and the reducing agent dithiothreitol (DTT), to reverse PKA phosphorylation and oxidation of the channel. Following this treatment, the mutant RyR1-RD linked RyR1-S4028L channels still exhibited increased sensitivity to Ca2+-dependent activation and showed increased activity at very low non-activating [Ca2+] ~ 150 nM, which is consistent with channel leak, albeit to a lesser extent than the phosphorylated and oxidized mutant channels (Additional file 1: Figure 1). These data indicate that the RyR1-RD linked mutation alone increases the sensitivity of the channel to Ca2+-dependent activation rendering the channel leaky and that stress-induced posttranslational modifications further exacerbate the channel dysfunction and resultant leak.

Single channel analysis and [3H]-binding of the RyR1-RM patient-related Ca2+ binding site mutants. A Representative single channel traces of WT RyR1 and Q3970K under 150 nM Ca2 + , 10 µM Ca2+, or 10 mM Ca2+. B Po versus Ca2+ concentration curve. C [3H]-ryanodine binding to ER microsomes of HEK293 cells expressing WT RyR1 and Q3970K mutant. Data are presented as mean ± S.E.M from 6 single channels for each group
Discussion
We previously solved the structure of RyR1 to near-atomic resolution using cryo-EM and identified ATP and Ca2+ binding sites [14, 76]. In the present study, we have characterized the function of the ATP and Ca2+ binding sites of RyR1 using mutagenesis and measurements of channel activity. Importantly, the RYR1-RD linked mutation T4980M, which is located in the ATP binding site, impairs ATP binding and ATP-dependent activation of the RyR1 channel. The RyR1-RD linked mutation Q3969K, which is located in the Ca2+ binding site, abolishes Ca2+-dependent activation. These findings suggest that interference with Ca2+- and ATP-dependent regulation of RyR1 may contribute to the pathophysiology of RyR1-RD including muscle weakness.
In skeletal muscle, the cytosolic ATP concentration is about 5 mM [25], but under physiological conditions, ATP regulation of RyR1 is influenced by Mg2+, a potent inhibitor of the RyR [13], and most cellular ATP is present as MgATP. Other nucleotides, such as CTP, GTP, ITP, and UTP, have no effect on RyR activity [46], which is consistent with our finding that the adenine base binding site is required for ATP binding to the RyR. We showed that T4979 is required for proper ATP-dependent regulation of RyR1 as RyR1-T4979F shows no binding with [α-32P] ATP. It is possible that the introduction of phenylalanine with a bulky hydrophobic group in the binding site of the adenine of ATP (T4979F) prevents the entry of ATP into that site, thus explaining why ATP binding to the T4979F mutant RyR1 is reduced. Other adenine nucleotides such as ADP, AMP, or adenosine can also increase the Po of RyR1, but with reduced efficacy [10, 30] suggesting that the phosphate groups of ATP are required for robust activation of RyR1. Previous work has reported that the triphosphate groups are the most important element for inducing a long open state in RyR2 [35], in the present study we have extended these studies by presenting data identifiying the specific amino acid residues in RyR1 that are responsible for binding to the triphosphate tail of ATP in it’s binding site. Indeed, in the present study, we demonstrate the structural basis for the critical role of the triphosphate tail of ATP in the activation of the RyR as the K4211S/K4214S/R4215S mutant RyR1, which replaces the positively charged lysine and arginine with a neutral serine, disrupts ATP-dependent activation of RyR1. The reduction in positively charged residues is presumed to decrease binding to the triphosphate tail of ATP in a way that mimics the reduced interaction of ADP and AMP with RyR1, both of which are much weaker activators of the channel [10, 30]. Since ATP is always present at mM levels in muscle it is reasonable to hypothesize that its binding to RyR1 is required for robust activation of the channel by Ca2+. The mutant channels which cannot bind ATP are likely less active and may contribute to impaired muscle contractility and weakness in RYR1-RD patients.
