
Abstract
It is recognized that the tumor microenvironment (TME) plays a critical role in the biology of cancer. To better understand the role of immune cell components in CNS tumors, we applied a deconvolution approach to bulk DNA methylation array data in a large set of newly profiled samples (n = 741) as well as samples from external data sources (n = 3311) of methylation-defined glial and glioneuronal tumors. Using the cell-type proportion data as input, we used dimensionality reduction to visualize sample-wise patterns that emerge from the cell type proportion estimations. In IDH-wildtype glioblastomas (n = 2,072), we identified distinct tumor clusters based on immune cell proportion and demonstrated an association with oncogenic alterations such as EGFR amplification and CDKN2A/B homozygous deletion. We also investigated the immune cluster-specific distribution of four malignant cellular states (AC-like, OPC-like, MES-like and NPC-like) in the IDH-wildtype cohort. We identified two major immune-based subgroups of IDH-mutant gliomas, which largely aligned with 1p/19q co-deletion status. Non-codeleted gliomas showed distinct proportions of a key genomic aberration (CDKN2A/B loss) among immune cell-based groups. We also observed significant positive correlations between monocyte proportion and expression of PD-L1 and PD-L2 (R = 0.54 and 0.68, respectively). Overall, the findings highlight specific roles of the TME in biology and classification of CNS tumors, where specific immune cell admixtures correlate with tumor types and genomic alterations.
Similar content being viewed by others

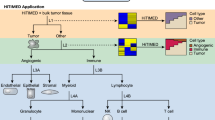

Introduction
Glial and glioneuronal tumors represent a wide range of tumor types with distinct biology and clinical outcomes and are currently assigned WHO grades 1–4 based on histopathologic and molecular features [1, 53]. The tumor microenvironment (TME) is a critical element in glioma biology and has been shown to alter sensitivity to immune-based therapies [17, 86]. In addition to immune cell burden, the specific cellular composition is also a critical feature of the TME and may influence key steps in tumor–immune interactions [39]. While the contribution of the TME to genomic instability in tumor cells has been previously investigated [20, 21, 71, 87], information is incomplete regarding the presence and/or activity of immunosuppressive or immunostimulatory cell types in gliomas, particularly in high-grade tumors [69]. Tumor infiltrating lymphocytes, or TILs, are often enriched in glioblastoma (GBM) with a mesenchymal gene expression signature and are strongly associated with mutations in NF1 and RB1 [75]. Conversely, TILs have been reported to be depleted in “classical” GBM (EGFR-amplified and PTEN-deleted) [75]. IDH-mutant gliomas often exhibit a less prominent TIL infiltration and decreased expression of the immune checkpoint molecule PD-L1 as compared to IDH wild types, reflecting a less immunosuppressive microenvironment [5]. Finally, recent evidence suggests tumor-associated macrophages (TAMs) may drive mesenchymal differentiation in GBM [76], consistent with reports that transcriptomic plasticity of GBM is significantly influenced by cellular heterogeneity in the tumor microenvironment [67, 78].
Aberrant DNA methylation is recognized as a key process for tumor development [42]. Distinct epigenetic profiles have been recognized in gliomas based on DNA methylation and, in combination with other somatic alterations, characterized clinically relevant subtypes: H3 K27, G34, IDH1, RTKI, RTKII, and mesenchymal [82]. An association with chromosomal alterations has been noted. For example, RTKI was found to be enriched for PDGFRA amplification and RTKII group showed combined chromosome 7 gain/chromosome 10 loss (+ 7/−10), CDKN2A loss and amplification of epidermal growth factor receptor (EGFR) [24, 81, 85]. IDH-wt subclass RTK III was predominantly reported in children and young adults, and correlates with pediatric high grade glioma, showing lower rates of TERT mutation and EGFR amplification as compared to adult-type GBMs [80]. Recently, DNA methylation profiling has emerged as a useful technique for tumor classification and the identification of novel subtypes in glial and glioneuronal tumors [10, 66].
Deconvolution of bulk tumors has typically involved in situ-based techniques, such as immunohistochemistry (IHC), as well as fluorescence-activated cell sorting (FACS), and more recently single-cell RNA sequencing (RNA-seq). Recent in silico techniques have allowed deconvolution on a much larger scale by utilizing high-throughput assays such as gene expression and DNA methylation microarrays [23, 33, 62, 63, 83, 84]. CIBERSORT is a widely-used reference-based deconvolution method used to de-convolve bulk transcriptomic data (e.g. RNA-seq) [37, 54, 95]. An adaptation of this method using DNA methylation data (MethylCIBERSORT) has been recently applied to pediatric CNS embryonal tumors and pediatric high-grade gliomas, revealing clinically and biologically relevant immune-based clusters in these select cohorts [12, 35]. Here, we sought to apply this deconvolution method to a large cohort of epigenetically defined, primarily adult glial and glioneuronal tumors with the aim of uncovering the relationship of tumor immune microenvironment with tumor subtype, canonical genetic alterations, and immune modulators.
Materials and methods
DNA methylation profiling
Samples (n = 741) were profiled as part of clinical methylation testing and analyzed as previously described by Capper et al. [10], and appropriate ethics approval was obtained for reporting. Briefly, bisulfite-converted genomic DNA was assayed on the Infinium MethylationEPIC kit (Illumina, USA), and the methylation profile was then classified using the Heidelberg methylation classifier. CNV profiles were derived from the R “conumee” package (http://bioconductor.org/packages/conumee/) as implemented in the classifier package. Batch effects were examined using variables of formalin-fixed, paraffin-embedded specimens (FFPE) versus frozen specimens. We also compared the 2 array types (450 k vs. EPIC). We did not observe major batch effects with respect to material type and array type.
Data collection
We collated published literature to collect additional data for glial/glioneuronal tumor subtypes. The following publicly-available datasets (GSE, GEO database; E-MTAB, ArrayExpress) were included for downstream analyses: GSE104293 [4], GSE109381 [10], GSE124617 [6], GSE128654 [79], E-MTAB-5528 [57], GSE140124 [59], GSE61160 [60], GSE103659 [46], GSE111165 [7], GSE122994 [45], GSE131482, GSE157397 [25], GSE123678 [19], E-MTAB-5552 [58], GSE122920 [91], E-MTAB-3476 [2], GSE143843 [45], GSE137845 [34], GSE136361 [89], GSE73895 [41], GSE92462 [96], GSE60274 [22], E-MTAB-7802 [18], E-MTAB-5797 [28], GSE125450 [50], GSE119774 [56], E-MTAB-7490 [27], GSE135017 [36], E-MTAB-8390 [31], GSE152653 [55], GSE92579 [47], GSE104723, GSE116298 [90], E-GEOD-73895 and E-MTAB-7804 [18]. Our final dataset (N = 4052) also included samples from TCGA (N = 530) as well as samples processed within our laboratory (N = 741). Raw intensity data files (IDAT) for both 450 K and EPIC arrays were combined into a matrix with a common probe set (452,453 probes). All samples were processed and normalized by single sample noob function provided in the Minfi R package [3]. Sample annotation for tumor methylation class was performed using DKFZ classifier calibrated scores (v11b4) [10]. The final dataset (N = 4052) consisted of three broad glioma/glioneuronal tumor types: IDH-wt GBM (N = 2072), IDH-mut (N = 1178) and low-to-intermediate grade glioneuronal tumors (LIGGNT) (N = 802). LIGGNT is a broad category of glio-neuronal tumors consisting of 11 tumor subtypes: ANA-PA (N = 57), LGG-DIG/DIA (N = 13), LGG- DNT (N = 83), LGG-GG (N = 42), LGG-MYB (N = 56), LGG-PA/MID (N = 85), LGG-PA/PF (N = 216), LGG-PAGG/ST (N = 72), LGG-RGNT (N = 26), LGG-SEGA (N = 23), PXA (N = 129).
