
Abstract
Cellular damage associated with traumatic brain injury (TBI) manifests in motor and cognitive dysfunction following injury. Experimental models of TBI reveal cell death in the granule cell layer (GCL) of the hippocampal dentate gyrus acutely after injury. Adult-born neurons residing in the neurogenic niche of the GCL, the subgranular zone, are particularly vulnerable. Injury-induced proliferation of neural progenitors in the subgranular zone supports recovery of the immature neuron population, but their development and localization may be altered, potentially affecting long-term survival. Here we show that increasing hippocampal levels of insulin-like growth factor-1 (IGF1) is sufficient to promote end-stage maturity of posttrauma-born neurons and improve cognition following TBI. Mice with conditional overexpression of astrocyte-specific IGF1 and wild-type mice received controlled cortical impact or sham injury and bromo-2′-deoxyuridine injections for 7d after injury to label proliferating cells. IGF1 overexpression increased the number of GCL neurons born acutely after trauma that survived 6 weeks to maturity (NeuN+BrdU+), and enhanced their outward migration into the GCL while significantly reducing the proportion localized ectopically to the hilus and molecular layer. IGF1 selectively affected neurons, without increasing the persistence of posttrauma-proliferated glia in the dentate gyrus. IGF1 overexpressing animals performed better during radial arm water maze reversal testing, a neurogenesis-dependent cognitive test. These findings demonstrate the ability of IGF1 to promote the long-term survival and appropriate localization of granule neurons born acutely after a TBI, and suggest these new neurons contribute to improved cognitive function.
Similar content being viewed by others


Introduction
TBI is the leading cause of death among people under 40 [1, 2]. Survivors of TBI often suffer from persisting learning and memory deficits [3,4,5,6], which may be related to the brain’s limited capacity to repair and replace lost neurons.
Throughout adulthood, neural stem cells in the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) proliferate and differentiate into new principal granule neurons of the granule cell layer (GCL) [7]. During normal adult neurogenesis, as many as half of all newly born neurons undergo apoptosis within 4 weeks of proliferating [8,9,10,11]. Surviving neurons form synapses to functionally integrate into the local hippocampal circuitry [12, 13]. Adult-born neurons have been shown to contribute to contextual fear memory and extinction [14, 15], spatial memory [16, 17], pattern separation [18], and cognitive flexibility [18,19,20,21].
Trauma has a pronounced impact on immature hippocampal neuron survival and development. Contusive brain injury results in the selective death of immature neurons in the GCL within days [22,23,24]. Proliferation of neural stem cells in the SGZ is concomitantly stimulated, resulting in recovery of immature neuron numbers within 1–2 weeks [23,24,25,26]. While a considerable subset survive to maturity [27,28,29,30,31,32], posttrauma-born hippocampal neurons develop with atypical dendritic arbors [25, 26, 33, 34], which may alter synapse-dependent maturation and survival [35,36,37]. Furthermore, TBI appears to stimulate outward migration of new neurons within the GCL [33, 38, 39] and may foster ectopic migration of a small percentage of newly born neurons from the GCL into either the molecular layer (ML) or hilar layer (HL) [34, 39, 40]. Mislocalization and aberrant development may interfere with the function and survival of neurons born following TBI.
Insulin-like growth factor-1 (IGF1) is a potent growth factor that is expressed at high levels during neuronal development and at lower levels in the adult CNS [41, 42]. IGF1 supports neurite outgrowth [43], is required for appropriate migration of neural progenitors during development [44], and promotes newborn neuron survival [45]. In the context of injury, we have shown that IGF1 increases the number of immature neurons born after trauma and restores their dendritic arborization to the levels of uninjured controls [26, 46]. Additionally, IGF1 has been shown to attenuate acute posttraumatic cognitive dysfunction [47,48,49].
Here, we utilized a transgenic mouse model in which conditional overexpression of IGF1 by astrocytes results in increased hippocampal levels of IGF1 acutely after contusive brain injury [48]. At 6 weeks post-injury, a time point beyond the window of maturation-dependent survival of adult-born DG granule cells [9], we examined the effects of IGF1 overexpression on the survival and localization of matured granule neurons born within the first week after TBI. IGF1 overexpression increased the number of posttrauma-born neurons that survived to end-stage maturity and reduced ectopic migration. IGF1 overexpression improved radial-arm water maze (RAWM) reversal learning after the 6-week period corresponding to enhanced stable neurogenesis, suggesting that IGF1 supports long-term cognitive recovery after TBI.
Materials and methods
Animal care
Transgenic mice with astrocyte-specific conditional overexpression of IGF1 were described previously [48, 50]. The mice were bred in-house by crossing heterozygous tTAGFAP mice with heterozygous IGF1pTRE mice, yielding double transgenic mice carrying both transgenes (tTAGFAP/IGF1pTRE) [26, 48]. IGF1 double transgenic mice (IGFtg) and their wildtype (WT) littermates were fed doxycycline supplemented chow (200 mg/kg) to suppress IGF1 expression from birth until 2 weeks prior to surgery, after which they received standard chow. The mice were provided with food and water ad libitum at the University of Kentucky Medical Center animal vivarium. They were housed in temperature-controlled rooms (23 ± 2 °C) with a 14/10-h light/dark cycle.
Controlled cortical impact injury
Isoflurane-anesthetized mice aged 13.3 ± 0.5 weeks old (n = 55) received a 5-mm diameter craniotomy over the left parietal cortex. For sham injury, a cranioplasty was then affixed to the skull to protect the exposed dura and the incision was closed. For controlled cortical impact (CCI) injury, an impactor with a rounded 3 mm diameter tip was used to deliver a rapid (3.5 m/s) impact to the intact dura, as previously described [26, 48]. For immunohistochemical analysis of hippocampal neurogenesis, a cohort of WT (5 female, 7 male) and IGFtg (6 female, and 5 male) mice were randomized for sham injury (3 per genotype) or CCI (8–9 per genotype) delivered using a pneumatically driven impactor device (Precision System Instruments). A second cohort designated for behavioral analysis consisted of WT (6 female, 7 male) and IGFtg (3 female, 8 male) mice that received CCI using a stereotaxic electromagnetic impactor (Leica Biosystems). As a behavioral analysis reference group, a third cohort of WT mice (3 female, 5 male) received sham injury. Impact depths were set for each device (0.8 mm for pneumatic, 1.1 mm for electronic) to produce comparable injury severity across the two cohorts as determined, prior to the onset of these studies, by the size of the cortical contusion.
BrdU administration
Beginning 24 h after CCI or sham injury each animal received two intraperitoneal injections of 5-Bromo-2′-deoxyuridine (BrdU; 50 mg/kg in saline; Fisher Scientific) at an 8 h interval each day until 7 days post-injury (dpi). Proliferating cells were labelled from one to 7 days after injury to capture the majority of the proliferative phase triggered by TBI [23, 26].
Histology and immunohistochemistry
Animals were deeply anesthetized by sodium pentobarbital (Fatal-Plus Solution, Vortech Pharmaceuticals) and transcardially perfused with heparinized saline followed by 10% buffered formalin at 42 dpi. Brains were removed 24 h after post-fixation in 10% formalin, cryoprotected using 30% sucrose solution, and snap frozen in cold isopentanes (≤ − 25 °C). Frozen brains were cut in a coronal plane at 40 μm thickness.