Global cytosolic [Ca2+]cyt in resting cells is approximately 100–150 nM and rises to at least 1 µM following Ca2+ release through RyR1 during EC coupling. Mutations of RyR1 residues E3893 and E3967 to either A or D significantly reduced the high affinity Ca2+-dependent-activation of RyR1 compared to the WT channel (Fig. 4), which is consistent with previous work showing that mutation of those 2 Glutamic acid residues to Glutamine (Q) or Valine (V) interfered with Ca2+ regulation of the channel [72]. However, unexpectedly, these same mutations also prevented the low affinity Ca2+-dependent deactivation of RyR1. This finding suggests that the Ca2+-dependent activation and inhibition involves a single Ca2+ binding site. One possible mechanism to explain this phenomenon is that at low Ca2+ concentration (from nM to µM), this Ca2+ binding site was occupied by one Ca2+, which forms a brige between the CTD and CSol to stabilize the open state of the RyR1 channel, whereas at high Ca2+ concentration (mM), CTD and CSol each bind to one Ca2+, which disrupts the CTD-CSol interface to make the channel close. A patient with a Q3970K mutation at this site exhibited the same impaired Ca2+-dependent activation and deactivation as the E3893A/D and E3967A/D mutant channels, which is consistent with a previous study showing that RyR1-Q3970K displayed low Ca2+ dependent channel activity [11]. It is likely that the additional positive charge from the lysine substitution in the Q3970K mutant channel reduces Ca2+-binding at this site. Similarly, the replacement of glutamic acid at 3967 or 3893 with the neutral alanine may reduce the affinity of the Ca2+ and thus impair both activation and deactivation, indicating the negative charges of glutamic acid are critical for the Ca2+ binding of RyR1. Replacing the glutamic acid at 3967 with an aspartic acid preserves the negative charge; however, since the side chain of glutamate is larger than that of aspartic acid, the interaction with Ca2+ may be weakened.
A previous study suggested that E4032 is part of the Ca2+ binding site of RyR1, as mutation of E4032 reduced Ca2+ activation in both RyR1 and RyR2 [20, 34]. However, our cryo-EM RyR1 structure indicates that E4032 is not close enough to the Ca2+ binding site to form a direct interaction with the bound Ca2+. Nevertheless, it may stabilize the CTD-CSol interface via hydrogen bonding to the amide nitrogens at the end of one of the CTD helices [14]. Interestingly, a mutant channel RyR1-S4028L which has been linked to RYR1-RM [27], exhibited increased Ca2+-dependent activation of RyR1 at low non-activating [Ca2+]cyt, which is consistent with channel leak [27], suggesting that the polar side chain of serine is necessary to stabilize the CTD-CSol interface. Moreover, blocking or reversing post-translational modifications of RyR1 (PKA phosphorylation and oxidation) revealed that the channel mutation alone is sufficient to cause Ca2+ leak and that the posttranslational modications are additive in terms of leak.
The present study identifies functional ATP and Ca2+-dependent regulatory sites in RyR1. Moreover, these are also the sites of RyR1-RM disease causing mutations, indicating that defective regulation of RyR1 by Ca2+ and ATP may be a component of the pathophysiology of this form of myopathy.
Materials and methods
Ryanodine receptor mutagenesis and expression
The recombinant RyR1 constructs T4979F, K4211S/K4214S/R4215S, E3893A, E3893D, E3967A, E3967D, T4979M, and Q3970K were generated by introducing the respective mutations into fragments of rabbit RyR1 using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent). Each fragment was subsequently subcloned into a full length RyR1 construct in pcDNA3.1 vector and confirmed by sequencing. The primers used to introduce specific mutants (codons in parentheses, mutated nucleotides in bold) are as follows: 5′-cacggcttcgagacccac(ttc)ctagaggagcacaatctg for T4979F, 5′-gtgggagatgccccaggtc(agc)gagtcc(agc)(agc)cagttcatcttc for K4211S-K4214S-R4215S, 5'-gcagctgctctgt(gcg)gggcacaacaacg for E3893A, 5′-ctgcagctgctctgt(gac)gggcacaac for E3893D, 5′-caacagcctcacc(gcg)tacatccagggcc for E3967A, 5′-acagcctcacc(gac)tacatccagggcc for E3967D, 5′-cacggcttcgagacccac(atg)ctagaggag for T4979M, and 5′-ctcaccgagtacatc(aag)ggcccctgcac for Q3970K. For all mutants, the second primer was the complementary reverse to the forward primer. HEK293 cells grown in DMEM supplemented with 10% (vol/vol) FBS (Invitrogen), penicillin (100 U/mL), streptomycin (100 µg/mL), and L-glutamine (2 mmol/L) were transfected with WT or mutant RyR1 cDNA using Lipofectamine 2000 (ThermoFisher Scientific). Cells were collected 48 h after transfection.