Signature matrix development and validation
To construct the reference immune cell signature matrix, we collected methylation profile of pure non-neoplastic cell types. Raw data files (idat) were obtained from publicly-available sources, including B-cells (GSE110554,GSE49618, GSE35069, GSE88824) [9, 44, 72, 77], CD8 T-cells (GSE110554, GSE35069, GSE88824) [44, 72, 77], endothelial cells (GSE82234, GSE144804) [29, 73], monocyte, neutrophil, NK cells, eosinophils (GSE35069, GSE88824) [44, 72], glia (GSE66351) [32], neurons (GSE98203,GSE66351) [32, 48], CD4 T-cells and Treg (GSE49667) [94]. Microglia methylation profile was obtained from in-house profiling. Methylation profiles from 61 cancer cell lines were obtained from various sources (GSE128654, GSE68379, GSE152035, GSE134165; E-MTAB-9297, GSE122809, GSE137845, E-MTAB-9282), representing select types and subtypes of glial/glioneuronal tumors. Methylation data used to generate the signature matrix was processed and normalized by single sample noob function provided in the Minfi R package [3]. For immune cell deconvolution, we used the MethylCIBERSORT R package using the procedure as described by Chakravarthy et al. [12]. For feature selection, we used a modified function of FeatureSelect.V4 as described in Williamson et al. [35]. A maximum of top 100 features per pairwise comparison were selected with a median β-value difference of 0.2 and false discovery rate of 0.01. We selected 1,290 probes from the generated signature matrix differentiating 13 cell types: B-cells, cancer, CD4T, CD8T, endothelial, eosinophil, glia, microglia, monocyte, neuron, neutrophil, NK cells and Treg. To validate the matrix represents a homogeneous signature for each putative cell type, we performed dimensionality reduction (t-SNE) of pure reference cell types and heatmap with unsupervised clustering of selected probes and all cancer cell lines used to create the signature matrix. We generated scatter plots between cancer fraction and two purity measures (ESTIMATE purity and ABSOLUTE purity) [70] to demonstrate a correlation as a validation step for true cancer fraction estimation. Finally, we ran CIBERSORT on bulk tumor data using a Docker container with 1000 permutations without quantile normalization [63]. To validate our findings, we compared MethylCIBERSORT fraction output with gene expression based CIBERSORT proportion of six major cell types in data subset (N = 394) by using LM6 signature matrix [14]. We observed significant positive Pearson’s correlation (Additional file 1: Fig. 1) between similar cell type fractions [93].
Dimensionality reduction and immune cell fraction estimation in tumor subtypes
We classified all samples (N = 4052) into three representative cohorts: IDH-wt (N = 2072), IDH-mut (N = 1178) and LIGGNT (N = 802). Our main objective was to investigate an immune cell fraction-based clustering pattern within these three major cohorts. We first investigated the clustering distribution by both including and excluding the inferred cancer proportion in each dataset (Additional file 2: Fig. 2). Prior to clustering we removed the cancer fraction and normalized the non-neoplastic cells (scaled from 0 to 1) and used immune cell fractions to generate UMAP for each cohort separately. The optimum numbers of clusters in each cohort was obtained using k-means clustering with Nbclust R package, providing 30 indices with which to determine the number of clusters [13]. Cluster selection process was based on the majority rule, which is available in the NbClust package. We compared three different methods; kmeans, ward.D and ward.D2 and finally selected kmeans clustering as a uniform approach to select optimum number of cluster (Additional file 3: Fig. 3). We next assessed overall immune cell proportions in each tumor subtype to determine methylation class-specific immune cell proportions (Additional file 4: Fig. 4a). We also investigated cancer proportion in each tumor subtype, and we found significant differences in cancer fraction of three major tumor cohorts (Additional file 4: Fig. 4b,c).
Copy number variants
Somatic copy number variants were computed from raw signal intensities (IDAT) using the Conumee R package [40]. This method combines the intensity values of the methylated and unmethylated probes of each of the CpG sites and normalizes them to a set of control samples. CNV calling using DNA methylation array data has its own limitation such as it cannot rely on allele frequencies to define a copy-number neutral state baseline as described by Capper et al. [11] but still it is useful for large DNA methylation datasets. To investigate the presence of oncogenic gene amplifications and homozygous deletions events, we adopted the methodology described by Capper et al. [10]. We focused our analysis on select diagnostic and/or prognostic oncogenic events described in glial/glioneuronal tumors: EGFR amplification, PTEN deletion, PDGFRA amplification, CDKN2A/B deletion, 1p/19q codeletion, MYCN and MDM2 amplification. Amplifications were defined as a log2ratio ≥ 1, and deletions defined as a log2ratio ≤ −0.8. We set these high cutoff values to identify true amplification/deletion events and to filter out small gain/loss. For 1p/19 codeletion, we considered co-deletion when both chromosome 1p and 19q showed log2ratio ≤ –0.20. MDM2 and CDK4 are frequently co-amplified genes in glioma, to validate our finding we examined all MDM2 amplified samples for CDK4 amplification. To investigate co-amplification we used average amplification cutoff (log2ratio ≥ 0.4) as described by Capper et al. [11] and we observed co-amplification of MDM2 and CDK4 in more than 68% of samples.
Immune cell proportion and PD-L1/2 gene expression
To investigate a possible association of monocyte proportion with gene expression and promoter methylation of the immune checkpoint ligands PD-L1 and PD-L2, we used a subset of the data (n = 594) which contained matched gene expression profiles (RNA-seq). RNA-seq trimmed mean M-values (TMM) was performed using the calcNormFactors function in the edgeR package [74]. Using the voom() function in limma, the counts were converted log2-counts-per-million (logCPM) and quantile normalized [49]. DNA methylation data was processed using single sample noob approach. We then selected promoter-associated probes in PD-L1/L2 and used the average beta value of all selected probes in these regions as the promoter methylation value.
Survival analysis
Survival data was collected from original source of data (if available) as described in data collection sub-section of material and methods. To examine the association of immune cell proportion with overall survival, we performed Kaplan–Meier survival analyses with the log-rank test using the survminer and survival packages in R [43]. Samples with high proportion (> median) and low proportion (< median) were compared to see survival differences. Survival analysis was also performed to assess the cluster based survival differences in some cases (if survival data was available e.g. O-IDH).
Results
Identification and verification of methylation-derived immune cell populations
A critical step in the deconvolution of bulk data is the appropriate choice of purified reference cell types and confirming a homogeneous population. Using MethylCIBERSORT, we constructed a signature matrix consisting of 1,290 probes distinguishing 13 specific cell types, as previously described: B-cells, CD4T, CD8T, Treg, NK cells, eosinophils, neutrophils, monocytes, microglia, endothelial cells, glial cells, neurons, and the cancer fraction. After generation of the signature matrix, we verified correct representation of cellular phenotypes by visualizing the profiles with t-SNE, which demonstrated an appropriate distinction between all purified reference groups (Fig. 1a). As confirmation, additional unsupervised hierarchical clustering of the signature matrix probes clearly distinguished these cell type clusters (Fig. 1b). We further investigated the association of the cancer fraction with two purity measures, ESTIMATE and ABSOLUTE (where available) and show a significant correlation with ESTIMATE-based (R = 0.59, p value < 2.2e−16) and ABSOLUTE purity (0.77, p value < 2.2e−16) (Fig. 1c).

Signature matrix generation a t-SNE plot showing distinct clusters of methylation profiles of the pure reference cell types used to generate signature matrix. b Heatmap generated by all selected probes of signature matrix. c Scatterplot showing the estimated CIBERSORT cancer fraction (N = 4052) correlates significantly with purity (Estimate purity; R = 0.59; p value < 2.2 × 10–16, Absolute purity; R = 0.76; p value < 2.2 × 10–16)
Immune cell deconvolution and clustering of IDH-wildtype diffuse gliomas
We first investigated the distribution of immune cell populations in a large set of IDH-wildtype diffuse glioblastoma (N = 2,072). This set consisted of seven methylation-defined glioma types: GBM-G34, GBM-MES, GBM-MID, GBM-MYCN, GBM-RTKI, GBM-RTKII and GBM-RTKIII that were defined using DKFZ DNA methylation classifier calibrated scores (version 11b4) [10]. From the deconvolution results, we calculated the overall mean proportion of immune cells (scaled from 0 to 1) in the IDH-wt dataset. The results indicated the average fraction of immune cells was proportionally highest for monocytes (31% of all non-cancer cells) and endothelial cells (11%) followed by CD8T (6%), B-cells (5%), neutrophils (5%), Treg (4%), microglia (3%), NK cells (2%), CD4T (1%) and eosinophils (1%). We also observed a significant contribution of non-immune cell such as glial cells (28%) and a minor contribution of neurons (1%).