Immunofluorescence was performed using published protocols for free-floating sections [26, 48] on three tissue sections selected at 400 μm intervals spanning the injury epicenter (between − 1.5 to − 2.5 mm bregma). To expose BrdU epitopes, the tissue was incubated in 2 N HCl (Fisher Scientific) at room temperature with agitation for 1 h, followed by 100 mM borate for 10 min to neutralize residual HCl. The tissue was rinsed overnight in TBS at 4 °C prior to a 30 min room temperature incubation in TBS with 0.1% Triton-X-100 and 5% Normal Horse Serum (NHS). Primary antibodies were diluted in TBS with 0.1% Triton-X-100 and 5% NHS and tissue was incubated overnight at 4 °C with agitation. Primary antibodies used were anti-NeuN (rabbit, 1:500, Millipore Sigma), anti-GFAP (rabbit polyclonal, 1:1000, Millipore Sigma), anti-Iba1 (rabbit polyclonal, 1:1000, Wako), and anti-BrdU (rat monoclonal, 1:1000, Abcam). Secondary antibodies were conjugated with Alexa-488, Cy-3 or Alexa-594 (Invitrogen). The omission of primary antibody served as a negative control. Sections damaged during processing were omitted from analysis for one mouse.
Image acquisition
Images were taken on a confocal epifluorescence microscope (C2 Tie Nikon confocal microscope) capturing the DG for each section per animal. BrdU+ and NeuN+BrdU+ cells in the densely packed GCL of the hippocampus ipsilateral to impact were imaged at 100x magnification under oil and at 0.75 μm step intervals through a 25 μm depth. Maximum intensity projection images of glia labelled with BrdU in the ML were generated from confocal images of the DG taken at 20x magnification at 1.5 μm step intervals through 20 μm of each section.
Cell quantification
The region of interest (ROI) area (mm2) and section thickness (mm) were auto-detected by NIS-Elements image analysis software using DAPI counterstain and confirmed by an observer and used to determine ROI volume (mm3). Cell counts were manually quantified in 3D using NIS-Elements image analysis software by an observer blinded to genotype and injury group. Absolute cell counts were normalized to the ROI volume (mm3) and expressed as density (count/mm3). To evaluate the effect of IGF1 on adult-born neuronal survival in the GCL, the numbers of BrdU+ and NeuN+BrdU+ cells in the GCL, ML, and HL were counted. Cells were determined to reside in the GCL if they were within one cell distance (0-10 μm) of the GCL border and the soma was adjacent to other NeuN+ cells along the border [34]. To determine the position within the GCL of neurons that were generated within the first week of injury (NeuN+BrdU+), the location of the center of the cell in relation to the GCL/HL border was determined as previously described [33, 51] and annotated using NIS-Elements, and cells were binned into the inner 1/3 (iGCL) or outer 2/3 (oGCL) of the GCL. As principal granule cells are the only type of neuron generated by adult neurogenesis in the DG, NeuN+BrdU+ neurons located in either the HL and ML were considered ectopic [7, 52]. To quantify the fraction of proliferated neurons localized ectopically, the combined number of proliferated neurons localized to the ML and HL were normalized to the total number of proliferated neurons in the DG (i.e. (ML + HL)/(ML + HL + GCL)).
Injury induces glial activation and proliferation within the DG [23, 32, 53,54,55]. To determine if IGF1 increased proliferation of glial cells, BrdU+, Iba1+BrdU+, and GFAP+BrdU+ cells were counted in the ML and normalized to volume. To determine if IGF1 overexpression altered the pattern of glial representation after injury, areas of GFAP and Iba1 immunoreactivity within the ML were quantified and normalized to the ML ROI area.
Radial arm water maze (RAWM)
The water maze behavior assessment describes a spatial learning test in which animals are trained to locate a fixed platform hidden beneath the surface of a pool. The water component serves as an aversive motivator that encourages rodents to escape quickly. Decreased exploration of areas that do not contain the platform over contiguous trials indicates learning. The reversal paradigm introduces a novel platform location after animals have learned to find the original location. How quickly rodents decrease preference for the old location and replace it with a novel location is an indicator of cognitive flexibility [20, 56]. Cognitive flexibility has been linked to an increase in the number of mature adult-born neurons [20].
To measure spatial reference memory and learning, mice were tested using an established 2-day RAWM acquisition protocol [57, 58] beginning 39 dpi by an observer blinded to animal genotype. The six-arm maze had an arm length of 30 cm and a common circular swim diameter of 40 cm within a 100 cm diameter pool. The pool was filled with 20–21 °C water, made opaque using tempura paint (Rich Art Co.), to a level approximately 2 cm above a clear (hidden) circular platform. The platform was placed at the end of an arm approximately 7 cm away from the side and back walls. During visible trials, a cue was placed on the edge of the platform. The pool was enclosed by a black curtain on which four unique geometric extra-maze visual cues were affixed.
On day 1 (acquisition day), mice were trained over 12 trials to identify the platform location, alternating between visible and hidden platform trials [58]. Trials 13–15 on day 1 consisted of 3 hidden platform trials, yielding a total of 15 trials. On day 1, mice unable to locate the platform within 60 s were gently guided to it. On day 2, reference memory was tested using 15 hidden platform trials. Mice received reversal training on the third day of testing, during which the platform was moved to a novel location, at least 2 arms away from the original location [57, 59]. On day 3, mice received 15 hidden platform trials. Training was conducted in 5 blocks of 3 trials each day of testing (days 1–3). Mice were tested in groups of 4–5 and never entered the same start arm as the previous trial. A mouse was considered to have entered the arm if its whole body crossed the threshold of the arm. Animals received an error point if they entered the wrong arm or did not make an arm choice for more than 15 s. Entries into the goal arm were not counted as errors even if the mouse did not locate the platform. Mice that have learned the RAWM exhibit performance errors of 1 or less, averaged over three trials near the end of the second day [57]. Control (WT sham mice, n = 8) learned the RAWM test by the third block of day 2, averaging 1.4 ± 0.3 errors. Because over-training animals is taxing and could influence performance [57, 60], the first 3 blocks of hidden platform trials during acquisition days 1 and 2 were used for analyses. The error data is presented as the sum of the errors during hidden platform trials per block.
Upon completion of RAWM testing, visual and swimming deficits were evaluated using an open pool visible platform test [57]. The latency to find the flagged platform in an open pool divided into 4 equal quadrants was assessed over 5 blocks of three 60-s trials. The start position remained constant throughout the testing, while the goal quadrant varied for each trial. Animals were excluded from RAWM analysis for failing the visible platform test [57] by receiving a latency of > 20 s in the last block of the test and ≥ 45 s latency across all 15 open pool trials (1 WT and 1 IGFtg). Two visually competent mice were excluded from analyses for non-participation, failing to reach the platform in every trial during the 3 days of testing (1 WT and 1 IGFtg).
All trials were recorded with a digital camera using EthoVision XT 8.0 video tracking software (Noldus Information Technology). The error and latency to platform data were manually analysed offline by blinded observers. Activity heat maps were generated by video analysis using Anymaze software (Stoelting Company).
Experimental design and statistical analyses
Data were acquired and assembled by an individual blinded to genotype and injury conditions. Statistical analyses and graph generation were performed using GraphPad Prism software 6.0. Cell counting and localization assays revealed no differences within groups as a function of the anterior-posterior coordinate of the sections; therefore, cell count data were summed across tissue sections. Cell counts and cell positioning data were analysed using a one-way analysis of variance (ANOVA), followed by post-hoc Bonferroni’s multiple comparison t-tests where appropriate. Post-hoc comparisons were limited a priori to four, to test for genotype effects across sham groups and across injured groups, and injury effects (sham versus CCI) for WT mice and for IGFtg mice. Where appropriate, Student’s T-tests were used to compare groups of two. RAWM (days 1–2) and RAWM reversal (day 3) data were analysed using a one-way ANOVA with repeated measures. Because the sham mice tested in the RAWM constituted a separate study and were not randomized with the WT and IGFtg CCI groups, the sham behavioral responses were not included in statistical analyses. Data are presented as mean + standard error of mean (SEM). For all comparisons p < 0.05 was considered statistically significant. Statistical analyses for data in all figures are presented in Tables 1 and 2.