ER vesicles preparation
ER vesicles from HEK293 cells expressing WT or mutant RyR1 were prepared by homogenizing cell pellets on ice using a Teflon-glass homogenizer with two volumes of solution containing 20 mmol/L (mM) Tris-maleate (pH 7.4), 1 mM EDTA, 1 mM DL-Dithiothreitol (DTT) and protease inhibitors (Roche). Homogenate was then centrifuged at 4,000 xg for 15 min at 4 °C and the resulting supernatant was centrifuged at 40,000 xg for 30 min at 4 °C. The final pellet, containing the ER fractions, was resuspended and aliquoted in 250 mM sucrose, 10 mM MOPS (pH 7.4), 1 mM EDTA, 1 mM DTT and protease inhibitors. Samples were frozen in liquid nitrogen and stored at −80 °C.
SR microsome preparation
Skeletal muscle SR microsomes were prepared as previsouly described [27]. Briefly, muscle samples were homogenized on ice using a Teflon-glass homogenizer with 2 volumes of: 20 mmol/L (mM) Tris-maleate (pH 7.4), 1 mM EDTA, 1 mM DL-dithiothreitol (DTT) and protease inhibitors (Roche). The resulting homogenate was then centrifuged at 4,000 g for 15 min at 4 °C and the supernatant was centrifuged at 50,000 g for 45 min at 4 °C. Pellets were resuspended in lysis buffer containing 300 mM sucrose.
[3H] Ryanodine and [α-32P]-ATP binding
Skeletal muscle SR microsomes or ER vesicles from HEK293 cells expressing WT or mutant RyR1 were incubated in media containing 5 nM [3H]-ryanodine or 5 nM [α-32P]-ATP, 1 M NaCl, 20 mM HEPES, and 0.5 mM EGTA at 37 °C for 2 h. The concentration of free Ca2+ was calculated with WinMaxC (version 2.50; www.stanford.edu/~cpatton/maxc.html). For ATP activation, 1 mM ATP and 1 mM free Ca2+ were added during incubation. The binding mix was then filtered through Whatman GF/B filters presoaked with 1% polyethyleneimine. The filters were washed three times with 5 mL of ice-cold washing buffer containing 0.2 M NaCl and 5 mM HEPES (pH 7.5) to remove unbound [3H]-ryanodine, and the amount of remaining [3H]-ryanodine was determined by liquid scintillation counting. Nonspecific binding was determined by measuring [3H]-ryanodine binding in the presence of 10 μM unlabeled ryanodine. All binding assays were done in duplicate.
Single-channel recordings
ER vesicles were fused to planar lipid bilayers formed by painting a lipid mixture of phosphatidylethanolamine and phosphatidylcholine (Avanti Polar Lipids) in a 5:3 ratio in decane across a 200 µm hole in polysulfonate cups (Warner Instruments) separating two chambers. The trans chamber (1.0 mL), representing the intra-SR (luminal) compartment, was connected to the head stage input of a bilayer voltage clamp amplifier. The cis chamber (1.0 mL), representing the cytoplasmic compartment, was held at virtual ground. Asymmetrical solutions used were as follows for the cis solution: 1 mM EGTA, 250/125 mM Hepes/Tris, 50 mM KCl, 0.64 mM CaCl2, pH 7.35; and for the trans solution: 53 mM Ca(OH)2, 50 mM KCl, 250 mM Hepes, pH 7.35. The concentration of free Ca2+ in the cis chamber was calculated as previously described [14]. ER vesicles were added to the cis side and fusion with the lipid bilayer was induced by making the cis side hyperosmotic by the addition of 400–500 mM KCl. After the appearance of potassium and chloride channels, the cis side was perfused with the cis solution. At the end of each experiment, 10 µM ryanodine was added to block the RyR channel. Single-channel currents were recorded at 0 mV using a Bilayer Clamp BC-525D (Warner Instruments), filtered at 1 kHz using a Low-Pass Bessel Filter 8 Pole (Warner Instruments), and digitized at 4 kHz. All experiments were performed at room temperature (23 °C). Data acquisition was performed by using Digidata 1322A and Axoscope 10.1 software (Axon Instruments). The recordings were analyzed using Clampfit 10.1 (Molecular Devices) and Graphpad Prism software.
