
Abstract
Diffuse intrinsic pontine gliomas (DIPGs) are high-grade tumors of the brainstem that often occur in children, with a median overall survival of less than one year. Given the fact that DIPGs are resistant to chemotherapy and are not amenable to surgical resection, it is imperative to develop new therapeutic strategies for this deadly disease. The p53 pathway is dysregulated by TP53 (~ 60%) or PPM1D gain-of-function mutations (~ 30%) in DIPG cases. PPM1D gain-of-function mutations suppress p53 activity and result in DIPG tumorigenesis. While MDM2 is a major negative regulator of p53, the efficacy of MDM2 inhibitor has not been tested in DIPG preclinical models. In this study, we performed a comprehensive validation of MDM2 inhibitor RG7388 in patient-derived DIPG cell lines established from both TP53 wild-type/PPM1D-mutant and TP53 mutant/PPM1D wild-type tumors, as well in TP53 knockout isogenic DIPG cell line models. RG7388 selectively inhibited the proliferation of the TP53 wild-type/PPM1D mutant DIPG cell lines in a dose- and time-dependent manner. The anti-proliferative effects were p53-dependent. RNA-Seq data showed that differential gene expression induced by RG7388 treatment was enriched in the p53 pathways. RG7388 reactivated the p53 pathway and induced apoptosis as well as G1 arrest. In vivo, RG7388 was able to reach the brainstem and exerted therapeutic efficacy in an orthotopic DIPG xenograft model. Hence, this study demonstrates the pre-clinical efficacy potential of RG7388 in the TP53 wild-type/PPM1D mutant DIPG subgroup and may provide critical insight on the design of future clinical trials applying this drug in DIPG patients.
Similar content being viewed by others


Introduction
Diffuse intrinsic pontine gliomas (DIPGs) are malignant brainstem gliomas. In the United States, there are approximately 200–300 new cases of DIPG every year [1]. DIPGs are resistant to conventional therapies, including radiation and chemotherapy, and are not amenable to surgical resection [2]. Consequently, DIPGs are universally lethal, with a median overall survival of less than one year [3, 4]. Novel therapies for DIPG are desperately needed.
The mutational landscape of DIPG has been well characterized. Recurrent somatic Lys-27-Met mutations in histone H3 genes (either in H3.1 or H3.3) have been identified as an oncogenic driver in ~ 80% of DIPGs [5,6,7,8,9]. In the latest 2016 World Health Organization classification, “diffuse midline glioma, H3K27M mutant” was introduced, and DIPGs are included in this new entity [10]. TP53 and the genetic components of p53 signaling are also frequently mutated in DIPGs. TP53 mutations were identified in around 60% H3K27M mutant DIPGs [11,12,13,14,15]. However, in TP53 wild-type DIPGs, approximately 80% have mutations in exon 6 of Protein Phosphatase, Mg2 + /Mn2 + Dependent 1D (PPM1D) mutations, which result in truncated PPM1D mutant proteins. The mutations stabilize the truncated PPM1D protein and lead to increases in its native dephosphorylation activity toward p53 and other proteins in DNA damage response (DDR) and cellular checkpoint pathways [11]. Recent studies in DIPGs have demonstrated that direct PPM1D inhibitor GSK2830371 inhibits p53 wild-type tumor cells growth and sensitizes cells to PARP inhibition and radiation through the re-activation of p53 pathway. These studies suggested that reactivation of p53 pathway is a feasible therapeutic strategy in DIPGs [16, 17].
Another approach to reactive p53 pathway is to reduce p53 protein ubiquitination and degradation by targeting Mouse double minute 2 homolog (MDM2), a key negative regulator of the p53 pathway. There are two major functions of MDM2: one as an E3 ubiquitin ligase of p53 and the other as an inhibitor of p53 transcriptional activation [18]. In addition, PPM1D dephosphorylates MDM2 at serine 395 and increases its affinity for p53. Dephosphorylated MDM2 protein inhibits p53 transcriptional activity and promotes p53 protein ubiquitination [19]. Several small molecule compounds have been recently developed targeting the MDM2-p53 axis to reactivate the tumor suppressor activity of p53. RG7388, also known as Idasanutlin, is a highly potent and selective second-generation MDM2 inhibitor which blocks the interaction between MDM2 and p53 [20]. Ongoing clinical trials using this orally bioavailable drug include a Phase III trial for acute myeloid leukemia (AML) and several Phase I/II trials in solid tumors [21, 22]. However, the preclinical and clinical evidence of using MDM2 inhibitors for treating DIPGs has not been well characterized. In this study, using a series of clinically and genetically relevant DIPG cell lines, we have investigated the therapeutic potential of using RG7388 for treating DIPGs.
Materials and methods
Analysis of cell lines CRISPR screening data
To identify the top dependent gene of TP53 wild-type/PPM1D-mutant tumors, we integrate data from Mutations, Copy Number Alterations (Cancer Cell Line Encyclopedia, Broad, 2019) and CRISPR screening data of 808 cancer cell lines (Broad’s institute Cancer Dependency Map Public 21Q1 (https://depmap.org/portal/) [23]. These cell lines were classified into different molecular subgroups based on TP53 and PPM1D status. We use Student’s t-test to compare the CERES score of each gene (18,116 genes total) in between molecular subgroups. These statistical analysis were performed in R, and 10% false-discovery rate (FDR, Benjamini-Hochberg) was applied using p.adjust function in R. Top dependency gene list were generated based on p value/adjusted p value and t value. CERES score of MDM2 across four subgroups were visualized with GraphPad Prism V9.0.