Results
IGF1 overexpression enhances posttraumatic neurogenesis without increasing cellular proliferation
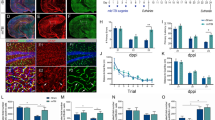
To determine the effect of IGF1 on long-term survival of cells that proliferate after TBI, we used BrdU to label cells dividing within the first 7 dpi and quantified the number of BrdU+ cells persisting until 42 dpi in IGFtg and WT mice. Proliferated cells were distributed throughout the DG and were more abundant in mice with TBI (Fig. 1a, c, e). Quantification in the GCL confirmed a significant increase in the density of acutely proliferated (BrdU+) cells that survived 6 weeks in both WT and IGFtg injured mice compared to sham controls (Fig. 1h). The injury-induced increase in proliferation within the GCL was equivalent in WT and IGFtg mice.

IGF1 overexpression enhances long-term posttraumatic neurogenesis and rescues trauma-induced reduction of differentiated mature neuron pool. Controlled cortical impact (CCI) enhanced proliferation in both wildtype (WT) and IGF1 overexpressing (IGFtg) mice. Cells dividing within 7 days following surgery were detected at 6 weeks after injury in the granule cell layer (GCL), molecular layer (ML) and hilus (HL) using bromodeoxyuridine (BrdU, red) immunolabelling of (a, b) sham controls, (c, d) injured WT, and (e, f) injured IGFtg mice. DAPI label is shown in blue. A subset of BrdU+ cells in the GCL colabelled with the mature neuron-specific protein, Neuronal Nuclei (NeuN, green) (b, d, f). Examples of colabelled cells are noted by arrows. (g) The phenotype of posttrauma-proliferated cells was confirmed by 3D reconstruction of confocal images, as demonstrated in high-magnification images for the cells in the red box in (f). The scale bar represents 50 μm in A, C and E, 10 μm in B, D and F, and 5 μm in G. (h) The density of BrdU+ cells (cells/mm3) within the GCL was increased in injured WT and injured IGFtg mice relative to their respective sham controls. (i) Following injury, IGF1 overexpression increased the density (cells/mm3) of surviving posttrauma-born neurons (NeuN+BrdU+) in the GCL. (j) Trauma reduced the proportion of proliferated progenitors that matured into end-stage neurons in WT mice but not in IGF1tg mice. Data are presented as mean + SEM; n = 3 sham /genotype, n = 8–9 CCI /genotype. One-way ANOVA, followed by Bonferroni’s selected comparisons post-hoc t-tests: *p < 0.05 and **p < 0.01 compared to respective sham group, and &&p < 0.01 and &&&p < 0.001 compared to injured WT
We previously showed that IGF1 stimulates an increase in numbers of immature neurons in the GCL early after CCI brain injury by promoting neuronal differentiation [26]. To determine if overexpression of IGF1 supported long-term survival of neurons born acutely after trauma, we co-labelled BrdU+ cells with NeuN (Fig. 1b, d, f, g) and quantified the density of proliferated neurons in the GCL at 42 dpi. In WT mice, brain injury did not result in a statistically significant increase in the density of NeuN+BrdU+ neurons in the GCL. In contrast, brain-injured IGFtg mice displayed a marked increase in NeuN+BrdU+ cell density when compared to either IGFtg shams or brain-injured WT mice (Fig. 1i). The density of surviving posttrauma-born neurons in brain-injured mice overexpressing IGF1 was nearly 3-fold higher than in injured WT mice, demonstrating that IGF1 stimulates stable hippocampal neurogenesis in the traumatically injured brain. In brain-injured WT mice, less than 30% of BrdU+ cells in the GCL expressed NeuN (Fig. 1j). IGF1 overexpression significantly increased the proportion of BrdU+ cells that exhibited a mature neuronal phenotype following injury to over 60%, restoring the balance of proliferated neurons a level equivalent to sham controls.
IGF1 overexpression does not potentiate posttraumatic gliogenesis in the DG
Experimental TBI is known to stimulate glial proliferation, producing long-lasting increases in numbers of astrocytes and microglia in the hippocampus. To examine whether IGF1 augmented trauma-induced gliosis, we quantified the numbers of cells generated within the first week after TBI within the ML of the DG, a region predominantly populated by glial cells, and identified glial phenotypes (astrocytes and microglia) of proliferated cells (Fig. 2a-d). As in the GCL, CCI brain injury was associated with a significant increase in the density of acutely proliferated (BrdU+) cells in the ML that persisted out to 6 weeks in both WT and IGFtg mice when compared to sham injury (Fig. 2e). Proliferation in the ML was not significantly altered by overexpression of IGF-1. Brain injury stimulated increases in both astrocyte (Fig. 2f) and microglia (Fig. 2g) proliferation, but the density of new glia surviving to 6 weeks post-injury was not significantly different in IGFtg mice as compared to WT mice. Injury-induced glial proliferation was also equivalent within the GCL (data not shown). While IGF1 overexpression did not increase numbers of new glia, the area of immunolabeling was significantly increased in the ML for both GFAP (Fig. 2h) and Iba1 (Fig. 2i) in IGFtg brain-injured mice when compared to sham controls. In sum, these data demonstrate that IGF1 strongly promotes the long-term survival of posttrauma-born hippocampal granule neurons, selectively increasing the density of new neurons without affecting the density of proliferated astrocytes or microglia in the DG after CCI brain injury.

IGF1 overexpression does not potentiate posttraumatic gliogenesis in the dentate gyrus. Cells that proliferated during the first week after controlled cortical impact (CCI) detected using bromodeoxyuridine (BrdU, red) were colabelled with (a, c) the astrocyte marker, glial fibrillary acidic protein (GFAP, green) or (b, d) the microglial marker, ionized calcium binding adaptor molecule 1 (Iba1, green) in the dentate gyrus of (a, b) wildtype (WT) and (c, d) IGF1 overexpressing (IGFtg) mice at 6 weeks after injury. DAPI label is shown in blue. The scale bar represents 10 μm. GCL, granule cell layer; ML, molecular layer. (e) Density (cells/mm3) of BrdU+ cells within the ML was increased in both WT and IGFtg mice relative to their respective sham controls. Brain injury resulted in an increased density (cells/mm3) of (f) GFAP+BrdU+ cells and (g) Iba1 + BrdU+ cells within the ML relative to sham controls. IGF1 overexpression increased the relative area of (h) GFAP and (i) Iba1 immunostaining within the ML of injured mice compared to controls. Data presented as mean + SEM; n = 3 sham /genotype, n = 6–9 CCI /genotype. One-way ANOVA, followed by Bonferroni’s selected comparisons post-hoc t-tests: *p < 0.05 and **p < 0.01 compared to respective sham group
IGF1 enhances outward migration of posttrauma-born neurons in the GCL and attenuates ectopic localization of posttrauma-born neurons in the DG
Earlier studies that characterized the development and migration of newborn neurons within the GCL identified two different regions, the inner and outer zones [11, 61,62,63,64]. Adult-born neural progenitors proliferate in the SGZ and primarily localize to the inner 1/3 of the GCL (iGCL), while a few localize within the outer 2/3 of the GCL (oGCL) as they mature [11, 65]. The majority of neurons born the first week after surgery localized to the iGCL in all groups (Fig. 3a). Attendant with the 3-fold increase in new mature neurons in the GCL, IGF1 overexpression increased the density of mature posttrauma-born neurons localized to both the iGCL (Fig. 3c) and the oGCL (Fig. 3d) at 6 weeks postinjury when compared to WT mice. To determine if this increase in new neurons in the oGCL was merely reflective of the overall IGF-1-mediated increase in neurogenesis, we examined the relative proportions of posttrauma-born neurons localized to the oGCL. While brain injury did not increase the proportion of NeuN+BrdU+ neurons that localized to the oGCL in WT mice compared to WT sham controls, IGF1 overexpression modestly increased the proportion that localized to the oGCL (Fig. 3e).