Immunoprecipitation and immunoblotting
RyR1 were immunoprecipitated from extracts of human patient muscle biopsy using anti- anti-RyR1-specific antibodies (2 μg) in 0.5 ml of a modified radioimmune precipitation assay buffer (50 mm Tris–HCl, pH 7.2, 0.9% NaCl, 5.0 mm NaF, 1.0 mm Na3VO4, 1% Triton X-100 and protease inhibitors) overnight at 4 °C as previously described [2]. The immune complexes were incubated with protein A-Sepharose beads (Sigma-Aldrich) at 4 °C for 1 h and the beads were washed three times with the modified radioimmune precipitation assay buffer. The immunoprecipitated proteins were size-fractionated on SDS–polyacrylamide gels (4–20% for RyR1) and transferred to nitrocellulose membranes for 2 h at a current of 200 mA. Immunoblots were probed with the following primary antibodies: anti-RyR1 (Affinity Bioreagents, 1:2,000 dilution), anti-Cys-NO (Sigma-Aldrich, 1:1,000 dilution), or anti-phospho-RyR-Ser(P)-2844 (Affinity Bioreagents, 1:5,000 dilution). To determine channel oxidation, the carbonyl groups in the protein side chains were derivatized to 2,4-dinitrophenol (DNP) by reaction with 2,4-dinitrophenylhydrazine. The DNP signal associated with total oxidized protein or with RyR was determined using a specific anti-DNP antibody according to the manufacturer's instructions (Millipore). All immunoblots were developed using an Odyssey system (LI-COR Biosciences), with infrared-labeled anti-mouse or anti-rabbit IgG (Abcam, 1:10,000 dilution) secondary antibodies.
Statistics
All results are presented as the mean ± SEM. Statistical analyses were performed using the unpaired Student’s t test, 2-tailed (for 2 groups), or the 1-way ANOVA with Tukey–Kramer post hoc correction (for groups of 3 or more) unless otherwise indicated. P < 0.05 was considered to be statistically significant.
Availability of data and materials
The data supporting the findings of this are documented within the paper and are available from the corresponding author upon request.
References
Amburgey K, McNamara N, Bennett LR, McCormick ME, Acsadi G, Dowling JJ (2011) Prevalence of congenital myopathies in a representative pediatric united states population. Ann Neurol 70:662–665. https://doi.org/10.1002/ana.22510
Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR (2011) Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 14:196–207. https://doi.org/10.1016/j.cmet.2011.05.014
Andersson DC, Meli AC, Reiken S, Betzenhauser MJ, Umanskaya A, Shiomi T, D’Armiento J, Marks AR (2012) Leaky ryanodine receptors in beta-sarcoglycan deficient mice: a potential common defect in muscular dystrophy. Skelet Muscle 2:9. https://doi.org/10.1186/2044-5040-2-9
Beard NA, Dulhunty AF (2015) C-terminal residues of skeletal muscle calsequestrin are essential for calcium binding and for skeletal ryanodine receptor inhibition. Skelet Muscle 5:6. https://doi.org/10.1186/s13395-015-0029-7
Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 15:325–330. https://doi.org/10.1038/nm.1916
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529. https://doi.org/10.1038/nrm1155
Bezprozvanny I, Watras J, Ehrlich BE (1991) Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351:751–754. https://doi.org/10.1038/351751a0
Blayney L, Beck K, MacDonald E, D’cruz L, Nomikos M, Griffiths J, Thanassoulas A, Nounesis G, Lai FA (2013) ATP interacts with the CPVT mutation-associated central domain of the cardiac ryanodine receptor. Biochim Biophys Acta 1830:4426–4432. https://doi.org/10.1016/j.bbagen.2013.05.038
Bussiere R, Lacampagne A, Reiken S, Liu X, Scheuerman V, Zalk R, Martin C, Checler F, Marks AR, Chami M (2017) Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J Biol Chem 292:10153–10168. https://doi.org/10.1074/jbc.M116.743070