We first examined clustering of IDH-wt GBM based on the calculated immune cell proportions. K-means consensus clustering was performed to identify an optimal number of clusters across all IDH-wt tumor samples (N = 2,072). The optimal number of clusters was determined by NbClust R package by providing 30 indices for determining the number of clusters [13]. Consensus clustering suggested five major clusters in the IDH-wt dataset (Fig. 2a,b). Cluster 1 was defined by a high proportion of neutrophils (median = 0.38), monocytes (median = 0.30), endothelial cells (median = 0.04) and B-cells (median = 0.02) (Fig. 2c). Interestingly, monocytes (median = 0.54) constituted the highest proportion in cluster 2 as compared to any other cluster, in addition to endothelial cells (median = 0.08) and B-cells (median = 0.04) (Fig. 2c). Cluster 3 demonstrated high levels of monocytes (median = 0.15), endothelial cells (median = 0.08), and CD8T (median = 0.07). Cluster 4 was defined by a high proportion of microglia (median = 0.41). Cluster 5 was similar to cluster 1 and was associated with high proportions of monocytes (median = 0.33), endothelial cells (median = 0.12), and B-cells (median = 0.05) (Fig. 2c). We observed that monocyte and endothelial proportions were dominant over any other cell type in the majority of clusters.

IDH-wt immune clustering showed five distinct clusters associated with immune cell proportions and key genomic aberrations. a UMAP clustering of seven tumor subtypes of IDH-wt tumor cohort (N = 2072) based on immune cell proportion (Non-cancer part scaled from 0 to 1) showed subtype specific clustering. b Five optimum number of cluster obtained by using k-means clustering with Nbclust method. c Six major Immune cell type proportions shown by boxplots for each cluster indicated significant difference between immune cells distribution across all cluster. Y axis represents non-cancer cell proportion of particular immune cell scaled from 0 to 1. Each box plots depicted, boxes indicate interquartile range with central bar indicating median and whiskers indicating the range. Blue dot represents mean value of the proportion. T-test and Wilcoxon test (p value shown) were used to calculate statistical significance. d Sample proportions of seven tumor subtypes in each cluster of IDH-wt tumor cohort (N = 2072). Distribution of tumor subtypes in each cluster calculated as percentage of samples represent specific tumor subtype. e Cluster specific genomic aberrations represented as proportion of samples undergoes for genomic changes. Horizontal bars between cluster bars with asterisks represent chi- square test based comparison with significant p value < (0.0001, 0.001, 0.01, 0.05, 1; symbols = "****", "***", "**", "*", "ns”). f Monocyte proportion shown by boxplots for various sample groups with genomic aberrations. T-test and Wilcoxon test (p value shown) were used to calculate statistical significance
Immune cell proportions showed a distinct relationship with GBM methylation classes. As shown in Fig. 2d, the majority of samples in cluster 1 were GBM-MES (70%) and GBM-RTKII (16%). Conversely, cluster 5 showed an inverse pattern, with highest proportion of GBM-RTKII (54%) and a relatively low proportion of GBM-MES (22%). Representation of several of the less-common GBM subtypes, including GBM-G34, GBM-RTKII and GBM MYCN, were observed in cluster 3. A Sankey diagram demonstrating this relationship between DNA methylation class and immune-based clustering is shown in Additional file 5: Fig. 5.
Immune clusters are associated with key oncogenic alterations in IDH-wt glioblastoma
Next, we investigated select copy number alterations in IDH-wt gliomas with respect to the defined immune clusters (Table. 1). We used conumee R package to analyze frequent genomic aberrations (amplifications/deletions) in GBM [40]. Cluster 5 showed a significantly higher proportion of samples harboring EGFR amplification and was approximately twice as frequent as tumors in clusters 3 and 4 (Fig. 2e). Tumors with homozygous deletion of PTEN were relatively enriched in cluster 4, and MYCN or PDGFRA amplification were enriched in cluster 3. To understand the significance and association of monocyte proportion in genomic aberrations we classified samples groups based on presence/absence of these alterations and compared the monocyte proportion between these groups (Fig. 2f). Interestingly, we found significant differences in monocyte proportions between sample groups (EGFR, p < 0.01; MYCN, p < 0.01, PTEN, p < 0.001, CDKN2AB, p < 0.05) based on genomic aberrations.
Immune clusters are associated with glioma cell states in IDH-wt GBM
Previously, four main cellular states for IDH-wt GBM (AC-like, OPC-like, MES-like and NPC-like) have been identified based on single-cell RNA sequencing data [61]. Here, we used TCGA samples based on the availability of matched mRNA and methylation data and used CIBERSORTx to estimate the relative proportion of these cellular states. We observed significant differences in the proportion of cell states versus our immune cell-defined clusters. Clusters 3 and 5 were associated with a high proportion of OPC-like and AC-like malignant states, respectively (Additional file 6: Fig. 6), and Clusters 1 and 2 were populated by tumors with a high proportion of tumors with predominant MES-like cell state. We also analyzed tumor-specific survival probabilities in relation to immune cell proportion in tumor subtypes of IDH-wt cohort (Additional file 7: Fig. 7). We compared two groups based on median monocyte proportion in GBM-MES (N = 121) and GBM-RTKI (N = 52) and we observed poor overall survival in monocyte-high samples (p < 0.01 and p = 0.05, respectively) (Additional file 7: Fig. 7a, b). Similarly, we compared high/low endothelial proportion groups based on median proportion and found that samples with a high proportion of endothelial cells showed improved survival probability (p = 0.032) (Additional file 7: Fig. 7c) in GBM-MES.
IDH-mutant diffuse gliomas
The IDH-mutant cohort (N = 1178) was next examined and comprised the methylation classes O-IDH (oligodendroglioma), A-IDH (astrocytoma) and A-IDH-HG (high-grade astrocytoma). The presence of an IDH mutation was inferred from the methylation data. Surprisingly, visualization (UMAP) revealed apparent discrimination of these three methylation classes according to relative immune cell proportion (Fig. 3a). K-means consensus clustering revealed n = 2 optimal number of clusters across all IDH-mutant tumor samples (Fig. 3b), and corresponding differences in immune cell proportion were noted (Fig. 3c): cluster 1 was relatively overrepresented by monocytes and cluster 2 showing increased representation of endothelial cells. Overall, cluster 1 showed higher infiltration of immune cells, and as expected based on the methylation classifier results, was enriched in non-1p19q codeleted samples (Fig. 3d). Cluster 1 contained 62 co-deleted and 359 non-codeleted samples, as compared to cluster 2, which contained 437 co-deleted and 320 non-codeleted samples, revealing a significant difference in immune cell composition in these genomically-defined groups (p < 2.2e−16, chi-square test). Our results are in line with a recent study described association of IDH mutation and 1p19q co-deletion with tumor immune microenvironment [52]. Interestingly, we found that the overall immune cell proportions in IDH mutant tumors was lower than IDH wild type GBMs, although A-IDH-HG tumors had increased immune cell infiltration as compared to other IDH mutant gliomas (Additional file 4: Fig. 4a).

IDH-mutant glioma immune clustering showed two distinct clusters associated with 1p/19q status and different immune cell proportions. a UMAP clustering of three tumor subtypes of IDH-mut tumor cohort (N = 1178) based on immune cell proportion (Non-cancer part scaled from 0 to 1) showed subtype specific clustering. b Two optimum number of cluster obtained in IDH-mut cohort by using k-means clustering with Nbclust method. c Six major Immune cell type proportions shown by boxplots for each cluster indicated significant difference between immune cells distribution between two clusters. Y axis represents non-cancer cell proportion of particular immune cell scaled from 0 to 1. Each box plots depicted, boxes indicate interquartile range with central bar indicating median and whiskers indicating the range. Blue dot represents mean value of the proportion. T-test and Wilcoxon test (p value shown) were used to calculate statistical significance. d Copy number analysis of DNA methylation profile of IDH-mut cohort showed distinct distribution of samples with 1p/19q codeletion
We next focused on oligodendroglioma samples in isolation (N = 444), where consensus clustering on immune cell proportion indicated four major clusters (Fig. 4a). Differences in individual cell types were noted (Fig. 4b). Cluster 2 and cluster 3 showed high proportion of grade 3 tumor samples as compared to cluster 1 and cluster 4, which consist of both grade 2 and grade 3 samples (Fig. 4c). Interestingly, significant survival differences were noted among immune-based groups (Fig. 4d).