IGF1 decreases ectopic localization of new neurons within the dentate gyrus after injury. (a) Posttrauma-born mature neurons immunolabeled for Neuronal Nuclei (NeuN, green) and the proliferation reporter bromodeoxyuridine (BrdU, red) primarily localized to the inner granule cell layer (iGCL) and the outer 2/3rd region of the granule cell layer (oGCL, white arrows) after controlled cortical impact (CCI). S, subgranular zone. (b) Mature neurons (NeuN+BrdU+) were occasionally observed in the hilar layer (HL) and the molecular layer (ML) after CCI. White arrows highlight double-labelled cells; the scale bar represents 10 μm. IGF1 overexpression was associated with a significant increase in the density (cells/mm3) of matured post-trauma proliferated neurons in both the iGCL (c) and oGCL (d) at 6 weeks after brain injury. (e) The proportion of proliferated GCL neurons that localized to the oGCL was increased by IGF1 overexpression. (f) Brain injury stimulated the ectopic migration of a small subset of new neurons into the ML and HL in both injured WT and injured IGFtg mice. The ML and HL NeuN+BrdU+ densities (cells/mm3) are separated for illustrative purposes. (g) However, the proportion of acutely born neurons that matured and localized ectopically to the ML or HL was reduced by IGF1 overexpression. Data presented as mean + SEM; Sham n = 3/genotype, CCI n = 8–9/genotype; One-way ANOVA, followed by Bonferroni’s selected comparisons post-hoc t-tests. *p < 0.05 and **p < 0.01 compared to sham controls. &p < 0.05 and &&p < 0.01 compared to injured WT
We next sought to determine whether increased posttraumatic neurogenesis in IGF1 overexpressing mice was associated with heightened risk for ectopic localization of adult-born neurons. As illustrated in Fig. 3b, new neurons were occasionally observed within the HL and ML in brain-injured mice. While absent in sham-injured mice, ectopically localized new neurons (localized outside the GCL, i.e. ML + HL) were observed in both WT and IGFtg injured mice at a similar density (Fig. 3f; Student’s T-test, p = 0.636). Despite a 3-fold increase in the density of BrdU+ GCL neurons mediated by IGF1 (Fig. 1i), the proportion of posttrauma-born DG neurons that were ectopically localized outside the GCL following injury was significantly reduced by IGF1 overexpression (Fig. 3g).
IGF1 improves reversal learning in RAWM 6 weeks after CCI
To elucidate the effect of IGF1 overexpression on acquisition and reversal learning after injury, mice were evaluated in a 3d RAWM test. On acquisition days 1 and 2, the numbers of errors did not differ significantly between injured WT and IGFtg mice, indicating a similar ability to learn the initial platform location (Fig. 4a). During day 3 RAWM reversal testing, the platform was moved to a novel goal arm, requiring mice to change their strategy and identify the new platform location. While injured WT and IGFtg mice made similar numbers of errors initially on day 3 (block 1), injured IGFtg mice showed a rapid decrease in errors by block 2 while injured WT mice appeared to take several blocks of training before learning the new location (Fig. 4c). Activity maps from RAWM trials during the reversal learning phase illustrate that injured WT mice had more activity in the vicinity of the old goal arm in blocks 1, 2, and 3 than did injured IGFtg mice (Fig. 4b). A decrease in time spent in the old (original) location can indicate an animals’ ability to extinguish prior memory and learn a new strategy [20, 66]. Therefore, we quantified the time spent in the old goal arm during the reversal learning phase of RAWM testing. Injured IGFtg mice spent significantly less time in the old goal arm during day 3 of RAWM testing compared to injured WT mice (Fig. 4d). Both WT and IGFtg mice performed similarly on a visual platform test (Fig. 4e), indicating that differences in old arm exploration times are not due to motor or visual deficits [57, 67].

Brain-injured mice overexpressing IGF1 more rapidly learn to find a novel platform location. (a) Brain-injured wildtype (WT) and IGF1 transgenic (IGFtg) mice learned to find a hidden platform during radial arm water maze (RAWM) acquisition testing (Days 1–2). Both WT and IGFtg mice show a decrease in numbers of errors over training days. Sham control WT mice from a separate study are shown as a reference. (b) Heat maps illustrate activity patterns for representative IGFtg and WT mice during the first three blocks of reversal testing on Day 3 in the RAWM. IGFtg mice extinguished memory of the old goal arm sooner, after the platform was moved from the original training location (old arm; denoted by a green square) to a novel location (novel arm; denoted by red asterisk). Heatmap blue to red coloration indicates increasing amount of time. (c) Both WT and IGFtg mice show a decrease in numbers of errors during RAWM reversal testing. (d) WT mice consistently spent more time than the IGFtg mice in the previous platform location during RAWM reversal testing. (e) Injured WT and IGFtg mice have similar times to locate a visible platform at 6 weeks after CCI, confirming that differences between groups during the hidden platform trials were not due to sensory or motor dysfunction. CCI n = 9–11/genotype; Repeated measures ANOVA. *p < 0.05, main effect of genotype (in D) or block (in E)
Discussion
The fate, localization, and function of posttrauma-born neurons in the hippocampal DG 6 weeks following TBI were examined in IGF1 overexpressing and WT mice. Three key findings from the studies are presented. First, of the hippocampal cell types that proliferate after trauma, the population of posttrauma-born neurons that survive to maturity is selectively increased by elevated brain levels of IGF1. IGF1 overexpression significantly increased the number of mature posttrauma-born neurons but not the number of astrocytes or microglia that proliferated within the first week of injury. Second, we show for the first time that IGF1 regulates migration of adult-born neurons following trauma. Brain-injured IGF1 mice had significantly more newly born neurons localized deep within the GCL. Although IGF1 promoted outward migration within the GCL, it did not potentiate injury-induced ectopic granule cell localization to the HL and ML. Third, we present a novel effect of IGF1 on cognitive flexibility following trauma. At 6 weeks, injured IGFtg mice had improved performance on the day of RAWM reversal testing, when the hidden platform was moved to a novel location. This data compels further evaluation of IGF1 for its therapeutic potential to promote hippocampal neurogenesis and cognitive flexibility following trauma.
Proliferation and survival
Brain injury drives extensive cell death of immature neurons in the GCL [22,23,24, 26]. Coincident with early neuron death, cellular proliferation increases markedly throughout the hippocampus, peaking around 3 days after TBI [27, 68, 69]. Proliferation in the ML and HL results predominantly in new glial cells, while the majority of proliferating cells within the GCL become neurons [23, 28, 32, 69]. Many groups have demonstrated, relative to sham controls, an increase in numbers of posttrauma-born GCL neurons which is sustained for up to 2 months after TBI, consistent with increased neurogenesis [23, 32, 34, 68]. However, it is unclear whether the milieu of the injured brain adversely affects the relative survival rate of newly born neurons. We show that, although TBI triggers an increase in cellular proliferation, the proportion of acutely proliferated cells that differentiate into neurons and survive to late-stage maturity is diminished by TBI in WT mice. The proportion of BrdU+ GCL cells that phenotype as mature neurons drops from approximately 60% in sham controls to less than 30% in WT mice 6 weeks after CCI, a decrease in close agreement with previous studies at 4–5 weeks after CCI [27] or lateral fluid percussion [23] TBI. Nonetheless, others have reported no change in the relative survival of posttrauma-born neurons in the GCL [28] or SGZ [70].
To examine the effect of IGF1 on proliferation and long-term survival of neurons born early after TBI, we utilized a transgenic mouse model in which IGF1 expression is driven through the GFAP promoter. We previously established that this construct leads to a significant elevation of IGF1 levels in the hippocampus as early as 1 day which increases further by 3 days after CCI brain injury, in parallel with robust hippocampal astrogliosis and upregulation of GFAP [48]. It is important to note that in sham controls, basal GFAP expression resulted in no detectable increase in human IGF1 levels. In the current study, no significant differences in proliferation or survival of posttrauma-born neurons or glia were found between WT and IGF1tg sham groups. Given the small number of mice available as sham controls, our current study was underpowered to detect small differences between sham groups; however, these findings are consistent with a larger study in which we also found no differences in early hippocampal proliferation or neurogenesis between IGFtg and WT sham controls (n = 8/genotype) [26].