Chan WM, Welch W, Sitsapesan R (2000) Structural factors that determine the ability of adenosine and related compounds to activate the cardiac ryanodine receptor. Br J Pharmacol 130:1618–1626. https://doi.org/10.1038/sj.bjp.0703459
Chirasani VR, Xu L, Addis HG, Pasek DA, Dokholyan NV, Meissner G, Yamaguchi N (2019) A central core disease mutation in the Ca(2+)-binding site of skeletal muscle ryanodine receptor impairs single-channel regulation. Am J Physiol Cell Physiol 317:C358–C365. https://doi.org/10.1152/ajpcell.00052.2019
Clarke OB, Hendrickson WA (2016) Structures of the colossal RyR1 calcium release channel. Curr Opin Struct Biol 39:144–152. https://doi.org/10.1016/j.sbi.2016.09.002
Copello JA, Barg S, Sonnleitner A, Porta M, Diaz-Sylvester P, Fill M, Schindler H, Fleischer S (2014) Differential activation by Ca2+, ATP and Caffeine of cardiac and skeletal muscle ryanodine receptors after block by Mg2+. J Membr Biol 187:51–64. https://doi.org/10.1007/s00232-001-0150-x
Desgeorges A, Clarke OB, Zalk R, Yuan Q, Condon KJ, Grassucci RA, Hendrickson WA, Marks AR, Frank J (2016) Structural basis for gating and activation of RyR1. Cell 167:145-157 e117. https://doi.org/10.1016/j.cell.2016.08.075
DeSouza N, Reiken S, Ondrias K, Yang YM, Matkovich S, Marks AR (2002) Protein kinase A and two phosphatases are components of the inositol 1,4,5-trisphosphate receptor macromolecular signaling complex. J Biol Chem 277:39397–39400. https://doi.org/10.1074/jbc.M207059200
Dridi H, Liu X, Yuan Q, Reiken S, Yehia M, Sittenfeld L, Apostolou P, Buron J, Sicard P, Matecki S et al (2020) Role of defective calcium regulation in cardiorespiratory dysfunction in Huntington’s disease. JCI Insight. https://doi.org/10.1172/jci.insight.140614
Dulhunty AF, Laver D, Curtis SM, Pace S, Haarmann C, Gallant EM (2001) Characteristics of irreversible ATP activation suggest that native skeletal ryanodine receptors can be phosphorylated via an endogenous CaMKII. Biophys J 81:3240–3252. https://doi.org/10.1016/S0006-3495(01)75959-0
Efremov RG, Leitner A, Aebersold R, Raunser S (2015) Architecture and conformational switch mechanism of the ryanodine receptor. Nature 517:39–43. https://doi.org/10.1038/nature13916
Farrell EF, Antaramian A, Benkusky N, Zhu X, Rueda A, Gomez AM, Valdivia HH (2004) Regulation of cardiac excitation-contraction coupling by sorcin, a novel modulator of ryanodine receptors. Biol Res 37:609–612
Fessenden JD, Chen L, Wang Y, Paolini C, Franzini-Armstrong C, Allen PD, Pessah IN (2001) Ryanodine receptor point mutant E4032A reveals an allosteric interaction with ryanodine. Proc Natl Acad Sci USA 98:2865–2870. https://doi.org/10.1073/pnas.041608898
Harnick DJ, Jayaraman T, Ma Y, Mulieri P, Go LO, Marks AR (1995) The human type 1 inositol 1,4,5-trisphosphate receptor from T lymphocytes. Structure, localization, and tyrosine phosphorylation. J Biol Chem 270:2833–2840
Huang F, Shan J, Reiken S, Wehrens XH, Marks AR (2006) Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: rescue of heart failure by calstabin2 in mice. Proc Natl Acad Sci USA 103:3456–3461
Klein A, Jungbluth H, Clement E, Lillis S, Abbs S, Munot P, Pane M, Wraige E, Schara U, Straub V et al (2011) Muscle magnetic resonance imaging in congenital myopathies due to ryanodine receptor type 1 gene mutations. Arch Neurol 68:1171–1179. https://doi.org/10.1001/archneurol.2011.188
Klein A, Lillis S, Munteanu I, Scoto M, Zhou H, Quinlivan R, Straub V, Manzur AY, Roper H, Jeannet PY et al (2012) Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum Mutat 33:981–988. https://doi.org/10.1002/humu.22056
Kushmerick MJ, Moerland TS, Wiseman RW (1992) Mammalian skeletal muscle fibers distinguished by contents of phosphocreatine, ATP, and Pi. Proc Natl Acad Sci USA 89:7521–7525
Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR (2010) Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci USA 107:10274–10279. https://doi.org/10.1073/pnas.1005843107
Kushnir A, Todd JJ, Witherspoon JW, Yuan Q, Reiken S, Lin H, Munce RH, Wajsberg B, Melville Z, Clarke OB et al (2020) Intracellular calcium leak as a therapeutic target for RYR1-related myopathies. Acta Neuropathol 139:1089–1104. https://doi.org/10.1007/s00401-020-02150-w
Lacampagne A, Liu X, Reiken S, Bussiere R, Meli AC, Lauritzen I, Teich AF, Zalk R, Saint N, Arancio O et al (2017) Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol: https://doi.org/10.1007/s00401-017-1733-7
Lau K, Van Petegem F (2014) Crystal structures of wild type and disease mutant forms of the ryanodine receptor SPRY2 domain. Nat Commun 5:1–11. https://doi.org/10.1038/ncomms6397
Laver DR, Lenz GK, Lamb GD (2001) Regulation of the calcium release channel from rabbit skeletal muscle by the nucleotides ATP, AMP, IMP and adenosine. J Physiol 537:763–778. https://doi.org/10.1111/j.1469-7793.2001.00763.x
Lawal TA, Todd JJ, Witherspoon JW, Bonnemann CG, Dowling JJ, Hamilton SL, Meilleur KG, Dirksen RT (2020) Ryanodine receptor 1-related disorders: an historical perspective and proposal for a unified nomenclature. Skelet Muscle 10:32. https://doi.org/10.1186/s13395-020-00243-4
Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR (2005) Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 123:25–35
Li L, Mirza S, Richardson SJ, Gallant EM, Thekkedam C, Pace SM, Zorzato F, Liu D, Beard NA, Dulhunty AF (2015) A new cytoplasmic interaction between junctin and ryanodine receptor Ca2+ release channels. J Cell Sci 128:951–963. https://doi.org/10.1242/jcs.160689
Li P, Chen SR (2001) Molecular basis of Ca(2)+ activation of the mouse cardiac Ca(2)+ release channel (ryanodine receptor). J Gen Physiol 118:33–44. https://doi.org/10.1085/jgp.118.1.33
Lindsay C, Sitsapesan M, Chan WM, Venturi E, Welch W, Musgaard M, Sitsapesan R (2018) Promiscuous attraction of ligands within the ATP binding site of RyR2 promotes diverse gating behaviour. Sci Rep 8:15011. https://doi.org/10.1038/s41598-018-33328-8
Liu X, Betzenhauser MJ, Reiken S, Meli AC, Xie W, Chen BX, Arancio O, Marks AR (2012) Role of leaky neuronal ryanodine receptors in stress-induced cognitive dysfunction. Cell 150:1055–1067. https://doi.org/10.1016/j.cell.2012.06.052
Maggi L, Scoto M, Cirak S, Robb SA, Klein A, Lillis S, Cullup T, Feng L, Manzur AY, Sewry CA et al (2013) Congenital myopathies–clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul Disord 23:195–205. https://doi.org/10.1016/j.nmd.2013.01.004
Marks AR (2013) Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Investig 123:46–52. https://doi.org/10.1172/JCI62834
Marks AR (2003) A guide for the perplexed: towards an understanding of the molecular basis of heart failure. Circulation 107:1456–1459
Marks AR (2002) Ryanodine receptors, FKBP12, and heart failure. Front Biosci 7:d970-977
Marks AR, Marx SO, Reiken S (2002) Regulation of ryanodine receptors via macromolecular complexes: a novel role for leucine/isoleucine zippers. Trends Cardiovasc Med 12:166–170
Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR (2001) Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res 88:1151–1158
Marx SO, Ondrias K, Marks AR (1998) Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science 281:818–821
Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, Yang YM, Rosemblit N, Marks AR (2001) Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol 153:699–708
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101:365–376
Meissner G (1984) Adenine nucleotide stimulation of Ca2+-induced Ca2+ release in sarcoplasmic reticulum. J Biol Chem 259:2365–2374