Cell lines
Ten cell lines were obtained from biopsy or autopsy tissues of DIPG patient tumors. TT10714, TT10728 and TT10630 cell lines were used in a previous study [24]. SF7761 was a gift from Dr. C David James [25]. HSJD-DIPG-007, HSJD-DIPG-012 and HSJD-DIPG-013 were gifts from Dr. Angel Montero Carcaboso [26]. SU-DIPG-VI, SU-DIPG-XIII and SU-DIPG-35 were gifts from Dr. Michelle Monje [27]. Cell line authentication (CLA) analysis was performed at the Duke University DNA Analysis Facility and the result is shown in Additional file 6: Table S1. Primers for PCR amplification and sequencing of H3F3A, TP53 and PPM1D genes are listed in Additional file 7: Table S2. HSJD-DIPG-007-NTC and HSJD-DIPG-TP53 KO isogenic lines were established and used in a previous study [16].
Western blot
Cell pellets were lysed and resolved using (4–12%) NuPAGE Bis–Tris gradient gel. Gels were soaked in NuPAGE protein transfer buffer and transferred to PVDF membranes. Membranes were blocked in room temperature for 1 h and incubated with primary antibodies at 4 degree overnight. Membranes were washed and then incubated with horseradish peroxidase (HRP) conjugated secondary antibody for one hour and HRP signals were detected by chemiluminescence using the BioRad ChemiDoc MP system. H3K27M (Cell Signaling Cat#74,829, 1:1000), p53 (Santa Cruz Cat#sc-126, 1:200), MDM2 (Cell Signaling Cat#86,934, 1:1000), p21 (Cell Signaling Cat#2947, 1:1000) and GAPDH (Santa Cruz Cat#sc-47724, 1:1000).
Cell viability assay
Cells were plated at a density of 3000 cells/well in 96-well white and clear bottom microplates (Greiner Bio-One) and were incubated for 24 h. For dose-dependent studies, cells were treated with RG7388 (Selleck Chemicals #S7205) diluted in DMSO, ranging from 7.62 nM to 50 uM, or DMSO alone as a control, for 3 days. For time-dependent studies, cells were treated with DMSO, 50 nM and 100 nM RG7388 and luminescence signal was measured at Days 0, 2 and 4. Cell viability was measured by CellTiter-Glo Luminescent Cell Viability Assay (Promega #G7571) per manufacturer’s instruction. The luminescence signals were recorded using a Tecan Infinite M200 PRO microplate reader. Relative viabilities were calculated by normalizing luminescence values for each treatment condition to DMSO treated wells.
RNA-seq
HSJD-DIPG-007-NTC and HSJD-DIPG-TP53 KO were treated with 100 nM RG7388 or DMSO for 24 h. Three samples for each condition were harvested and RNA extractions were done using Maxwell RSC simplyRNA Cells Kit (Promega #AS1390). RNA samples were sent to for RNA-Seq and the resulting data was analyzed using the Galaxy/Europe platform [28].
Cell apoptosis analysis
Cells were plated at a density of 3000 cells/well in 96-well white and clear-bottom microplates (Greiner Bio-One). After 24-h incubation, cells were treated with 100 nM RG7388 or DMSO for 24 h. Cell viability was measured by Caspase-Glo® 3/7 Assay (Promega #G8091) per manufacturer’s instruction. The luminescence signals were recorded using a Tecan Infinite M200 PRO microplate reader. Relative viabilities were calculated by normalizing luminescence values for each treatment condition to DMSO treated wells.
Cell cycle analysis
HSJD-DIPG-007-NTC and HSJD-DIPG-TP53 KO were treated with DMSO and 100 nM RG7388 for 24 h. 1 × 106 Cells were harvested and fixed in cold 70% ethanol for 30 min at 4℃. After centrifugation and washing with PBS, 0.5 ml FxCycle PI/RNAse Staining Solution (ThermoFisher #F10797) was added to the cell pellet and incubated for 15–30 min at room temperature in the dark. Flow cytometry analysis was performed using 532-nm excitation with a 585/42-nm bandpass filter. Data was analyzed using FlowJo software.
In vivo efficacy on mouse models
To access the pharmacokinetic of RG7388, 12 outbred athymic nude mice (J:NU Stock#007850) were treated with RG7388 delivered by oral gavage at 50 mg/kg. Plasma and tissues were collected and analyzed by Mass Spectrometry (AB Sciex 5500). To further access the in vivo efficacy of RG7388, 5 × 105 of HSJD-DIPG-007 cells with CMV-Firefly luciferase lentivirus (Cellomics Technology cat#C839R47) were implanted stereotactically into the nude mouse brain stem region (n = 18). On the 21st-day post injection, mice were randomly assigned to two groups. For each group, nine mice were treated by oral gavage with 50 mg/kg of RG7388 (AstaTech cat#40916) or vehicle once per day, five days per week for three weeks. Mice were assessed for neurological symptoms and weight loss and sacrificed when either 20% weight loss was measured or when they exhibited neurologic symptoms. Kaplan–Meier curves were used for survival analysis.
H&E and immunofluorescence staining
Six mice were injected with DIPG cells and treated with RG7388 (n = 3) or vehicle (n = 3) beginning on day 21 as previously described. These mice were sacrificed on day 42 and tumor samples were collected for immunofluorescence staining. The mouse brain was fixed in 10% formalin, embedded, and cut into sections. Standard H&E staining procedures and immunofluorescent staining procedures were performed. Immunofluorescent images were imaged on the Zeiss 880 at Duke Light Microscopy Core Facility. Positively stained cells were both manually counted and counted with ImageJ. Those assigned to counting were blinded to animal genotype and were given designated quadrants of specific size and magnification to count.