Immune clustering of IDH-mut oligodendroglioma (N = 444) showed four distinct clusters associated with tumor grade and patient outcome. a UMAP clustering of OIDH tumor cohort (N = 444) based on immune cell proportion (Non-cancer part scaled from 0 to 1) showed distinct clusters. b Six major Immune cell type proportions shown by boxplots for each cluster indicated significant difference between immune cells proportion across all cluster. Y axis represents non-cancer cell proportion of particular immune cell scaled from 0 to 1. Each box plots depicted, boxes indicate interquartile range with central bar indicating median and whiskers indicating the range. Blue dot represents mean value of the proportion. T-test and Wilcoxon test (p value shown) were used to calculate statistical significance. c Tumor grade distribution of samples across all clusters. Significance based on Fisher's exact test (cluster1-cluster3; p value = 0.000017, cluster3-cluster4; p value = 0.014). d Kaplan–Meier plot showing significant difference in overall survival between cluster 1 cluster 3 and 4
In IDH-mutant, non-1p/19q co-deleted gliomas (N = 734), we identified five optimal immune-based clusters (Fig. 5a, b). Significant differences were apparent for monocytes, CD8 T-cells, CD4 T-cells and microglia (Fig. 5c). When examining the distribution of histologic grade (Fig. 5d), there was a notable absence of low-grade (2) in cluster 2, defined by a high proportion of CD8 T-cells (Fig. 5c). As an additional analysis, we analyzed the proportion of tumors with homozygous loss of CDKN2A/B (a clinically relevant marker in these tumors), showing a higher proportion in cluster 2 consistent with the higher overall tumor grade in this cluster (Fig. 5e). Similarly, we also investigated heterozygous and homozygous deletion by selecting a range of log2ratio(homozygous < −0.8; heterozygous < −0.3) as separate event [26] and we found cluster 2 with higher number heterozygous/homozygous deletions as compared to other clusters (Additional file 8: Fig. 8). Overall, increased numbers of CD8 T-cells were associated with higher grade and CDKN2A/B homozygous deletion, highlighting the relationship of the TME with somatic alterations.

IDH-mut astrocytoma immune clustering showed five distinct clusters associated with tumor grade and CDKN2A/B deletion. a UMAP clustering of IDH-mutant astrocytoma cohort (N = 734) based on immune cell proportion (Non-cancer part scaled from 0 to 1) showed subtype specific clustering. b Five optimum number of cluster obtained in IDH-mut cohort by using k-means clustering with Nbclust method. c Six major Immune cell type proportions shown by boxplots for each cluster indicated significant difference between immune cells distribution across all cluster. Y axis represents non-cancer cell proportion of particular immune cell scaled from 0 to 1. Each box plots depicted, boxes indicate interquartile range with central bar indicating median and whiskers indicating the range. Blue dot represents mean value of the proportion. T-test and Wilcoxon test (p value shown) were used to calculate statistical significance. d Sample proportions as tumor grade 1, 2 & 3 in each cluster (N = 370; significance based on Fisher's exact test; p value < (0.0001, 0.001, 0.01, 0.05, 1; symbols = "****", "***", "**", "*", "ns”). e Cluster 2 and 5 showed significant difference between sample proportion with CDKN2A/B deletion. Horizontal bars between cluster bars with asterisks represent chi- square test based comparison with significant p value (< 0.001)
Circumscribed and low-to-intermediate grade glial/glioneuronal tumors
Immune-based clustering of low-to-intermediate grade glioneuronal tumors (LIGGNT) (N = 802) consisted of 11 DNA methylation-based types: anaplastic pilocytic astrocytoma/high grade astrocytoma with piloid features (ANA-PA), desmoplastic infantile ganglioglioma and astrocytoma (DIG-DIA), dysembryoplastic neuroepithelial tumor (DNT), ganglioglioma (GG), low-grade glioma with MYB/MYBL1 alteration (MYB), midline pilocytic astrocytoma (PA-MID), pilocytic astrocytoma, posterior fossa (PA-PF), supratentorial pilocytic astrocytoma/ganglioglioma (PA/GG-ST), rosette-forming glioneuronal tumor (RGNT), subependymal giant cell astrocytoma (SEGA), and pleomorphic xanthoastrocytoma (PXA) (Fig. 6a). K-means clustering and Nbclust showed an optimum number of two clusters in the LIGGNT cohort (Fig. 6b). There were distinct differences in the proportion of immune cells according to cluster group (Fig. 6c), including monocytes, neutrophils, and NKcells. Cluster 1 was enriched for MYB, DNT and GG samples, while cluster 2 was enriched for PA/PF, PXA, PA/MID and ANA-PA.

Immune clustering of Low-to-intermediate grade glioneuronal tumors (LIGGNT) showed distinct clusters associated with tumor type and different immune cell proportions. a UMAP clustering of eleven tumor subtypes of the LIGGNT cohort (N = 802) based on immune cell proportion (Non-cancer part scaled from 0 to 1) showed subtype specific clustering. b Two optimum number of cluster obtained in the LIGGNT cohort by using k-means clustering with Nbclust method. c Six major Immune cell type proportions shown by boxplots for each cluster indicated significant difference between immune cells distribution between two clusters. T-test and Wilcoxon test (p value shown) were used to calculate statistical significance
Monocyte proportion is associated with PD-L1 and PD-L2 gene expression and promoter methylation in bulk methylation data
Consistent with prior studies [15, 35] we found that cells of the monocytic lineage were among the most abundant immune cell types in glial/glioneuronal tumors. Furthermore, the success of immune checkpoint inhibitors in lung carcinoma and melanoma [64, 65] has led to interest in characterizing the immunologic milieu in brain tumors. We therefore investigated the association between monocyte proportion and gene expression and promoter methylation of the immune checkpoints PD-L1 and PD-L2. Using a subset of samples (n = 594) with matched gene expression profiles, we observed a significant positive correlation between monocyte proportion and expression of PD-L1 and PD-L2 (R = 0.54; p < 2.2e−16 and 0.68; p < 2.2e−16, respectively). Consistently, we found high negative correlations (R = − 0.51 and − 0.61, respectively) between monocytes and promoter methylation of PD-L1 (Fig. 7a, b,c) and PD-L2 (Fig. 7d, e, f). After sorting samples in decreasing order of PD-L1 and PD-L2 expression, we found an expected trend of promoter methylation (low to high) and monocyte proportion (high to low) (Fig. 7).

Association of monocyte proportion with PD-L1 and PD-L2 gene expression and gene promoter methylation (N = 594). a Scatterplot of Monocyte proportion against the gene expression of PD-L1 showed significant positive correlation. b Scatterplot of Monocyte proportion against the promoter methylation of PD-L1 showed significant negative correlation. c Bar plot of 594 sample arranged from high to low expression of PD-L1. Arranged samples showed probe methylation low to high (Opposite to gene expression) and monocyte proportion high to low, similar to gene expression. d Scatterplot of Monocyte proportion against the gene expression of PD-L2 showed significant positive correlation. e Scatterplot of Monocyte proportion against the promoter methylation of PD-L2 showed significant negative correlation as compared to PD-L1. f Bar plot of 594 sample arranged from high to low expression of PD-L2. Arranged samples showed probe methylation low to high (Opposite to gene expression) and monocyte proportion high to low, similar to gene expression
Discussion
Here we report results of a large-scale deconvolution analysis of > 4000 methylation-defined, primarily glial and glioneuronal tumors. We observed significant diversity in the tumor microenvironment composition among tumor subtypes and demonstrate an association with specific genomic aberrations, tumor grade and prognosis. Similar to prior studies, we found monocytes constitute one of the most abundant cell types in the immune microenvironment of gliomas [8, 16, 35, 68, 69, 88]. This was particularly evident in IDH-wt tumors, where an association with EGFR and MDM2 amplification and CDKN2A/B deletion was observed in monocyte-rich clusters. Conversely, we observed a low monocyte proportion in MYCN amplified and PTEN deleted groups. The proportion of CD8 T-cells, another prominent immune cell type in high-grade gliomas, was associated with oncogene amplifications in MYCN, PDGFRA, and MDM2.