Meissner G (1994) Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol 56:485–508. https://doi.org/10.1146/annurev.ph.56.030194.002413
Meissner G (2017) The structural basis of ryanodine receptor ion channel function. J Gen Physiol 149:1065–1089. https://doi.org/10.1085/jgp.201711878
Moore CP, Rodney G, Zhang JZ, Santacruz-Toloza L, Strasburg G, Hamilton SL (1999) Apocalmodulin and Ca2+ calmodulin bind to the same region on the skeletal muscle Ca2+ release channel. Biochemistry 38:8532–8537. https://doi.org/10.1021/bi9907431
Popova OB, Baker MR, Tran TP, Le T, Serysheva II (2012) Identification of ATP-binding regions in the RyR1 Ca(2)(+) release channel. PLoS ONE 7:e48725. https://doi.org/10.1371/journal.pone.0048725
Porter Moore C, Zhang JZ, Hamilton SL (1999) A role for cysteine 3635 of RYR1 in redox modulation and calmodulin binding. J Biol Chem 274:36831–36834
Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N et al (2003) PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol 160:919–928
Rodney GG, Williams BY, Strasburg GM, Beckingham K, Hamilton SL (2000) Regulation of RYR1 activity by Ca(2+) and calmodulin. Biochemistry 39:7807–7812
Rosemblit N, Moschella MC, Ondriasa E, Gutstein DE, Ondrias K, Marks AR (1999) Intracellular calcium release channel expression during embryogenesis. Dev Biol 206:163–177
Rossi D, Bencini C, Maritati M, Benini F, Lorenzini S, Pierantozzi E, Scarcella AM, Paolini C, Protasi F, Sorrentino V (2014) Distinct regions of triadin are required for targeting and retention at the junctional domain of the sarcoplasmic reticulum. Biochem J 458:407–417. https://doi.org/10.1042/BJ20130719
Santulli G, Marks AR (2015) Essential roles of intracellular calcium release channels in muscle, brain, metabolism, and aging. Curr Mol Pharmacol 8:206–222
Santulli G, Pagano G, Sardu C, Xie W, Reiken S, D’Ascia SL, Cannone M, Marziliano N, Trimarco B, Guise TA et al (2015) Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J Clin Investig 125:4316. https://doi.org/10.1172/JCI84937
Seo M-D, Velamakanni S, Ishiyama N, Stathopulos PB, Rossi AM, Khan SA, Dale P, Li C, Ames JB, Ikura M et al (2012) Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483:108–112. https://doi.org/10.1038/nature10751
Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR (2010) Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Investig 120:4375–4387. https://doi.org/10.1172/JCI37649
Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ, Marks AR (2010) Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Investig 120:4388–4398. https://doi.org/10.1172/JCI32726
Sharma P, Ishiyama N, Nair U, Li W, Dong A, Miyake T, Wilson A, Ryan T, MacLennan DH, Kislinger T et al (2012) Structural determination of the phosphorylation domain of the ryanodine receptor. FEBS J 279:3952–3964. https://doi.org/10.1111/j.1742-4658.2012.08755.x
Snoeck M, van Engelen BG, Kusters B, Lammens M, Meijer R, Molenaar JP, Raaphorst J, Verschuuren-Bemelmans CC, Straathof CS, Sie LT et al (2015) RYR1-related myopathies: a wide spectrum of phenotypes throughout life. Eur J Neurol 22:1094–1112. https://doi.org/10.1111/ene.12713
Sonnleitner A, Fleischer S, Schindler H (1997) Gating of the skeletal calcium release channel by ATP is inhibited by protein phosphatase 1 but not by Mg2+. Cell Calcium 21:283–290
Sudhof TC (2012) Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol 4:a011353. https://doi.org/10.1101/cshperspect.a011353
Tencerova B, Zahradníková A, Gaburjakova J, Gaburjakova M (2012) Luminal Ca2+ controls activation of the cardiac ryanodine receptor by ATP. J Gen Physiol 140:93–108. https://doi.org/10.1074/jbc.272.37.23389
Tripathy A, Xu L, Mann G, Meissner G (1995) Calmodulin activation and inhibition of skeletal muscle Ca2+ release channel (ryanodine receptor). Biophys J 69:106–119