More method details are in Supplementary Methods
Results
RG7388 selectively suppresses proliferation of TP53 wild-type DIPG cells
To validate MDM2 as a potential therapeutic target for TP53 wild-type/PPM1D mutant cell lines, we combined data from Mutations, Copy Number Alterations (Cancer Cell Line Encyclopedia, Broad, 2019) and CRISPR screening data of 808 cancer cell lines (https://depmap.org/portal/; https://depmap.org/portal/; https://depmap.org/portal/) [23]. We determined that 543 cell lines harbor TP53 mutation while 28 cell lines contain activated PPM1D either through PPM1D amplification or exon 6 truncations. Notably, among these 28 cell lines, 11 cell lines are TP53 wild-type. To identify the top candidate genes for survival dependence in TP53 wild-type cell lines, Student’s t-test was performed to compare gene effect (CERES score) across 18,116 genes in-between different molecular subgroups. Compared to all TP53 mutant cell lines, MDM2 is among the top ten dependent genes in TP53 wild-type, PPM1D amplified or mutant cell lines (Fig. 1A). The MDM2 gene effect CERES score is significant lower in TP53 wild-type cell lines with/without PPM1D amplification mutation (Fig. 1B).

RG7388 selectively suppresses proliferation of TP53 wild-type DIPG cells. A Top dependent genes of TP53 wildtype, PPM1D amplification or truncated mutant cell lines. B MDM2 Gene Effect (CERES) across four subgroups. C Ten DIPG cell lines were treated for three days with RG7388 at varying concentrations. Relative viabilities were calculated by normalizing luminescence values for each treatment condition to DMSO treated wells. D The viabilities of the five TP53 wild-type DIPG lines treated with DMSO, 50 nM and 100 nM RG7388 for 2 and 4 days. Relative viabilities were calculated by normalizing luminescence values for each time point to Day 0 at the same treatment conditions. Mean ± SEM, n = 3 independent studies for each condition. P values based on Two-way ANOVA. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.0001. ns = non-significant. E Relative Caspase-3/7 activity after treatment with RG7388 for 24 h, normalized to DMSO treatment in TP53 wild-type DIPG cell lines. Mean ± SEM, n = 3 independent studies for each condition. P values based on Student’s t-test. *P < 0.05. **P < 0.01. ****P < 0.0001. ns = non-significant
To further investigate the potential of targeting MDM2 in vitro, we tested RG7388, a second-generation inhibitor of MDM2 in ten DIPG patient-derived cell lines that were characterized genetic mutations common to DIPGs, including H3K27M, TP53, and PPM1D mutations (Additional file 1: Fig. S1). These experiments revealed that the TP53 wild-type cell lines were significantly more sensitive to RG7388 (IC50 of 67 nM; range: 28–177 nM) compared to the TP53 mutant cell lines (IC50 of 11.7 µM; range: 3.4–21.1 µM) (Student’s t-test, P < 0.0001, Fig. 1C, Additional file 2: Fig. S2A). Moreover, measured at two time points following treatment, both doses of RG7388 (50 nM and 100 nM) selectively suppressed the proliferation of the TP53 wild-type cell lines, as compared to DMSO control (Fig. 1D). In contrast, the TP53 mutant cell lines were resistant to both doses of RG7388, with no difference in cell numbers between treatment and DMSO control (Additional file 2: Fig. S2B). Treatments with RG7388 led to cell apoptosis in TP53 wild-type DIPG cell lines but not in those without functional TP53, as indicated by the RG7388-induced activation of caspase-3/7 activities in the former but not the latter group of cell lines (Student’s t-test, p < 0.05, Fig. 1E). Collectively, these results established MDM2 as a top candidate for therapeutic targeting in TP53 wild-type/PPM1D mutant cell lines, and suggested a strategy of leveraging MDM2 inhibitors to target the subset of DIPG with functional TP53.
The susceptibility of DIPG cells to RG7388 is TP53-dependent
To determine the roles of functional TP53 in mediating the suppression effects of RG7388 on DIPG cells, we used an isogenic pair of HSJD-DIPG-007 cell line derivative with or without functional TP53 [16], namely, a non-targeting control (NTC) sgRNA and TP53 KO, respectively (Fig. 2A) and tested their susceptibility to RG7388. These experiments revealed that TP53 KO conferred resistance to RG7388, as evidenced by the > 100 × IC50 for the TP53 KO line when compared to the IC50 for the NTC line (8.931 µM vs. 86 nM, Student’s t-test, p < 0.01, Fig. 2B, Additional file 2: Fig. S2C). Consistent with these findings, RG7388 exerted both dose and time-dependent suppression effects on the control cell lines, but displayed no effects on the TP53 KO cell line (Fig. 2C, Additional file 2: Fig. S2D). In further supporting the essential role of functional TP53 in conferring the susceptibility to RG7388, HSJD-DIPG-007-NTC cells, but not the HSJD-DIPG-007-TP53 KO counterparts, underwent both apoptosis (Fig. 2D) and G1 arrest (Fig. 2E) in response to RG7388 treatment.