By linking IGF1 expression to the GFAP promoter, IGF1 levels are amplified selectively in mice following CCI through posttraumatic astrogliosis [48]. Importantly, posttraumatic IGF1 overexpression stimulated a 3-fold increase in numbers of neurons that reached late-stage maturity 6 weeks after injury, restoring the proportion of proliferated cells that became mature neurons to a level equivalent to uninjured controls. IGF1 did not significantly enhance cellular proliferation, but may promote increased generation of mature neurons by promoting neuronal differentiation and attenuating apoptosis.
Our previous work demonstrated that IGF1 overexpression resulted in an increase in neuronal differentiation of SGZ neural stem cells and greater immature neuron dendritic complexity within 10 days of CCI brain injury [26]. We and others have demonstrated that posttrauma-born GCL neurons develop with abnormal, stunted dendritic arborization [26, 33, 71]. Improved dendritic complexity may protect against apoptosis during synapse-dependent competitive survival of adult-born neurons [8,9,10,11]. In vitro studies and developmental studies suggest that IGF1 supports the survival of newborn neurons to end-stage maturity through cooperative inhibition of programmed cell death and enhanced immature neuron dendritic arborization [45, 48, 72,73,74,75,76]. It is not yet known whether IGF1 overexpression decreases the rate of apoptosis of newly proliferated neurons in the GCL. However, IGF1 overexpression has been shown to protect against hilar neuron and CA3 pyramidal neuron loss after TBI [48].
IGF1 overexpression facilitated the long-term survival of posttrauma-born neurons without increasing numbers of new astrocytes or microglia, suggesting a selective effect on neuronal survival. Our data corroborates previous studies describing acute proliferation and long-term survival of glial cells in the ML after injury [32]. It is possible, then, that the selective effect of IGF1 on newborn neuronal survival may be due in part to inhibition of apoptosis pathways that are activated after TBI to a greater extent in neurons as compared to glia [77]. Because the IGF1 receptor is ubiquitously expressed on all neural cell types [41, 78], it would be expected that astrocyte-derived IGF1 overexpression affects multiple cell types. IGF1 appeared to act on both astrocytes and microglia, potentiating injury-induced hypertrophy, as suggested by increases in the area of labelled glia. While we previously showed acutely elevated hippocampal IGF1 levels and an IGF1-mediated increase in astrocyte hypertrophy in IGFtg mice after CCI brain injury [48], it is not yet known whether the chronic astrocyte hypertrophy noted in the current study supports increased in brain IGF1 levels driven through the GFAP promoter as long as 6 weeks post-injury.
Migration
In naïve animals, the majority of adult-born neurons migrate from the neurogenic niche of the SGZ within the first 4 weeks of their birth and remain within the iGCL [11, 79, 80]. Consistent with previous reports [33, 34, 38], we demonstrate that TBI promotes a modest increase in the number of posttrauma-born neurons that migrate further outward to the oGCL. The functional implications of this enhanced migration are not yet clear. Posttrauma-born GCL neurons appear to synaptically integrate within the DG [34] and connect to appropriate CA3 targets [69, 81]. However, newly matured neurons located in the oGCL exhibit more pronounced deficits in dendritic arbor development even 5 weeks post-trauma [33]. Here we show that, in addition to improving dendritic arborization of immature neurons following injury [26], IGF1 regulates outward migration of posttrauma-born neurons, consistent with a developmentally born phenotype. IGF1 regulates known factors of neuronal migration including GSK3B, PSA-NCAM, Cdk5 and reelin [12, 44, 82]. Signalling studies have revealed that inhibiting the activity of GSK3B, a protein inactivated downstream of IGF1 signalling, is sufficient to increase the number of adult-born neurons localized to the oGCL [83]. This may be an indication that IGF1 can accelerate development of newborn neurons following trauma.
As immature neurons develop in the postnatal DG, they are rarely found localized to regions outside the GCL. In the uninjured brain, during the process of adult neurogenesis, immature DG granule neurons that ectopically localize within the DG undergo cell-mediated apoptosis before they reach maturity [84, 85]. Although quantification of newborn neurons is often restricted to the SGZ and GCL [23, 86], ectopic migration to either the ML [33, 34, 39] or the HL [33, 40] has been reported after experimental TBI. Although a relatively small number of posttrauma proliferated neurons localize ectopically to the HL and ML at 6 weeks post-CCI in WT and IGFtg mice, their physiological relevance remains unclear. Ectopic hilar neurons are thought to contribute to hippocampal hyperexcitability [87, 88] and seizure development [89, 90]. Furthermore, the ectopic localization of immature adult-born granule neurons associated with the neuropathology induced by epilepsy suggests a contribution to maladaptive plasticity [89, 91]. The CCI model of brain trauma does produce delayed mossy fiber sprouting and seizure activity, particularly with more severe injuries [92, 93]. While we did not observe seizure behaviour in injured mice, our study was designed to examine posttraumatic hippocampal neuroplasticity that develops prior to the window of seizure onset, which is estimated to be as late as 8 weeks after CCI [92].
While key factors regulating posttraumatic newborn neuron migration are not fully known, IGF1 may play a role in inhibiting ectopic hilar migration. We found that IGF1 overexpression inhibits the localization of mature posttrauma-born granule neurons to regions outside the DG GCL. IGF1 may regulate immature neuron migration indirectly through an effect on reelin, a migratory stop signal expressed by hilar basket cells [85] which prevents granule cells from migrating ectopically into the hilus [94]. Increasing neurogenesis after TBI has been suggested to enhance the likelihood of seizure activity [86]; however, IGF1 treatment has been shown to reduce seizure behavior in a rodent model of temporal lobe epilepsy [75].
Reversal learning
Systemic IGF-1 administration and astrocyte-specific IGF1 overexpression have both been shown to improve cognitive function within the first 2 weeks after TBI [47,48,49]. Our study provides the first evidence that increasing brain levels of IGF1 following injury can influence cognition in the chronic phase after TBI, using a reversal water maze task shown to be sensitive to alterations in hippocampal neurogenesis. At 6 weeks after TBI, during the reversal phase of a RAWM test when the hidden platform was moved to a new location, injured IGFtg mice spent less time in the original goal arm than did injured WT mice. Inhibition of adult neurogenesis using the anti-proliferation drug temozolomide caused a strong and lasting preference for the old goal arm during Morris water maze testing when the goal arm was moved to a novel location [20]. Thus, it is possible that injured WT mice had an increased preference for the original goal location due to impaired neurogenesis. Conversely, injured IGFtg mice exhibited significant enhancement of posttraumatic hippocampal neurogenesis with concomitant improvement in RAWM reversal learning. Increasing the number of mature adult-born neurons is sufficient to improve the integration of novel information upon introduction of a novel platform location during water maze assessment in mice, demonstrating cognitive flexibility and memory extinction [14, 15, 18, 95].
Acquisition learning may be less sensitive than reversal learning to modulation of hippocampal neurogenesis, as suggested by previous studies demonstrating that an attenuation of adult neurogenesis does not prevent acquisition of the escape platform location during water maze training [20, 96]. Brain-injured WT and brain-injured IGF overexpressing mice showed equivalent abilities to learn the location of a hidden platform during the acquisition phase (days 1–2) of RAWM testing. Although we and others have demonstrated that moderate or severe CCI results in persistent learning and memory dysfunction in mice [97,98,99,100,101,102], it is possible that the lack of effect of IGF-1 during acquisition may be related to an inadequate behavioral dysfunction at 6 weeks postinjury.
Our data show that increasing brain levels of IGF1 enhances hippocampal plasticity by promoting maturation and long-term survival of neurons born after TBI and improving cognitive flexibility. These findings have important implications for treatment of TBI-related cognitive dysfunction. Over 65% of patients with moderate to severe TBI report long-term cognitive deficits [103]. Modulation of adult hippocampal neurogenesis is a potential therapeutic target for TBI recovery. Collectively our findings support IGF1 supplementation following brain injury as a potential means to enhance appropriate and persistent posttraumatic hippocampal neurogenesis.