Tung C-C, Lobo PA, Kimlicka L, Van Petegem F (2010) The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature 468:585–588. https://doi.org/10.1038/nature09471
Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, Chiechi A, Wright LE, Umanskaya A, Niewolna M et al (2015) Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat Med 21:1262–1271. https://doi.org/10.1038/nm.3961
Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N et al (2003) FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 113:829–840
Wehrens XH, Lehnart SE, Reiken SR, Marks AR (2004) Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res 94:e61-70
Xiong L, Zhang J-Z, He R, Hamilton SL (2006) A Ca2+-binding domain in RyR1 that interacts with the calmodulin binding site and modulates channel activity. Biophys J 90:173–182. https://doi.org/10.1529/biophysj.105.066092
Xu L, Chirasani VR, Carter JS, Pasek DA, Dokholyan NV, Yamaguchi N, Meissner G (2018) Ca(2+)-mediated activation of the skeletal-muscle ryanodine receptor ion channel. J Biol Chem 293:19501–19509. https://doi.org/10.1074/jbc.RA118.004453
Yan Z, Bai X-c, Yan C, Wu J, Li Z, Xie T, Peng W, Yin C-C, Li X, Scheres SHW et al (2015) Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517:50–55. https://doi.org/10.1038/nature14063
Yuan Q, Chen Z, Santulli G, Gu L, Yang ZG, Yuan ZQ, Zhao YT, Xin HB, Deng KY, Wang SQ et al (2014) Functional role of Calstabin2 in age-related cardiac alterations. Sci Rep 4:7425. https://doi.org/10.1038/srep07425
Yuchi Z, Yuen SMWK, Lau K, Underhill AQ, Cornea RL, Fessenden JD, Van Petegem F (2015) Crystal structures of ryanodine receptor SPRY1 and tandem-repeat domains reveal a critical FKBP12 binding determinant. Nat Commun 6:7947. https://doi.org/10.1038/ncomms8947
Zalk R, Clarke OB, des Georges A, Grassucci RA, Reiken S, Manica F, Hendrickson WA, Frank J, Marks AR (2015) Structure of a mammalian ryanodine receptor. Nature 517:44–49. https://doi.org/10.1038/nature13950
Zalk R, Lehnart SE, Marks AR (2007) Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem 76:367–385. https://doi.org/10.1146/annurev.biochem.76.053105.094237
Zhang JZ, Wu Y, Williams BY, Rodney G, Mandel F, Strasburg GM, Hamilton SL (1999) Oxidation of the skeletal muscle Ca2+ release channel alters calmodulin binding. Am J Physiol 276:C46-53
Acknowledgements
This study was is supported by R01HL145473, R01DK118240, R01HL142903, R01HL140934, R01AR070194, T32HL120826, and R25 NS076445 to ARM.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
ARM is on the scientific advisory board, board of directors and is an equity owner in ARMGO, Inc. a biotech company targeting RyR for therapeutic purposes, Columbia University also owns equity in ARMGO, Inc.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure 1.
RyR1-S4028L patient mutation causes RyR1 channel leak. (A) The mutant RyR1-S4028L channel was PKA phosphorylated at Ser2844 and oxidized (DNP) compared to control. PP1 and DTT were used to reverse the oxidation and phosphorylation. (B) The mutant RyR1-S4028L channels exhibited increased sensitivity to Ca2+-dependent activation consistent with channel leak as determined by 3[H]-ryanodine binding at the indicated [Ca2+]cyt. Data are presented as mean ± S.E.M from 4 for each group *P < 0.05 vs. WT; #P < 0.05 vs. RyR1-S4028L, ANOVA, Tukey-Kramer with post hoc correction.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yuan, Q., Dridi, H., Clarke, O.B. et al. RyR1-related myopathy mutations in ATP and calcium binding sites impair channel regulation. acta neuropathol commun 9, 186 (2021). https://doi.org/10.1186/s40478-021-01287-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01287-3