The susceptibility of DIPG cells to RG7388 is TP53-dependent. A The p53 expression of HSJD-DIPG-007-NTC (TP53 wild-type) and HSJD-DIPG-007-TP53 KO lines by western blot. B Cell viability of HSJD-DIPG-007-NTC and HSJD-DIPG-007-TP53 KO lines measured after three days of RG7388 treatment. Relative viabilities were calculated by normalizing luminescence values for each treatment condition to DMSO treated wells. C The viabilities of HSJD-DIPG-007-NTC line treated with DMSO, 50 nM and 100 nM RG7388 for two and four days. Relative viabilities were calculated by normalizing luminescence values for each time point to Day 0 at the same drug concentration. Mean ± SEM, n = 3 independent studies for each condition. P-values based on Two-way ANOVA. **P < 0.01. D Relative Caspase-3/7 activity after treatment with RG7388 for 24 h, normalized to DMSO treatment in HSJD-DIPG-007-NTC and HSJD-DIPG-007-TP53 KO lines. Mean ± SEM, n = 3 independent studies for each condition. P-values based on Student’s t-test. **P < 0.01. ns = non-significant. E Percentage of G1, S and G2 phases and representative images of HSJD-DIPG-007-NTC and HSJD-DIPG-007-TP53 KO treated with DMSO or RG7388 as measured by PI staining. Mean ± SEM, n = 3 independent studies for each condition. P values based on chi-squared test. ***P < 0.001. ns = non-significant
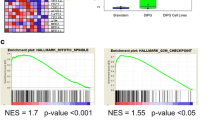
To gain further insight into the mechanism underlying the effects of RG7388, we performed RNA-Seq analysis comparing the isogenic pair of derivatives, HSJD-DIPG-007-NTC and HSJD-DIPG-007-TP53 KO, after treatment with DMSO or 100 nM RG7388 for 24 h. The differences in transcript level (FPKM) between RG7388 treatment and DMSO control were assessed, and differentially expressed genes (adjusted P-value < 0.05, fold change > 1.5) were selected. While the global gene expression was mostly unaffected in the HSJD-DIPG-007-TP53 KO cell line, 476 genes were significantly changed in the HSJD-DIPG-007-NTC line (Fig. 3A). Gene sets enrichment analysis revealed the top five most significantly altered KEGG pathways in this cell line in response to RG7388 treatment (adjusted P-values ranging from 4.49 × 10–29 to 1.03 × 10–8) included the down-regulation of the pathways involving DNA replication, cell cycle, mismatch repair and homologous recombination and most notably, the upregulation of the p53 signaling pathway [29] (Fig. 3B). Hierarchical clustering analysis of genes in these pathways (adjusted P-value < 0.05, fold change > 1.5) (DESeq2-normalized counts + 1) revealed those related to cell cycle arrest and apoptosis were significantly changed, in that apoptosis related genes (e.g., BAX, BBC3 (PUMA), TNFRSF10B (DR5) and ZMAT3 (PAG608) were up-regulated while cell cycle progression related genes (e.g., CDK1, CDK2, CCNB1 and CCNB2) were down-regulated (Fig. 3C, Additional file 3: Fig. S3A and S3B). Notably, CDKN1A, the gene encoding p21, exhibited the lowest adjusted P-value and displayed one of the highest fold-changes in response to treatment of genes in the p53 pathway (Additional file 3: Fig. S3C). To further corroborate the activation of the p53 pathway in the p53-intact cells, we treated HSJD-DIPG-007-NTC and TP53 KO lines with RG7388 determined the expression of proteins in the p53 pathway at different time points post the treatment (Fig. 3D). These experiments revealed a rapid induction of the p53, p21 and MDM2 in response to RG7388, reaching peak levels at 6 h post the treatment, while the induction of these proteins was not detectable in the HSJD-DIPG-007-TP53 KO after treatment with RG7388 (Additional file 4: Fig. S4A).

P53 pathway was up-regulated after RG7388 treatment. A Hierarchical clustering of significantly changed genes (adjusted P-values < 0.05, fold change > 1.5) of HSJD-DIPG-007-NTC and HSJD-DIPG-007-TP53 KO lines after treatment with DMSO and RG7388. Columns in the heat map represent individual replicates (three for each condition). Rows represent genes, colored by log-transformed transcript intensity (DESeq2-normalized counts + 1) in z-score. Blue shows replicates with low expression (z-score < 0); red shows replicates with high expression (z-score > 0). B Top five most differentially regulated KEGG pathways of HSJD-DIPG-007-NTC and HSJD-DIPG-007-TP53 KO lines response to RG7388. C Hierarchical clustering of significantly changed genes (adjusted P-values < 0.05, fold change > 1.5) within the p53 pathway of HSJD-DIPG-007-NTC response to RG7388. Columns in the heat map represent individual replicates (three for each condition). Rows represent genes, colored by log-transformed transcript intensity (DESeq2-normalized counts + 1) in z-score. Blue shows replicates with low expression (z-score < 0); red shows replicates with high expression (z-score > 0). Volcano plot of p53 pathway genes with significant change (adjusted P-values < 0.05, fold change > 1.5). Blue, red and gray show down-regulated, up-regulated and not significantly changed genes, respectively. D Protein expression level of MDM2, p53 and p21 in HSJD-DIPG-007-NTC and HSJD-DIPG-007-TP53 KO treated with DMSO or 25 nM RG7388 for 6 h, 12 h, 24 h and 48 h. E Protein expression level of MDM2, p53 and p21 in five TP53 wild-type (Top) and five TP53 mutant (Bottom) DIPG cell lines treated with DMSO and 100 nM RG7388 for 24 h
Finally, to further confirm the activation of the p53 pathway in response to RG7388 in DIPG cell lines with functional TP53, we treated the panel of the original DIPG cell lines with RG7388 for 24 h and determined the levels of MDM2, p53 and p21 proteins. These experiments confirmed that in the TP53 mutant DIPG cell lines, the expression levels of MDM2, p53 and p21 displayed only minimal alterations, and in contrast, the levels of these proteins were all markedly increased compared to DMSO control in five TP53 wild-type DIPG lines, with a mean fold change ranging from 19 to 63 (MDM2: Student’s t-test, P < 0.0001), 4 to 71 (p53: Student’s t-test, P < 0.0001) and 7 to 32 (p21: Student’s t-test, P < 0.0001) (Fig. 3E, Additional file 4: Fig. S4B).