Conclusions
This work is critical to our understanding of adult-born neuron development in the context of brain injury. Stimulation of neural stem cell proliferation by TBI contributes to recovery of the vulnerable immature hippocampal neuron population, but the extent of long-term survival and functional integration of neurons born in the aftermath of trauma is debated. Further, TBI alters the morphological development and localization of posttrauma-born neurons, raising concerns about detrimental consequences of implementing therapies that augment posttraumatic neurogenesis. We show that IGF1 (a) promotes acute neurogenesis without increasing gliogenesis, (b) enhances migration within the GCL but decreases aberrant migration outside the GCL, (c) supports end-stage maturity of newborn neurons, and (d) promotes hippocampus-dependent cognitive flexibility. These data support the continued evaluation of IGF1 as a potential therapeutic for neurological disorders and disease.
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author upon reasonable request.
Abbreviations
- ANOVA:
-
Analysis of variance
- BrdU:
-
Bromodeoxyuridine
- CA3:
-
Cornu ammonis field-3 of the hippocampus
- CCI:
-
Controlled cortical impact
- Cdk5:
-
Cell division protein kinase 5
- CNS:
-
Central nervous system
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DG:
-
Dentate gyrus
- dpi:
-
Days post-injury
- GCL:
-
Granule cell layer
- GFAP:
-
Glial fibrillary acidic protein
- GSK3B:
-
Glycogen synthase kinase 3 beta
- HL:
-
Hilar layer
- Iba1:
-
Ionized calcium-binding adapter molecule 1
- iGCL:
-
Inner 1/3 of the GCL
- IGF1:
-
Insulin-like growth factor-1
- IGFtg:
-
IGF1 double transgenic mice
- ML:
-
Molecular layer
- NeuN:
-
Neuronal nuclei
- NHS:
-
Normal horse serum
- oGCL:
-
Outer 2/3 of the GCL
- PSA-NCAM:
-
Polysialylated neuronal cell adhesion molecule
- RAWM:
-
Radial arm water maze
- ROI:
-
Region of interest
- SEM:
-
Standard error of mean
- SGZ:
-
Subgranular zone
- TBI:
-
Traumatic brain injury
- TBS:
-
Tris-buffered saline
- WT:
-
Wildtype
References
Popescu C, Anghelescu A, Daia C, Onose G (2015) Actual data on epidemiological evolution and prevention endeavours regarding traumatic brain injury. J Med Life 8(3):272–277
Fleminger S, Ponsford J (2005) Long term outcome after traumatic brain injury. BMJ 331(7530):1419–1420
Flanagan SR (2015) Invited commentary on "Centers for Disease Control and Prevention report to congress: traumatic brain injury in the United States: epidemiology and rehabilitation". Arch Phys Med Rehabil 96(10):1753–1755
Schachar RJ, Park LS, Dennis M (2015) Mental health implications of traumatic brain injury (TBI) in children and youth. J Can Acad Child Adolesc Psychiatry 24(2):100–108
Ratcliff G, Colantonio A, Escobar M, Chase S, Vernich L (2005) Long-term survival following traumatic brain injury. Disabil Rehabil 27(6):305–314
Watson MR, Fenton GW, McClelland RJ, Lumsden J, Headley M, Rutherford WH (1995) The post-concussional state: neurophysiological aspects. Br J Psychiatry 167(4):514–521
Amaral DG, Scharfman HE, Lavenex P (2007) The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies). Prog Brain Res 163:3–22
Ryu JR, Hong CJ, Kim JY, Kim EK, Sun W, Yu SW (2016) Control of adult neurogenesis by programmed cell death in the mammalian brain. Mol Brain 9:43
Dayer AG, Ford AA, Cleaver KM, Yassaee M, Cameron HA (2003) Short-term and long-term survival of new neurons in the rat dentate gyrus. J Comp Neurol 460(4):563–572
Cameron HA, McKay RD (2001) Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol 435(4):406–417
Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH (2003) Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development 130(2):391–399
Jessberger S, Aigner S, Clemenson GD Jr, Toni N, Lie DC, Karalay O, Overall R, Kempermann G, Gage FH (2008) Cdk5 regulates accurate maturation of newborn granule cells in the adult hippocampus. PLoS Biol 6(11):e272
van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH (2002) Functional neurogenesis in the adult hippocampus. Nature 415(6875):1030–1034
Snyder JS, Choe JS, Clifford MA, Jeurling SI, Hurley P, Brown A, Kamhi JF, Cameron HA (2009) Adult-born hippocampal neurons are more numerous, faster maturing, and more involved in behavior in rats than in mice. J Neurosci 29(46):14484–14495
Ishikawa R, Fukushima H, Frankland PW, Kida S (2016) Hippocampal neurogenesis enhancers promote forgetting of remote fear memory after hippocampal reactivation by retrieval. Elife 5:e17464
Farioli-Vecchioli S, Saraulli D, Costanzi M, Pacioni S, Cina I, Aceti M, Micheli L, Bacci A, Cestari V, Tirone F (2008) The timing of differentiation of adult hippocampal neurons is crucial for spatial memory. PLoS Biol 6(10):e246
Lu D, Qu C, Goussev A, Jiang H, Lu C, Schallert T, Mahmood A, Chen J, Li Y, Chopp M (2007) Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J Neurotrauma 24(7):1132–1146
Sahay A, Scobie KN, Hill AS, O'Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R (2011) Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 472(7344):466–470
Burghardt NS, Park EH, Hen R, Fenton AA (2012) Adult-born hippocampal neurons promote cognitive flexibility in mice. Hippocampus 22(9):1795–1808
Garthe A, Behr J, Kempermann G (2009) Adult-generated hippocampal neurons allow the flexible use of spatially precise learning strategies. PLoS One 4(5):e5464
Garthe A, Kempermann G (2013) An old test for new neurons: refining the Morris water maze to study the functional relevance of adult hippocampal neurogenesis. Front Neurosci 7:63
Gao X, Deng-Bryant Y, Cho W, Carrico KM, Hall ED, Chen J (2008) Selective death of newborn neurons in hippocampal dentate gyrus following moderate experimental traumatic brain injury. J Neurosci Res 86(10):2258–2270
Rola R, Mizumatsu S, Otsuka S, Morhardt DR, Noble-Haeusslein LJ, Fishman K, Potts MB, Fike JR (2006) Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Exp Neurol 202(1):189–199
Yu TS, Zhang G, Liebl DJ, Kernie SG (2008) Traumatic brain injury-induced hippocampal neurogenesis requires activation of early nestin-expressing progenitors. J Neurosci 28(48):12901–12912
Ngwenya LB, Mazumder S, Porter ZR, Minnema A, Oswald DJ, Farhadi HF (2018) Implantation of neuronal stem cells enhances object recognition without increasing neurogenesis after lateral fluid percussion injury in mice. Stem Cells Int 2018:4209821
Carlson SW, Madathil SK, Sama DM, Gao X, Chen J, Saatman KE (2014) Conditional overexpression of insulin-like growth factor-1 enhances hippocampal neurogenesis and restores immature neuron dendritic processes after traumatic brain injury. J Neuropathol Exp Neurol 73(8):734–746
Gao X, Chen J (2013) Moderate traumatic brain injury promotes neural precursor proliferation without increasing neurogenesis in the adult hippocampus. Exp Neurol 239:38–48
Wang X, Gao X, Michalski S, Zhao S, Chen J (2016) Traumatic brain injury severity affects neurogenesis in adult mouse Hippocampus. J Neurotrauma 33(8):721–733