Studies of cell states in glioblastoma have previously demonstrated an association with chromosomal alterations [61]. We therefore investigated whether the immune-based clusters identified in the present study were associated with these cellular states. In the current study, we found the AC-like cell state was associated with immune clusters harboring EGFR amplification, while the OPC-like cell state was associated with PDGFRA amplification. The association of immune-based clusters with somatic alterations and cell states described by Suva et al. suggest a role of tumor microenvironment cellular constituents (monocytes, T/B-cells, endothelial cells) in contributing to these malignant cell states, suggesting cross-talk between tumor cell states and the immune microenvironment.
A recent study showed that IDH wild-type tumors associated with high degree of immune cell infiltration, and were suggested as immune-hot phenotype, whereas IDH mut-tumors with 1p/19q codeletion cohort showed low degree of immune cell infiltration as immune-cold phenotype [51]. Our Immune cell based clustering of IDH-mutant tumors also revealed low infiltration of immune cells in IDH-mut-codel associated with1p/19q codeletion.
Recently, it has been shown that CDKN2A homozygous deletion was associated with poorer outcome among IDH-mutant gliomas lacking 1p/19q codeletion (IDH-mutant astrocytoma) as well as among anaplastic oligodendrogliomas, IDH-mutant-1p/19q codeleted [3]. We also observed two major clusters with significant difference in CDKN2A/B deletion in IDH mutant astrocytoma cohort associated with high proportion of CD8 T, B-cell, and endothelial cells. We found in IDH-wt cohort that the cluster with high proportion of endothelial cells and B-cells prominently showed a greater number of samples with CDKN2A/B deletion and similarly in IDH-mut astrocytomas we found that in cluster 2 significant number of samples presented CDKN2A/B deletion.
Immune clustering of low-to-intermediate grade glioneuronal tumors (LIGGNT) (N = 802, 11 tumor subtypes) showed some promising outputs regarding tumor subtype specific overall infiltration of immune cells. We observed that several tumor subtypes, including MYB, DNT, GG and RGNT showed lower infiltration of immune cells as compared to other subtypes and these observations are in line with a recent study [35].
Given that cells of the monocyte lineage are one of the most dominant and key modulator of tumor microenvironment in glio-neuronal tumors, we examined the association of monocytes with programmed cell death ligands such as PD-L1 and PD-L2 gene expression, which plays crucial role in immunotherapy. It has been shown in literature that GBM EVs (extracellular vesicles) induce immunosuppressive monocytes, including myeloid-derived suppressor cells (MDSCs) and nonclassical monocytes (NCMs) [38] and glioblastoma stem cell-derived exosomes can induce M2 macrophages and PD-L1 expression on human monocytes [30]. Accordingly, it has been suggested that blocking PD-1/PD-L1 pathway could be potential new treatment study in glioma [92]. We investigated the possible role of monocyte proportion-relative gene expression and promoter methylation of the immune checkpoint PD-L1 and PD-L2 genes, we used a data subset (n = 594) sample with matched gene expression profiles. We observed significantly high positive correlations (R = 0.54 and 0.68, respectively) between monocyte proportion and expression of PD-L1 and PD-L2, in line with prior reports that monocytic cells can express these immune markers [38, 97]. Consistent with this, we found high negative correlations (R = − 0.51 and − 0.61, respectively) between monocytes and promoter methylation of PD-L1 and PD-L2 respectively. These results suggest a role of monocytic-lineage cells in immunotherapy response of gliomas, which warrants further investigation.
Conclusion
In conclusion, our analysis relies on a large sample set of glial/glioneuronal tumors to demonstrate relationships of tumor immune microenvironmental factors with tumor type and key genomic aberrations. We also highlight the prominent role of monocytic lineage cells in these tumors, including associations with expression of immune checkpoint ligand PD-L1 and PD-L2 in these tumors. Results of this investigation provides insights for future investigation into glioma biology and immunotherapeutic approaches in gliomas.
Availability of data and materials
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the Materials and Methods section. Newly profiled data is available from authors upon reasonable request.
References
Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A (2015) Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol 129:829–848. https://doi.org/10.1007/s00401-015-1432-1
Alentorn A, Durán-Peña A, Malousi A, Marie Y, Mokhtari K, Sanson M, Hoang-Xuan K, Delattre JY, Idbaih A, Vecht C (2015) Differential gene methylation in paired glioblastomas suggests a role of immune response pathways in tumor progression. J Neurooncol 124:385–392. https://doi.org/10.1007/s11060-015-1869-z
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363–1369. https://doi.org/10.1093/bioinformatics/btu049
Bady P, Kurscheid S, Delorenzi M, Gorlia T, van den Bent MJ, Hoang-Xuan K, Vauléon É, Gijtenbeek A, Enting R, Thiessen B et al (2018) The DNA methylome of DDR genes and benefit from RT or TMZ in IDH mutant low-grade glioma treated in EORTC 22033. Acta Neuropathol 135:601–615. https://doi.org/10.1007/s00401-018-1810-6
Berghoff AS, Kiesel B, Widhalm G, Wilhelm D, Rajky O, Kurscheid S, Kresl P, Wöhrer A, Marosi C, Hegi ME et al (2017) Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol 19:1460–1468. https://doi.org/10.1093/neuonc/nox054
Bledea R, Vasudevaraja V, Patel S, Stafford J, Serrano J, Esposito G, Tredwin LM, Goodman N, Kloetgen A, Golfinos JG et al (2019) Functional and topographic effects on DNA methylation in IDH1/2 mutant cancers. Sci Rep 9:16830. https://doi.org/10.1038/s41598-019-53262-7
Braun PR, Han S, Hing B, Nagahama Y, Gaul LN, Heinzman JT, Grossbach AJ, Close L, Dlouhy BJ, Howard MA, Kawasaki H (2019) Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl Psychiatry 9:47. https://doi.org/10.1038/s41398-019-0376-y
Busch S, Talamini M, Brenner S, Abdulazim A, Hänggi D, Neumaier M, Seiz-Rosenhagen M, Fuchs T (2019) Circulating monocytes and tumor-associated macrophages express recombined immunoglobulins in glioblastoma patients. Clin Transl Med 8:18–18. https://doi.org/10.1186/s40169-019-0235-8
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW et al (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368:2059–2074. https://doi.org/10.1056/NEJMoa1301689
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Capper D, Stichel D, Sahm F, Jones DTW, Schrimpf D, Sill M, Schmid S, Hovestadt V, Reuss DE, Koelsche C et al (2018) Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol 136:181–210. https://doi.org/10.1007/s00401-018-1879-y
Chakravarthy A, Furness A, Joshi K, Ghorani E, Ford K, Ward MJ, King EV, Lechner M, Marafioti T, Quezada SA et al (2018) Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun 9:3220. https://doi.org/10.1038/s41467-018-05570-1