Collectively, these results confirmed that RG7388 induced activation of the p53 pathway and that MDM2 inhibition-induced suppression of the DIPG cell lines was TP53 dependent.
RG7388 was able to reach the brainstem and exerted therapeutic efficacy in an orthotopic DIPG xenograft model
Permeability to the brainstem poses a major challenge for a drug’s application for treating DIPGs. To determine the applicability of RG7388 for treating DIPG, we determined the permeability of orally administered RG7388 into the brainstem in mice. We administered RG7388 via oral gavage (50 mg/kg) and collected blood samples, cerebrospinal fluid (CSF), cerebral hemispheres tissues and brainstem tissues from the mice at different time points (0.5/1/2/4/8/24 h) afterward and analyzed the presence and concentrations of RG7388 by mass spectrometry. The brains were perfused to remove blood before the submission for mass spectrometry. RG7388 was detectable in plasma with mean concentration of 263 ng/ml 30 min after the drug administration. Plasma concentration increased to the maximum (mean 570 ng/ml) after 1 h, remained relatively steady at least for the following 7 h, and decreased dramatically by 24 h (4.26 ng/ml) (Fig. 4A). Unlike in the plasma, RG7388 was mostly not detectable in the CSF, as it was detected at a low concentration (average 1.17 ng/ml) after 8 h on in mouse. Notably, RG7388 was present in the cerebral hemispheres and in the brainstem rapidly after its administration, measured at an average of ~ 18 ng and 8.1 ng per gram of tissues in the cerebral hemispheres and in the brainstem, respectively, within one hour post its administration. The accumulation of RG7388 subsequently increased to the maximum level of 105 ng and 112 ng per gram of tissues in the cerebral hemispheres and brainstem, respectively (Fig. 4B). The peak permeability of the drug to the brainstem reached at the 8-h time point, with a ratio of RG7388 concentration in the brainstem to plasma of 0.292. RG7388 was not detectable either in brainstem or cerebral hemispheres at the 24-h time point. Collectively, these results from the pharmacokinetics assays suggest RG7388 is permeable to the brainstem independent of tumor-mediated blood–brain–barrier disruption.

RG7388 was able to reach the brainstem and exert therapeutic efficacy in an orthotopic DIPG xenograft model. A The pharmacokinetic profile shows the RG7388 concentration level in plasma and CSF (mean ± SEM, n = 2 per time point). B The pharmacokinetic profile shows the RG7388 concentration level in cerebral hemispheres and brainstem (mean ± SEM, n = 2 per time point). C Log values of bioluminescence signals for each group on day 21 and day 42 respectively (left). Acquired on day 42, representative bioluminescence signal of two mice assigned to treatment and vehicle group (right). P-values based on Student’s t-test. ***P < 0.001. ns = non-significant. D Kaplan–Meier curve of treatment and vehicle group. N = 9 for each group. P-values based on log-rank test. ****P < 0.0001. E The expression of p21 of mouse brain samples from vehicle or RG7388 treatment group on day 42. Scale bar: 100 μm. Mean ± SEM, n = 3 for each group. P-values based on chi-squared test. ***P < 0.001
We then assessed the in vivo therapeutic efficacy of RG7388 in a DIPG orthotopic model derived from the HSJD-DIPG-007 cell line. Tumor cells expressing luciferase were injected into the brainstem of NSG mice, and the tumor development was monitored by the Luciferase imaging system. Host mice displaying similar bioluminescence signal on day 21 post-injection were randomly assigned to vehicle or RG7388 treatment groups. RG7388 or vehicle was orally administered once per day, five days per week for three weeks (Day 42 post-implantation) and the progression of tumors was determined. No overt toxicity to other organs was observed during the experiment. Notably, while the bioluminescence of orthotopic tumors was comparable between groups before the treatment (on Day 21), three weeks of RG7388 treatment resulted in measurably suppressed tumor progression, as demonstrated by reduced bioluminescence signals (Student’s t-test, p < 0.001) (Fig. 4C). Subsequently, mice were sacrificed when either 20% weight loss was measured or when they exhibited neurologic symptoms. Kaplan–Meier survival analysis found mice in the RG7388 treatment group had a significantly better survival than those in the vehicle group (Fig. 4D, median survival 65 days vs. 51 days, p < 0.0001).
To assess the P53 pathway status after drug treatment, three mice for each RG7388 and vehicle group were sacrificed on day 42 after injection, and their tumor tissues were analyzed by H&E staining and immunofluorescence. H&E and H3K27M immunofluorescence staining confirm the presence of tumor cells in the brainstem region (Additional file 4: Fig. S4C). Furthermore, the activation of the p53 pathway was confirmed in the RG7388-treated tumors, as evidenced by the increased expression of p21 in RG7388-treated tumors in comparison to the control tumors (Fig. 4E). Collectively, results from these in vivo experiments suggest that RG7388 treatment activates the p53 pathway in orthotopic DIPG and displays therapeutic efficacy by prolonging survival of animals relative to vehicle treated controls.