Bye N, Carron S, Han X, Agyapomaa D, Ng SY, Yan E, Rosenfeld JV, Morganti-Kossmann MC (2011) Neurogenesis and glial proliferation are stimulated following diffuse traumatic brain injury in adult rats. J Neurosci Res 89(7):986–1000
Lu D, Mahmood A, Qu C, Goussev A, Schallert T, Chopp M (2005) Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J Neurotrauma 22(9):1011–1017
Ning R, Xiong Y, Mahmood A, Zhang Y, Meng Y, Qu C, Chopp M (2011) Erythropoietin promotes neurovascular remodeling and long-term functional recovery in rats following traumatic brain injury. Brain Res 1384:140–150
Kernie SG, Erwin TM, Parada LF (2001) Brain remodeling due to neuronal and astrocytic proliferation after controlled cortical injury in mice. J Neurosci Res 66(3):317–326
Ibrahim S, Hu W, Wang X, Gao X, He C, Chen J (2016) Traumatic brain injury causes aberrant migration of adult-born neurons in the Hippocampus. Sci Rep 6:21793
Villasana LE, Kim KN, Westbrook GL, Schnell E (2015) Functional integration of adult-born hippocampal neurons after traumatic brain injury (1,2,3). eNeuro 2(5):ENEURO.0056-15.2015
Ge S, Sailor KA, Ming GL, Song H (2008) Synaptic integration and plasticity of new neurons in the adult hippocampus. J Physiol 586(16):3759–3765
Deng W, Aimone JB, Gage FH (2010) New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11(5):339–350
Tashiro A, Sandler VM, Toni N, Zhao C, Gage FH (2006) NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature 442(7105):929–933
Blaiss CA, Yu TS, Zhang G, Chen J, Dimchev G, Parada LF, Powell CM, Kernie SG (2011) Temporally specified genetic ablation of neurogenesis impairs cognitive recovery after traumatic brain injury. J Neurosci 31(13):4906–4916
Villasana LE, Peters A, McCallum R, Liu C, Schnell E (2019) Diazepam inhibits post-traumatic neurogenesis and blocks aberrant dendritic development. J Neurotrauma 36(16):2454–2467
Shapiro LA (2017) Altered hippocampal neurogenesis during the first 7 days after a fluid percussion traumatic brain injury. Cell Transplant 26(7):1314–1318
Ocrant I, Valentino KL, Eng LF, Hintz RL, Wilson DM, Rosenfeld RG (1988) Structural and immunohistochemical characterization of insulin-like growth factor I and II receptors in the murine central nervous system. Endocrinology 123(2):1023–1034
Nieto-Estevez V, Defterali C, Vicario-Abejon C (2016) IGF-I: a key growth factor that regulates neurogenesis and synaptogenesis from embryonic to adult stages of the brain. Front Neurosci 10:52
Kim B, Leventhal PS, Saltiel AR, Feldman EL (1997) Insulin-like growth factor-I-mediated neurite outgrowth in vitro requires mitogen-activated protein kinase activation. J Biol Chem 272(34):21268–21273
Hurtado-Chong A, Yusta-Boyo MJ, Vergano-Vera E, Bulfone A, de Pablo F, Vicario-Abejon C (2009) IGF-I promotes neuronal migration and positioning in the olfactory bulb and the exit of neuroblasts from the subventricular zone. Eur J Neurosci 30(5):742–755
Baker NL, Carlo Russo V, Bernard O, D'Ercole AJ, Werther GA (1999) Interactions between bcl-2 and the IGF system control apoptosis in the developing mouse brain. Brain Res Dev Brain Res 118(1–2):109–118
Carlson SW, Saatman KE (2018) Central infusion of insulin-like growth Factor-1 increases hippocampal neurogenesis and improves neurobehavioral function after traumatic brain injury. J Neurotrauma 35(13):1467–1480
Saatman KE, Contreras PC, Smith DH, Raghupathi R, McDermott KL, Fernandez SC, Sanderson KL, Voddi M, McIntosh TK (1997) Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp Neurol 147(2):418–427
Madathil SK, Carlson SW, Brelsfoard JM, Ye P, D'Ercole AJ, Saatman KE (2013) Astrocyte-specific overexpression of insulin-like growth Factor-1 protects hippocampal neurons and reduces behavioral deficits following traumatic brain injury in mice. PLoS One 8(6):e67204
Rubovitch V, Edut S, Sarfstein R, Werner H, Pick CG (2010) The intricate involvement of the insulin-like growth factor receptor signaling in mild traumatic brain injury in mice. Neurobiol Dis 38(2):299–303
Ye P, Popken GJ, Kemper A, McCarthy K, Popko B, D'Ercole AJ (2004) Astrocyte-specific overexpression of insulin-like growth factor-I promotes brain overgrowth and glial fibrillary acidic protein expression. J Neurosci Res 78(4):472–484
Han J, Kim HJ, Schafer ST, Paquola A, Clemenson GD, Toda T, Oh J, Pankonin AR, Lee BS, Johnston ST et al (2016) Functional implications of miR-19 in the migration of newborn neurons in the adult brain. Neuron 91(1):79–89
Scharfman HE (2016) The enigmatic mossy cell of the dentate gyrus. Nat Rev Neurosci 17(9):562–575
Dietrich WD, Truettner J, Zhao W, Alonso OF, Busto R, Ginsberg MD (1999) Sequential changes in glial fibrillary acidic protein and gene expression following parasagittal fluid-percussion brain injury in rats. J Neurotrauma 16(7):567–581
Cortez SC, McIntosh TK, Noble LJ (1989) Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res 482(2):271–282
Hinkle DA, Baldwin SA, Scheff SW, Wise PM (1997) GFAP and S100beta expression in the cortex and hippocampus in response to mild cortical contusion. J Neurotrauma 14(10):729–738
Anacker C, Hen R (2017) Adult hippocampal neurogenesis and cognitive flexibility - linking memory and mood. Nat Rev Neurosci 18(6):335–346
Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D (2006) Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat Protoc 1(4):1671–1679
Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, Ferguson AR, Rosi S (2015) CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci 35(2):748–760
Selenica ML, Benner L, Housley SB, Manchec B, Lee DC, Nash KR, Kalin J, Bergman JA, Kozikowski A, Gordon MN et al (2014) Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimers Res Ther 6(1):12
Diamond DM, Park CR, Heman KL, Rose GM (1999) Exposing rats to a predator impairs spatial working memory in the radial arm water maze. Hippocampus 9(5):542–552
Martin LA, Tan SS, Goldowitz D (2002) Clonal architecture of the mouse hippocampus. J Neurosci 22(9):3520–3530
Altman J, Bayer SA (1990) Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J Comp Neurol 301(3):365–381
Zhao C, Teng EM, Summers RG Jr, Ming GL, Gage FH (2006) Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J Neurosci 26(1):3–11
Hayashi K, Kubo K, Kitazawa A, Nakajima K (2015) Cellular dynamics of neuronal migration in the hippocampus. Front Neurosci 9:135
Cheng X, Li Y, Huang Y, Feng X, Feng G, Xiong ZQ (2011) Pulse labeling and long-term tracing of newborn neurons in the adult subgranular zone. Cell Res 21(2):338–349
Saha R, Knapp S, Chakraborty D, Horovitz O, Albrecht A, Kriebel M, Kaphzan H, Ehrlich I, Volkmer H, Richter-Levin G (2017) GABAergic synapses at the axon initial segment of basolateral amygdala projection neurons modulate fear extinction. Neuropsychopharmacology 42(2):473–484
Bromley-Brits K, Deng Y, Song W (2011) Morris water maze test for learning and memory deficits in Alzheimer's disease model mice. J Vis Exp 53:2920
Dash PK, Mach SA, Moore AN (2001) Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res 63(4):313–319
Sun D, McGinn MJ, Zhou Z, Harvey HB, Bullock MR, Colello RJ (2007) Anatomical integration of newly generated dentate granule neurons following traumatic brain injury in adult rats and its association to cognitive recovery. Exp Neurol 204(1):264–272