Charrad M, Ghazzali N, Boiteau V, Niknafs A (2014) NbClust: An R package for determining the relevant number of clusters in a data set. J Stat Soft 61:36. https://doi.org/10.18637/jss.v061.i06
Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA (2018) Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol 1711:243–259. https://doi.org/10.1007/978-1-4939-7493-1_12
Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, Rasmussen R, Dwivedi B, Seby S, Wolf SA et al (2017) Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Can Res 77:2266–2278. https://doi.org/10.1158/0008-5472.Can-16-2310
Chen Z, Hambardzumyan D (2018) Immune microenvironment in glioblastoma subtypes. Front Immunol. https://doi.org/10.3389/fimmu.2018.01004
Chuntova P, Chow F, Watchmaker PB, Galvez M, Heimberger AB, Newell EW, Diaz A, DePinho RA, Li MO, Wherry EJ et al (2021) Unique challenges for glioblastoma immunotherapy—discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro Oncol 23:356–375. https://doi.org/10.1093/neuonc/noaa277
Clarke M, Mackay A, Ismer B, Pickles JC, Tatevossian RG, Newman S, Bale TA, Stoler I, Izquierdo E, Temelso S et al (2020) Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov 10:942–963. https://doi.org/10.1158/2159-8290.Cd-19-1030
Court F, Le Boiteux E, Fogli A, Müller-Barthélémy M, Vaurs-Barrière C, Chautard E, Pereira B, Biau J, Kemeny JL, Khalil T et al (2019) Transcriptional alterations in glioma result primarily from DNA methylation-independent mechanisms. Genome Res 29:1605–1621. https://doi.org/10.1101/gr.249219.119
D’Anselmi F, Masiello MG, Cucina A, Proietti S, Dinicola S, Pasqualato A, Ricci G, Dobrowolny G, Catizone A, Palombo A et al (2013) Microenvironment promotes tumor cell reprogramming in human breast cancer cell lines. PLoS ONE 8:e83770. https://doi.org/10.1371/journal.pone.0083770
Dakhova O, Ozen M, Creighton CJ, Li R, Ayala G, Rowley D, Ittmann M (2009) Global gene expression analysis of reactive stroma in prostate cancer. Clin Cancer Res 15:3979–3989. https://doi.org/10.1158/1078-0432.Ccr-08-1899
De Meyer T, Bady P, Trooskens G, Kurscheid S, Bloch J, Kros JM, Hainfellner JA, Stupp R, Delorenzi M, Hegi ME et al (2015) Genome-wide DNA methylation detection by MethylCap-seq and Infinium HumanMethylation450 BeadChips: an independent large-scale comparison. Sci Rep 5:15375. https://doi.org/10.1038/srep15375
Decamps C, Privé F, Bacher R, Jost D, Waguet A, Achard S, Amblard E, Bacher R, Bergmann F, Blum M et al (2020) Guidelines for cell-type heterogeneity quantification based on a comparative analysis of reference-free DNA methylation deconvolution software. BMC Bioinform 21:16. https://doi.org/10.1186/s12859-019-3307-2
Dejaegher J, Solie L, Hunin Z, Sciot R, Capper D, Siewert C, Van Cauter S, Wilms G, van Loon J, Ectors N et al (2020) DNA methylation based glioblastoma subclassification is related to tumoral T-cell infiltration and patient survival. Neuro Oncol 23:240–250. https://doi.org/10.1093/neuonc/noaa247
Dodgshun AJ, Fukuoka K, Edwards M, Bianchi VJ, Das A, Sexton-Oates A, Larouche V, Vanan MI, Lindhorst S, Yalon M et al (2020) Germline-driven replication repair-deficient high-grade gliomas exhibit unique hypomethylation patterns. Acta Neuropathol 140:765–776. https://doi.org/10.1007/s00401-020-02209-8
Engqvist H, Parris TZ, Biermann J, Rönnerman EW, Larsson P, Sundfeldt K, Kovács A, Karlsson P, Helou K (2020) Integrative genomics approach identifies molecular features associated with early-stage ovarian carcinoma histotypes. Sci Rep 10:7946. https://doi.org/10.1038/s41598-020-64794-8
Euskirchen P (2019) Methylation profiling of adult astroblastoma, arrayexpress-repository. V1:
Euskirchen P, Bielle F, Labreche K, Kloosterman WP, Rosenberg S, Daniau M, Schmitt C, Masliah-Planchon J, Bourdeaut F, Dehais C et al (2017) Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol 134:691–703. https://doi.org/10.1007/s00401-017-1743-5
Franzen J, Zirkel A, Blake J, Rath B, Benes V, Papantonis A, Wagner W (2017) Senescence-associated DNA methylation is stochastically acquired in subpopulations of mesenchymal stem cells. Aging Cell 16:183–191. https://doi.org/10.1111/acel.12544
Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, Wang F, Hawke D, Yu J, Healy LM et al (2018) Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 7:e1412909–e1412909. https://doi.org/10.1080/2162402X.2017.1412909
Gareton A, Tauziède-Espariat A, Dangouloff-Ros V, Roux A, Saffroy R, Castel D, Kergrohen T, Fina F, Figarella-Branger D, Pagès M et al (2020) The histomolecular criteria established for adult anaplastic pilocytic astrocytoma are not applicable to the pediatric population. Acta Neuropathol 139:287–303. https://doi.org/10.1007/s00401-019-02088-8
Gasparoni G, Bultmann S, Lutsik P, Kraus TFJ, Sordon S, Vlcek J, Dietinger V, Steinmaurer M, Haider M, Mulholland CB et al (2018) DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin 11:41. https://doi.org/10.1186/s13072-018-0211-3
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD et al (2015) The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21:938–945. https://doi.org/10.1038/nm.3909
Golebiewska A, Hau AC, Oudin A, Stieber D, Yabo YA, Baus V, Barthelemy V, Klein E, Bougnaud S, Keunen O et al (2020) Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol 140:919–949. https://doi.org/10.1007/s00401-020-02226-7
Grabovska Y, Mackay A, O’Hare P, Crosier S, Finetti M, Schwalbe EC, Pickles JC, Fairchild AR, Avery A, Cockle J et al (2020) Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat Commun 11:4324. https://doi.org/10.1038/s41467-020-18070-y
Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C, Bridge T, Kim B, Arnoldo A, Kowalski PE et al (2019) Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 10:4343. https://doi.org/10.1038/s41467-019-12187-5
Haddad AF, Chen J-S, Oh T, Pereira MP, Joshi RS, Aghi MK (2020) Higher cytolytic score correlates with an immunosuppressive tumor microenvironment and reduced survival in glioblastoma. Sci Rep 10:17580. https://doi.org/10.1038/s41598-020-73793-8
Himes BT, Peterson TE, de Mooij T, Garcia LMC, Jung MY, Uhm S, Yan D, Tyson J, Jin-Lee HJ, Parney D et al (2020) The role of extracellular vesicles and PD-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro Oncol 22:967–978. https://doi.org/10.1093/neuonc/noaa029
Hirata E, Sahai E (2017) Tumor microenvironment and differential responses to therapy. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a026781
Hovestadt V, Zapatka M (2017) Conumee: Enhanced copy-number variation analysis using Illumina DNA methylation arrays., R package version 1.9.0.:
Johnson KC, Houseman EA, King JE, von Herrmann KM, Fadul CE, Christensen BC (2016) 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat Commun 7:13177. https://doi.org/10.1038/ncomms13177
Karlow JA, Miao B, Xing X, Wang T, Zhang B (2021) Common DNA methylation dynamics in endometriod adenocarcinoma and glioblastoma suggest universal epigenomic alterations in tumorigenesis. Commun Biol 4:607. https://doi.org/10.1038/s42003-021-02094-1
Kassambara A, Kosinski M, Biecek P, Fabian S, Survminer: Drawing survival curves using ‘ggplot2’R package version 0.4.4.