Discussion
Given the key role of MDM2 in p53 regulation, targeting p53-MDM2 axis to stabilize and activate p53 has been explored as a novel therapeutic strategy for human cancers [30]. The pyrrolidine compound RG7388 is a second-generation MDM2 small molecular inhibitor, which blocks the physical interaction between MDM2 and p53 and is undergoing pre-clinical and clinical evaluation as a potential anti-cancer therapeutic [20]. However, due to the scarcity of pre-clinical DIPG models, especially the TP53 wild-type subtype, the in vitro and in vivo efficacy of RG7388 had not been studied in DIPG models. Hence in this study, we performed a comprehensive validation of RG7388 in five TP53 wild-type and five TP53 mutant patient-derived DIPG cell lines, as well as the engineered TP53 knockout isogenic lines. Nanomolar levels of RG7388 suppressed cell proliferation, altered gene expression patterns, and induced cell apoptosis and G1 arrest in the TP53 wild-type DIPG cell lines, indicating its high potency. TP53 mutant/knockout DIPG cell lines were resistant to RG7388 with approximately 100 times higher IC50 and did not show significant alterations in gene expression patterns, cell apoptosis or cell cycle variation in response to the inhibitor treatment, representing RG7388’s high selectivity. These results demonstrate the promising clinically therapeutic potential of RG7388 in treating TP53 wild-type DIPG tumors.
Reactivation of the p53 pathway is a critical indicator for the effectiveness of MDM2 inhibitors and a potential pharmacodynamic biomarker for MDM2 inhibitor response. In this study, we evaluated the re-activation of p53 function in response to RG7388 by various perspectives. First, gene expression level analysis was performed on RNA-seq data, showing that the p53 pathway was one of the top three most differentially regulated pathways after RG7388 treatment in the TP53-NTC line. Notably, CDKN1A, which encodes the key p53 pathway downstream marker p21, was dramatically up-regulated after the drug treatment. Second, protein expression level of MDM2, p53 and p21 was assessed and indicated that all of these proteins were up-regulated in response to RG7388 in TP53 wild-type lines. Interestingly, while the protein level of p53 was significantly increased, mRNA expression of TP53 was unchanged after drug treatment, indicating that p53 protein expression is likely regulated through post-transcriptional mechanisms [31]. In contrast, MDM2 and p21 demonstrated significant up-regulation in both gene and protein expression levels upon MDM2-p53 interaction inhibition with RG7388. Third, induction of apoptosis, as one of the hallmarks of the re-activation of p53 pathway, was evaluated [32]. RNA-seq data demonstrated increased apoptosis-related genes such as BAX, PUMA, DR5 and ZMAT3 in the TP53-NTC cell line compared to the KO line. Similarly, all patient-derived TP53 wild-type/PPM1D mutant DIPG cells lines displayed significantly high level of caspase-3/7 activity in response to RG7388 treatment, compared to their TP53 mutant counterparts. Finally, cell cycle alteration, another characteristic phenotype of p53 re-activation, was assessed [32]. Cell cycle regulation was shown to be the second most differentially altered pathway by RNA-seq gene analysis and key G2/M markers such as CDK1, CDK2, CCNB1 and CCNB2 were all down-regulated after RG7388 treatment in the TP53-NTC line. In the same cell line, indications of G1 arrest by PI staining were observed, which was absent in the TP53-KO setting. In summary, these data confirm the efficacy of RG7388 in re-activating the p53 pathway in TP53 wild-type DIPG cell lines.
One of major concerns for brain tumor drug treatment is the penetration of blood–brain barrier (BBB). A previous study has shown the effects of RG7388 treatment in glioblastoma orthotopic models [30]. However, the BBB in the brainstem area is generally regarded as more intact for DIPG as compared with the BBB in supratentorial tumors [33], thus the penetration of RG7388 to brainstem remained unclear. In this study, we confirmed that RG7388 was able to reach brainstem even without the tumor-mediated blood–brain–barrier disruption and maintained a relatively high level of concentration for at least 8 h. Moreover, we used DIPG orthotopic models to assess the in vivo efficacy of RG7388 and demonstrated its ability to inhibit tumor growth through a three-week drug administration period. As a result, treatment with RG7388 not only prolonged survival time, but also increased the expression of p21, the hallmark of p53 pathway activation.
Based on our data, RG7388 could be a valuable therapeutic approach to develop clinically in the treatment of TP53 wild-type/PPM1D-mutant DIPG subgroup as a p53 pathway re-activation drug. One of the merits of RG7388 is that its safety and feasibility have already been confirmed by a variety of clinical trials, including a Phase III trial. RG7388 is currently undergoing clinical trials alone or in combination with other drugs for acute myeloid leukemia (phase I/II: NCT02670044, NCT03850535; phase III: NCT02545283), myeloma (phase I/II: NCT02633059), lymphoma (phase I/II: NCT03135262), breast cancer (phase I/II: NCT03566485), a combination of acute leukemias and solid tumors (phase I/II: NCT04029688) and glioblastoma bearing an unmethylated MGMT promoter (phase I/II: NCT03158389). In general, RG7388 is well-tolerated by patients and the most common adverse events are limited to gastrointestinal system symptoms (diarrhea and vomiting), infections and myelosuppression [21, 22].
Several potential limitations of this MDM2-targeting therapeutic strategy are noted. First, in TP53 wild-type DIPGs, the mutant PPM1D can stabilize MDM2 and promotes MDM2-mediated p53 ubiquitination and degradation. Thus, inhibition of PPM1D may further potentiate the efficacy of RG7388. A small-molecule PPM1D inhibitor, GSK2830371, was recently shown to enhance DNA damage response and suppress cell growth in DIPGs [16]. Further studies testing the combination therapy of GSK2830371 and RG7388 are warranted. Second, we speculate that the pressure from the prolonged treatment with RG7388 may lead to the selection and expansion of p53 mutant cells and thus the development of secondary resistance [34]. Additional combination treatment approaches, including radiation therapy, chemotherapeutic agents [35], the CDK4 inhibitors palbociclib [36], the PARP inhibitors rucaparib [37] and the MEK inhibitor trametinib [30], may be needed to maximize the eradication of tumor cells and mitigate the tumor recurrence. Finally, instead of oral administration, other novel drug delivery techniques, such as nanoparticle application [38] and convection-enhanced delivery (CED) [39], may offer superior local drug deliveries to further improve the therapeutic efficacy of RG7388.
Availability of data and materials
The datasets used and analyzed during the current study available from the corresponding author on reasonable request.
References
Warren KE (2012) Diffuse intrinsic pontine glioma: poised for progress. Front Oncol 2:205
Vanan MI, Eisenstat DD (2015) DIPG in children—what can we learn from the past? Front Oncol 5:237
Jones C, Perryman L, Hargrave D (2012) Paediatric and adult malignant glioma: close relatives or distant cousins? Nat Rev Clin Oncol 9(7):400–413
Kebudi R, Cakir FB (2013) Management of diffuse pontine gliomas in children: recent developments. Paediatr Drugs 15(5):351–362
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44(3):251–253
Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E et al (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124(3):439–447
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482(7384):226–231
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46(5):444–450
Gielen GH, Gessi M, Hammes J, Kramm CM, Waha A, Pietsch T (2013) H3F3A K27M mutation in pediatric CNS tumors: a marker for diffuse high-grade astrocytomas. Am J Clin Pathol 139(3):345–349
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK et al (2016) The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6):803–820
Zhang L, Chen LH, Wan H, Yang R, Wang Z, Feng J et al (2014) Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet 46(7):726–730
Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C et al (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46(5):457–461
Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M et al (2014) Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46(5):451–456
Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M et al (2016) Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 7:11185
Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR et al (2017) Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32(4):520
Wang ZH, Xu C, Diplas BH, Moure CJ, Chen CPJ, Chen LH et al (2020) Targeting mutant PPM1D sensitizes diffuse intrinsic pontine glioma cells to the PARP inhibitor olaparib. Mol Cancer Res 18(7):968–980
Akamandisa MP, Nie K, Nahta R, Hambardzumyan D, Castellino RC (2019) Inhibition of mutant PPM1D enhances DNA damage response and growth suppressive effects of ionizing radiation in diffuse intrinsic pontine glioma. Neuro Oncol 21(6):786–799
Iwakuma T, Lozano G (2003) MDM2, an introduction. Mol Cancer Res 1(14):993–1000
Lu XB, Ma O, Nguyen TA, Joness SN, Oren M, Donehower LA (2007) The Wip1 phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell 12(4):342–354
Ding QJ, Zhang ZM, Liu JJ, Jiang N, Zhang J, Ross TM et al (2013) Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem 56(14):5979–5983
Yee K, Martinelli G, Vey N, Dickinson MJ, Seiter K, Assouline S et al (2014) Phase 1/1b Study of RG7388, a Potent MDM2 Antagonist, in Acute Myelogenous Leukemia (AML) Patients (Pts). Blood 124(21):116
Siu LL, Italiano A, Miller WH, Blay JY, Gietema JA, Bang YJ et al (2014) Phase 1 dose escalation, food effect, and biomarker study of RG7388, a more potent second-generation MDM2 antagonist, in patients (pts) with solid tumors. J Clin Oncol 32(15):2535
Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H et al (2017) Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49(12):1779
Xu C, Liu XQ, Geng YB, Bai QR, Pan CC, Sun Y et al (2017) Patient-derived DIPG cells preserve stem-like characteristics and generate orthotopic tumors. Oncotarget 8(44):76644–76655
Hashizume R, Smirnov I, Liu S, Phillips JJ, Hyer J, McKnight TR et al (2012) Characterization of a diffuse intrinsic pontine glioma cell line: implications for future investigations and treatment. J Neuro-Oncol 110(3):305–313
Tsoli M, Liu J, Franshaw L, Shen H, Cheng C, Jung M et al (2018) Dual targeting of mitochondrial function and mTOR pathway as a therapeutic strategy for diffuse intrinsic pontine glioma. Oncotarget 9(7):7541–7556
Grasso CS, Tang YJ, Truffaux N, Berlow NE, Liu LN, Debily MA et al (2015) Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 21(6):555–559
Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M et al (2018) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46(W1):W537–W544
Alhamdoosh M, Ng M, Wilson NJ, Sheridan JM, Huynh H, Wilson MJ et al (2017) Combining multiple tools outperforms individual methods in gene set enrichment analyses. Bioinformatics 33(3):414–424
Berberich A, Kessler T, Thome CM, Pusch S, Hielscher T, Sahm F et al (2019) Targeting resistance against the MDM2 inhibitor RG7388 in glioblastoma cells by the MEK inhibitor trametinib. Clin Cancer Res 25(1):253–265
Dai C, Gu W (2010) p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med 16(11):528–536
Shen H, Maki CG (2011) Pharmacologic activation of p53 by small-molecule MDM2 antagonists. Curr Pharm Design 17(6):560–568
Bradley KA, Pollack IF, Reid JM, Adamson PC, Ames MM, Vezina G et al (2008) Motexafin gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: a children’s oncology group phase I study. Neuro Oncol 10(5):752–758
Skalniak L, Kocik J, Polak J, Skalniak A, Rak M, Wolnicka-Glubisz A et al (2018) Prolonged idasanutlin (RG7388) treatment leads to the generation of p53-mutated cells. Cancers 10(11):396
Chen LD, Rousseau RF, Middleton SA, Nichols GL, Newell DR, Lunec J et al (2015) Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget 6(12):10207–10221
Laroche-Clary A, Chaire V, Algeo MP, Derieppe MA, Loarer FL, Italiano A (2017) Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol. https://doi.org/10.1186/s13045-017-0482-3
Zanjirband M, Curtin N, Edmondson RJ, Lunec J (2017) Combination treatment with rucaparib (Rubraca) and MDM2 inhibitors, Nutlin-3 and RG7388, has synergistic and dose reduction potential in ovarian cancer. Oncotarget 8(41):69779–69796
Bredlau AL, Dixit S, Chen C, Broome AM (2017) Nanotechnology applications for diffuse intrinsic pontine glioma. Curr Neuropharmacol 15(1):104–115
Zhou ZP, Singh R, Souweidane MM (2017) Convection-enhanced delivery for diffuse intrinsic pontine glioma treatment. Curr Neuropharmacol 15(1):116–128
Acknowledgements
We acknowledge Dr. C David James (Northwestern University), Dr. Angel Montero Carcaboso (Barcelona) and Dr. Michelle Monje (Stanford University) for kindly providing us the patient-derived DIPG cell lines.