Kennedy DW, White NM, Benton MC, Fox A, Scott RJ, Griffiths LR, Mengersen K, Lea RA (2018) Critical evaluation of linear regression models for cell-subtype specific methylation signal from mixed blood cell DNA. PLoS ONE 13:e0208915. https://doi.org/10.1371/journal.pone.0208915
Kessler T, Berberich A, Sadik A, Sahm F, Gorlia T, Meisner C, Hoffmann DC, Wick A, Kickingereder P, Rübmann P et al (2020) Methylome analyses of three glioblastoma cohorts reveal chemotherapy sensitivity markers within DDR genes. Cancer Med 9:8373–8385. https://doi.org/10.1002/cam4.3447
Kickingereder P, Neuberger U, Bonekamp D, Piechotta PL, Götz M, Wick A, Sill M, Kratz A, Shinohara RT, Jones DTW et al (2018) Radiomic subtyping improves disease stratification beyond key molecular, clinical, and standard imaging characteristics in patients with glioblastoma. Neuro Oncol 20:848–857. https://doi.org/10.1093/neuonc/nox188
Kling T, Wenger A, Beck S, Carén H (2017) Validation of the MethylationEPIC BeadChip for fresh-frozen and formalin-fixed paraffin-embedded tumours. Clin Epigenetics 9:33. https://doi.org/10.1186/s13148-017-0333-7
Kozlenkov A, Jaffe AE, Timashpolsky A, Apontes P, Rudchenko S, Barbu M, Byne W, Hurd YL, Horvath S, Dracheva S (2017) DNA methylation profiling of human prefrontal cortex neurons in heroin users shows significant difference between genomic contexts of hyper- and hypomethylation and a younger epigenetic age. Genes (Basel). https://doi.org/10.3390/genes8060152
Law CW, Chen Y, Shi W, Smyth GK (2014) voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15:R29. https://doi.org/10.1186/gb-2014-15-2-r29
Lehman NL, Usubalieva A, Lin T, Allen SJ, Tran QT, Mobley BC, McLendon RE, Schniederjan MJ, Georgescu MM, Couce M et al (2019) Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7:42. https://doi.org/10.1186/s40478-019-0689-3
Lin W, Qiu X, Sun P, Ye Y, Huang Q, Kong L, Lu JJ (2021) Association of IDH mutation and 1p19q co-deletion with tumor immune microenvironment in lower-grade glioma. Mol Therapy Oncolytics 21:288–302. https://doi.org/10.1016/j.omto.2021.04.010
Lin W, Qiu X, Sun P, Ye Y, Huang Q, Kong L, Lu JJ (2021) Association of IDH mutation and 1p19q co-deletion with tumor immune microenvironment in lower-grade glioma. Mol Ther Oncolytics, City, pp 288–302
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. https://doi.org/10.1007/s00401-007-0243-4
Lu J, Li H, Chen Z, Fan L, Feng S, Cai X, Wang H (2019) Identification of 3 subpopulations of tumor-infiltrating immune cells for malignant transformation of low-grade glioma. Cancer Cell Int 19:265. https://doi.org/10.1186/s12935-019-0972-1
Lucas CG, Gupta R, Doo P, Lee JC, Cadwell CR, Ramani B, Hofmann JW, Sloan EA, Kleinschmidt-DeMasters BK, Lee HS et al (2020) Comprehensive analysis of diverse low-grade neuroepithelial tumors with FGFR1 alterations reveals a distinct molecular signature of rosette-forming glioneuronal tumor. Acta Neuropathol Commun 8:151. https://doi.org/10.1186/s40478-020-01027-z
Mack SC, Singh I, Wang X, Hirsch R, Wu Q, Villagomez R, Bernatchez JA, Zhu Z, Gimple RC, Kim LJY et al (2019) Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J Exp Med 216:1071–1090. https://doi.org/10.1084/jem.20190196
Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M et al (2017) Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32:520-537.e525. https://doi.org/10.1016/j.ccell.2017.08.017
Mackay A (2018) Illumina Infinium HumanMethylation450 BeadChip profiling of paediatric high grade gliomas from the HERBY clinical trial, arrayexpress-repository. V1:
Mondal G, Lee JC, Ravindranathan A, Villanueva-Meyer JE, Tran QT, Allen SJ, Barreto J, Gupta R, Doo P, Van Ziffle J et al (2020) Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol 139:1071–1088. https://doi.org/10.1007/s00401-020-02155-5
Mur P, Mollejo M, Ruano Y, de Lope ÁR, Fiaño C, García JF, Castresana JS, Hernández-Laín A, Rey JA, Meléndez B (2013) Codeletion of 1p and 19q determines distinct gene methylation and expression profiles in IDH-mutated oligodendroglial tumors. Acta Neuropathol 126:277–289. https://doi.org/10.1007/s00401-013-1130-9
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM et al (2019) An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178:835-849.e821. https://doi.org/10.1016/j.cell.2019.06.024
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA (2015) Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12:453–457. https://doi.org/10.1038/nmeth.3337
Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D et al (2019) Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37:773–782. https://doi.org/10.1038/s41587-019-0114-2
Onoi K, Chihara Y, Uchino J, Shimamoto T, Morimoto Y, Iwasaku M, Kaneko Y, Yamada T, Takayama K (2020) Immune checkpoint inhibitors for lung cancer treatment: a review. J Clin Med 9:1362. https://doi.org/10.3390/jcm9051362
Ottaviano M, De Placido S, Ascierto PA (2019) Recent success and limitations of immune checkpoint inhibitors for cancer: a lesson from melanoma. Virchows Arch 474:421–432. https://doi.org/10.1007/s00428-019-02538-4
Perez E, Capper D (2020) Invited review: DNA methylation-based classification of paediatric brain tumours. Neuropathol Appl Neurobiol 46:28–47. https://doi.org/10.1111/nan.12598
Perrin SL, Samuel MS, Koszyca B, Brown MP, Ebert LM, Oksdath M, Gomez GA (2019) Glioblastoma heterogeneity and the tumour microenvironment: implications for preclinical research and development of new treatments. Biochem Soc Trans 47:625–638. https://doi.org/10.1042/bst20180444
Pinton L, Masetto E, Vettore M, Solito S, Magri S, D’Andolfi M, Del Bianco P, Lollo G, Benoit J-P, Okada H et al (2019) The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J Immunother Cancer 7:58–58. https://doi.org/10.1186/s40425-019-0536-x
Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA (2020) Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife 9:e52176. https://doi.org/10.7554/eLife.52176
Qin Y, Feng H, Chen M, Wu H, Zheng X (2018) InfiniumPurify: an R package for estimating and accounting for tumor purity in cancer methylation research. Genes Diseases 5:43–45. https://doi.org/10.1016/j.gendis.2018.02.003
Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA et al (2005) Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436:123–127. https://doi.org/10.1038/nature03688
Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, Söderhäll C, Scheynius A, Kere J (2012) Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS ONE 7:e41361. https://doi.org/10.1371/journal.pone.0041361
Rhead B, Shao X, Quach H, Ghai P, Barcellos LF, Bowcock AM (2020) Global expression and CpG methylation analysis of primary endothelial cells before and after TNFa stimulation reveals gene modules enriched in inflammatory and infectious diseases and associated DMRs. PLoS ONE 15:e0230884. https://doi.org/10.1371/journal.pone.0230884
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Rutledge WC, Kong J, Gao J, Gutman DA, Cooper LA, Appin C, Park Y, Scarpace L, Mikkelsen T, Cohen ML et al (2013) Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin Cancer Res 19:4951–4960. https://doi.org/10.1158/1078-0432.Ccr-13-0551
Sa JK, Chang N, Lee HW, Cho HJ, Ceccarelli M, Cerulo L, Yin J, Kim SS, Caruso FP, Lee M et al (2020) Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol 21:216. https://doi.org/10.1186/s13059-020-02140-x
Salas LA, Koestler DC, Butler RA, Hansen HM, Wiencke JK, Kelsey KT, Christensen BC (2018) An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol 19:64. https://doi.org/10.1186/s13059-018-1448-7
Schiffer D, Annovazzi L, Casalone C, Corona C, Mellai M (2019) Glioblastoma: microenvironment and niche concept. Cancers. https://doi.org/10.3390/cancers11010005
Shen Y, Grisdale CJ, Islam SA, Bose P, Lever J, Zhao EY, Grinshtein N, Ma Y, Mungall AJ, Moore RA et al (2019) Comprehensive genomic profiling of glioblastoma tumors, BTICs, and xenografts reveals stability and adaptation to growth environments. Proc Natl Acad Sci U S A 116:19098–19108. https://doi.org/10.1073/pnas.1813495116
Stichel D, Ebrahimi A, Reuss D, Schrimpf D, Ono T, Shirahata M, Reifenberger G, Weller M, Hänggi D, Wick W et al (2018) Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol 136:793–803. https://doi.org/10.1007/s00401-018-1905-0
Sturm D, Bender S, Jones DTW, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF et al (2014) Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer 14:92–107. https://doi.org/10.1038/nrc3655
Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones David TW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. https://doi.org/10.1016/j.ccr.2012.08.024
Teschendorff AE, Breeze CE, Zheng SC, Beck S (2017) A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinform 18:105. https://doi.org/10.1186/s12859-017-1511-5