Funding
This study was supported by the Duke Glioma Translational Fund and the Zachem Family Fund. They are granted through Duke’s The Preston Robert Tisch Brain Tumor Center.
Author information
Authors and Affiliations
Contributions
Project design: H.Y., Y.H., C.X., M.S.W., L.Z. Experiment carry-out: C.X., H.L., C.J.P. Data analysis: L.H.C., H.L., C.X., B.D. Project administration: P.K.G. Supervision: H.Y., Y.H. Original draft preparation: C.X., H.L. Reviewing and Editing: C.J.P., L.H.C., P.K.G., B.D., L.Z., M.S.W., Y.H., H.Y. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Animal studies were approved by the Institutional Animal Care and Use Committee of Duke University (accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), International).
Consent for publication
Not applicable.
Competing interests
H.Y. is the chief scientific officer and has owner interest in Genetron Holdings, and receives royalties from Genetron, Agios, and Personal Genome Diagnostics (PGDX).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
(A) The characterization of ten patient-derived DIPG cell lines. Five cell lines (on the top) harbored H3K27M mutation and PPM1D truncating mutations, but were TP53 wild-type. The other five cell lines (on the bottom) harbored mutant H3K27M and TP53, but did not contain mutation in exon 6 of PPM1D. (B) Protein expression level of H3K27M and p53 by Western Blot in ten patient-derived DIPG cell lines. (C) Sanger sequencing result of H3K27M mutation. (D) Sanger sequencing result of TP53 mutation in SU-DIPG-XIII line. (E) Sanger sequencing result of PPM1D mutation in five TP53 wild-type DIPG cell lines.
Additional file 2: Figure S2.
(A) IC50 of RG7388 in five TP53 wild-type and five TP53 mutant DIPG lines. (B) The viabilities of the five TP53 mutant DIPG lines treated with DMSO, 50nM and 100nM RG7388 for two and four days. Relative viabilities were calculated by normalizing luminescence values for each time point to Day 0 at the same treatment conditions. Mean ± SEM, n=3 independent studies for each condition. P-values based on Two-way ANOVA. ns=non-significant. (C) IC50 of RG7388 in HSJD-DIPG-007 NTC and TP53 KO lines. (D) The viabilities of HSJD-DIPG-007 TP53 KO line treated with DMSO, 50nM and 100nM RG7388 for two and four days. Relative viabilities were calculated by normalizing luminescence values for each time point to Day 0 at the same treatment conditions. Mean ± SEM, n=3 independent studies for each condition. P-values based on Two-way ANOVA. ns=non-significant.
Additional file 3: Figure S3.
(A) Top five most differentially regulated KEGG pathways of HSJD-DIPG-007-NTC and HSJD-DIPG-007-TP53 KO lines response to RG7388. (B) Hierarchical clustering of significantly changed genes (adjusted P-values < 0.05, fold change > 1.5) within the DNA replication and cell cycle pathway of HSJD-DIPG-007-NTC response to RG7388. Columns in the heat map represent individual replicates (three for each condition). Rows represent genes, colored by log-transformed transcript intensity (DESeq2-normalized counts + 1) in z-score. Blue shows replicates with low expression (z-score < 0); red shows replicates with high expression (z-score > 0). (C) Transcriptional levels of MDM2, TP53 and CDKN1A after RG7388 treatment in HSJD-DIPG-007-NTC line.
Additional file 4: Figure S4.
(A) Quantification of protein expression level of MDM2, p53 and p21 in HSJD-DIPG-007-NTC and HSJD-DIPG-007-TP53 KO treated with DMSO or 25nM RG7388 for 6 hours, 12 hours, 24 hours and 48 hours. Mean ± SEM, n=3 independent studies for each condition. P-values based on Two-way ANOVA. *P < 0.05. ***P < 0.001. ****P < 0.0001. (B) Quantification of protein expression level of MDM2, p53 and p21 in five TP53 wild-type and five TP53 mutant DIPG cell lines treated with DMSO and 100nM RG7388 for 24 hours. Mean ± SEM, n=3 independent studies for each condition. P-values based on Student’s t-test. ****P < 0.0001. (C) Representative H&E and immunofluorescence staining of brainstem xenograft. Scale bar: 100μm.
Additional file 5:
Supplementary Methods
Additional file 6: Supplymentary Table S1.
CLA analysis of tem DIPG cell lines
Additional file 7: Supplymentary Table S2.
Primers for Sanger sequencing
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, C., Liu, H., Pirozzi, C.J. et al. TP53 wild-type/PPM1D mutant diffuse intrinsic pontine gliomas are sensitive to a MDM2 antagonist. acta neuropathol commun 9, 178 (2021). https://doi.org/10.1186/s40478-021-01270-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01270-y