Teschendorff AE, Zheng SC (2017) Cell-type deconvolution in epigenome-wide association studies: a review and recommendations. Epigenomics 9:757–768. https://doi.org/10.2217/epi-2016-0153
Tilak M, Holborn J, New LA, Lalonde J, Jones N (2021) Receptor tyrosine kinase signaling and targeting in glioblastoma multiforme. Int J Mol Sci 22:1831. https://doi.org/10.3390/ijms22041831
Tomaszewski W, Sanchez-Perez L, Gajewski TF, Sampson JH (2019) Brain tumor microenvironment and host state: implications for immunotherapy. Clin Cancer Res 25:4202–4210. https://doi.org/10.1158/1078-0432.Ccr-18-1627
Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, Richardson M, Rigaill G, Parrini MC, Lucchesi C et al (2010) Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med 2:211–230. https://doi.org/10.1002/emmm.201000073
Uneda A, Kurozumi K, Fujimura A, Fujii K, Ishida J, Shimazu Y, Otani Y, Tomita Y, Hattori Y, Matsumoto Y et al (2021) Differentiated glioblastoma cells accelerate tumor progression by shaping the tumor microenvironment via CCN1-mediated macrophage infiltration. Acta Neuropathol Commun 9:29. https://doi.org/10.1186/s40478-021-01124-7
Wefers AK, Stichel D, Schrimpf D, Coras R, Pages M, Tauziède-Espariat A, Varlet P, Schwarz D, Söylemezoglu F, Pohl U et al (2020) Isomorphic diffuse glioma is a morphologically and molecularly distinct tumour entity with recurrent gene fusions of MYBL1 or MYB and a benign disease course. Acta Neuropathol 139:193–209. https://doi.org/10.1007/s00401-019-02078-w
Wenger A, Ferreyra Vega S, Kling T, Bontell TO, Jakola AS, Carén H (2019) Intratumor DNA methylation heterogeneity in glioblastoma: implications for DNA methylation-based classification. Neuro Oncol 21:616–627. https://doi.org/10.1093/neuonc/noz011
Wick A, Kessler T, Platten M, Meisner C, Bamberg M, Herrlinger U, Felsberg J, Weyerbrock A, Papsdorf K, Steinbach JP et al (2020) Superiority of temozolomide over radiotherapy for elderly patients with RTK II methylation class, MGMT promoter methylated malignant astrocytoma. Neuro Oncol 22:1162–1172. https://doi.org/10.1093/neuonc/noaa033
Xue S, Hu M, Iyer V, Yu J (2017) Blocking the PD-1/PD-L1 pathway in glioma: a potential new treatment strategy. J Hematol Oncol 10:81. https://doi.org/10.1186/s13045-017-0455-6
Zenkova D, Kamenev V, Sablina R, Artyomov M, Sergushichev A (2019) Phantasus: visual and interactive gene expression analysis. https://genome.ifmo.ru/phantasus. https://doi.org/10.18129/B9.bioc.phantasus
Zhang Y, Maksimovic J, Naselli G, Qian J, Chopin M, Blewitt ME, Oshlack A, Harrison LC (2013) Genome-wide DNA methylation analysis identifies hypomethylated genes regulated by FOXP3 in human regulatory T cells. Blood 122:2823–2836. https://doi.org/10.1182/blood-2013-02-481788
Zhong Q-Y, Fan E-X, Feng G-Y, Chen Q-Y, Gou X-X, Yue G-J, Zhang G-h (2019) A gene expression-based study on immune cell subtypes and glioma prognosis. BMC Cancer 19:1116. https://doi.org/10.1186/s12885-019-6324-7
Zhou D, Alver BM, Li S, Hlady RA, Thompson JJ, Schroeder MA, Lee JH, Qiu J, Schwartz PH, Sarkaria JN et al (2018) Distinctive epigenomes characterize glioma stem cells and their response to differentiation cues. Genome Biol 19:43. https://doi.org/10.1186/s13059-018-1420-6
Zhu Z, Zhang H, Chen B, Liu X, Zhang S, Zong Z, Gao M (2020) PD-L1-mediated immunosuppression in glioblastoma is associated with the infiltration and M2-polarization of tumor-associated macrophages. Front Immunol. https://doi.org/10.3389/fimmu.2020.588552
Acknowledgements
This work was supported and funded by the Intramural Research Program (IRP) of the National Cancer Institute (NCI) and utilized the computational resources of the NIH HPC Biowulf cluster. (http://hpc.nih.gov). The authors declare no competing interests.
Funding
Open Access funding provided by the National Institutes of Health (NIH).
Author information
Authors and Affiliations
Contributions
OS performed the majority of data analysis and DP collected samples from various published studies. OS, DP and KA contributed to the writing and editing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: To validate our MethylCIBERSORT output we selected a data subset (N=394) with available gene expression data. CIBERSORTx method was used with LM6 signature matrix to get fraction of six major cell types. We found significant positive correlations between MethylCIBERSORT fraction and CIBERSORTx derived fraction of six major cell types.
Additional file 2
: Assessment of clustering pattern in all three major cohorts IDH-wt (N=2072), IDH-mutant (N=1178) and Low-to-intermediate grade glioneuronal tumors (LIGGNT) (N=802) by including and excluding cancer part and scaling non-cancer part from 0 to 1. (a) UMAP clustering for IDH-wt cohort with all cell type (including cancer). (b) UMAP clustering for IDH-wt cohort with all normal cell type scaled 0 to 1 (non-cancer part). (c) UMAP clustering for IDH-wt cohort with all immune cells type scaled 0 to 1 (non-cancer part). (d) UMAP clustering for IDH-mutant cohort with all cell type(including cancer). (e) UMAP clustering for IDH-mutant cohort with all normal cell type scaled 0 to 1 (non-cancer part). (f) UMAP clustering for IDH-mutant cohort with all immune cells type scaled 0 to 1 (non-cancer part). (g) UMAP clustering for LIGGNT cohort with all cell type (including cancer). (h) UMAP clustering for LIGGNT cohort with all normal cell type scaled from 0 to 1 (non-cancer part). (i) UMAP clustering for LIGGNT cohort with all immune cells type scaled 0 to 1 (non-cancer part).
Additional file 3
: Cluster selection process was based on the majority rule, which is available in the NbClust package. We compared three different methods; kmeans, ward.D and ward.D2. (a) For each method we selected optimum number of clusters (given in brackets) proposed by maximum number of indices out of 30. We compared output of each method and found similar results. Finally, we selected kmeans clustering as a uniform approach to select optimum number of cluster (proposed by maximum number of indices out of 30) in each cohort. (b) In IDH wild type 11 indices proposed five cluster. (c) In IDH mutant type 8 indices proposed two cluster. (d) In O-IDH cohort, 6 indices proposed two and four clusters respectively. In this case we selected four cluster as ward.D2 method also suggested four clusters in O-IDH cohort. (e) In A-IDH/A-IDH-HG 8 indices proposed five clusters. (f) In LIGGNT 10 indices proposed two clusters.
Additional file 4
: (a) Tumor subtype specific bar plot distribution of mean Immune cell proportion (Non-cancer part scaled 0 to 1, Glia and Neuron excluded). (b) Overall cell fractions of each cell type (Including Cancer, Glia and Neuron). (c) Cancer proportion shown by boxplots for each tumor subtype indicated significant differences. T-test and Wilcoxon test (p value shown) were used to calculate statistical significance.
Additional file 5
: Sankey diagram-based associations between immune cells and tumor subtype in IDH-wt cohort (N=2072). Sankey plot showing proportions shared between each immune clusters and tumor subtype.
Additional file 6
: Analysis of the IDH-wt glioblastoma cohort (N=100) shows cluster specific distribution of AC-like, OPC-like, MES-like and NPC-like cellular states, respectively. Tumor cell states were derived from single cell data of IDH-wild type GBMs (PMID: 31327527). A signature matrix was derived and applied to TCGA samples for which gene expression data were available. Cell state estimations for each sample were performed using CIBERSORTx. T-test and Wilcoxon test (p-value shown) were used to calculate statistical significance.
Additional file 7
: Kaplan-Meier plot of (a) GBM-MES for Monocyte proportion high and low groups (High>median, Low<median). (b) GBM-RTK-I dataset with high and low proportion of monocytes (High>median, Low<median). (c) Kaplan-Meier plot for Endothelial cell high and low groups in GBM-MES (High>median, Low<median).
Additional file 8
: To investigate homozygous and heterozygous deletion of CDKN2AB as a separate event we selected a range of log2ratio and annotated samples as an event of homozygous deletion, heterozygous deletion, and no deletion. Significance was calculated by applying Fisher's exact test; p-value <(0.0001, 0.001, 0.01, 0.05, 1; symbols ="****", "***", "**", "*", "ns”).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Singh, O., Pratt, D. & Aldape, K. Immune cell deconvolution of bulk DNA methylation data reveals an association with methylation class, key somatic alterations, and cell state in glial/glioneuronal tumors. acta neuropathol commun 9, 148 (2021). https://doi.org/10.1186/s40478-021-01249-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01249-9