Sun D, Colello RJ, Daugherty WP, Kwon TH, McGinn MJ, Harvey HB, Bullock MR (2005) Cell proliferation and neuronal differentiation in the dentate gyrus in juvenile and adult rats following traumatic brain injury. J Neurotrauma 22(1):95–105
Wu H, Li J, Xu D, Zhang Q, Cui T (2018) Growth differentiation factor 5 improves neurogenesis and functional recovery in adult mouse Hippocampus following traumatic brain injury. Front Neurol 9:592
Kim S, Lee SH, Kim JH, Jeong YW, Hashem MA, Koo OJ, Park SM, Lee EG, Hossein MS, Kang SK et al (2006) Anti-apoptotic effect of insulin-like growth factor (IGF)-I and its receptor in porcine preimplantation embryos derived from in vitro fertilization and somatic cell nuclear transfer. Mol Reprod Dev 73(12):1523–1530
Nieto-Estevez V, Oueslati-Morales CO, Li L, Pickel J, Morales AV, Vicario-Abejon C (2016) Brain insulin-like growth factor-I directs the transition from stem cells to mature neurons during postnatal/adult hippocampal neurogenesis. Stem Cells 34(8):2194–2209
Huat TJ, Khan AA, Pati S, Mustafa Z, Abdullah JM, Jaafar H (2014) IGF-1 enhances cell proliferation and survival during early differentiation of mesenchymal stem cells to neural progenitor-like cells. BMC Neurosci 15:91
Miltiadous P, Stamatakis A, Koutsoudaki PN, Tiniakos DG, Stylianopoulou F (2011) IGF-I ameliorates hippocampal neurodegeneration and protects against cognitive deficits in an animal model of temporal lobe epilepsy. Exp Neurol 231(2):223–235
Rong Z, Pan R, Chang L, Lee W (2015) Combination treatment with ethyl pyruvate and IGF-I exerts neuroprotective effects against brain injury in a rat model of neonatal hypoxic-ischemic encephalopathy. Int J Mol Med 36(1):195–203
Miller AP, Shah AS, Aperi BV, Kurpad SN, Stemper BD, Glavaski-Joksimovic A (2017) Acute death of astrocytes in blast-exposed rat organotypic hippocampal slice cultures. PLoS One 12(3):e0173167
Muhic M, Vardjan N, Chowdhury HH, Zorec R, Kreft M (2015) Insulin and insulin-like growth factor 1 (IGF-1) modulate cytoplasmic glucose and glycogen levels but not glucose transport across the membrane in astrocytes. J Biol Chem 290(17):11167–11176
Kempermann G, Gage FH (2002) Genetic influence on phenotypic differentiation in adult hippocampal neurogenesis. Brain Res Dev Brain Res 134(1–2):1–12
Yu TS, Washington PM, Kernie SG (2016) Injury-induced neurogenesis: mechanisms and relevance. Neuroscientist 22(1):61–71
Emery DL, Fulp CT, Saatman KE, Schutz C, Neugebauer E, McIntosh TK (2005) Newly born granule cells in the dentate gyrus rapidly extend axons into the hippocampal CA3 region following experimental brain injury. J Neurotrauma 22(9):978–988
Wang JM, Hayashi T, Zhang WR, Sakai K, Shiro Y, Abe K (1999) Insulin-like growth factor-1 affects expressions of cyclin-dependent kinase 5 and its activator p35 in reperfused rat brain. Neurosci Lett 277(1):17–20
Ng T, Hor CH, Chew B, Zhao J, Zhong Z, Ryu JR, Goh EL (2016) Neuropilin 2 signaling is involved in cell positioning of adult-born neurons through glycogen synthase kinase-3beta (GSK3beta). J Biol Chem 291(48):25088–25095
Stallock J, Molyneaux K, Schaible K, Knudson CM, Wylie C (2003) The pro-apoptotic gene Bax is required for the death of ectopic primordial germ cells during their migration in the mouse embryo. Development 130(26):6589–6597
Myers CE, Bermudez-Hernandez K, Scharfman HE (2013) The influence of ectopic migration of granule cells into the hilus on dentate gyrus-CA3 function. PLoS One 8(6):e68208
Neuberger EJ, Swietek B, Corrubia L, Prasanna A, Santhakumar V (2017) Enhanced dentate neurogenesis after brain injury undermines long-term neurogenic potential and promotes seizure susceptibility. Stem Cell Rep 9(3):972–984
Althaus AL, Sagher O, Parent JM, Murphy GG (2015) Intrinsic neurophysiological properties of hilar ectopic and normotopic dentate granule cells in human temporal lobe epilepsy and a rat model. J Neurophysiol 113(4):1184–1194
Scharfman HE, Pierce JP (2012) New insights into the role of hilar ectopic granule cells in the dentate gyrus based on quantitative anatomic analysis and three-dimensional reconstruction. Epilepsia 53(Suppl 1):109–115
Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH (1997) Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 17(10):3727–3738
Scharfman H, Goodman J, Macleod A, Phani S, Antonelli C, Croll S (2005) Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp Neurol 192(2):348–356
Scharfman H, Goodman J, McCloskey D (2007) Ectopic granule cells of the rat dentate gyrus. Dev Neurosci 29(1–2):14–27
Hunt RF, Boychuk JA, Smith BN (2013) Neural circuit mechanisms of post-traumatic epilepsy. Front Cell Neurosci 7:89
Hunt RF, Scheff SW, Smith BN (2009) Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol 215(2):243–252
Gong C, Wang TW, Huang HS, Parent JM (2007) Reelin regulates neuronal progenitor migration in intact and epileptic hippocampus. J Neurosci 27(8):1803–1811
Idris N, Neill J, Grayson B, Bang-Andersen B, Witten LM, Brennum LT, Arnt J (2010) Sertindole improves sub-chronic PCP-induced reversal learning and episodic memory deficits in rodents: involvement of 5-HT(6) and 5-HT (2A) receptor mechanisms. Psychopharmacology 208(1):23–36
Horster H, Garthe A, Walker TL, Ichwan M, Steiner B, Khan MA, Lie DC, Nicola Z, Ramirez-Rodriguez G, Kempermann G (2017) p27kip1 is required for functionally relevant adult hippocampal neurogenesis in mice. Stem Cells 35(3):787–799
Ritzel RM, Li Y, He J, Khan N, Doran SJ, Faden AI, Wu J (2020) Sustained neuronal and microglial alterations are associated with diverse neurobehavioral dysfunction long after experimental brain injury. Neurobiol Dis 136:104713
Sirbulescu RF, Chung JY, Edmiston Wj III, Poznansky SA, Poznansky MC, Whalen MJ (2019) Intraparenchymal application of mature B lymphocytes improves structural and functional outcome after contusion traumatic brain injury. J Neurotrauma 36(17):2579–2589
Pischiutta F, Micotti E, Hay JR, Marongiu I, Sammali E, Tolomeo D, Vegliante G, Stocchetti N, Forloni G, De Simoni MG et al (2018) Single severe traumatic brain injury produces progressive pathology with ongoing contralateral white matter damage one year after injury. Exp Neurol 300:167–178
Dixon CE, Kochanek PM, Yan HQ, Schiding JK, Griffith RG, Baum E, Marion DW, DeKosky ST (1999) One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma 16(2):109–122
Scheff SW, Baldwin SA, Brown RW, Kraemer PJ (1997) Morris water maze deficits in rats following traumatic brain injury: lateral controlled cortical impact. J Neurotrauma 14(9):615–627
Washington PM, Forcelli PA, Wilkins T, Zapple DN, Parsadanian M, Burns MP (2012) The effect of injury severity on behavior: a phenotypic study of cognitive and emotional deficits after mild, moderate, and severe controlled cortical impact injury in mice. J Neurotrauma 29(13):2283–2296
Rabinowitz AR, Levin HS (2014) Cognitive sequelae of traumatic brain injury. Psychiatr Clin North Am 37(1):1–11
Acknowledgements
We thank Adam Bachstetter, PhD and Richard Kryscio, PhD for consultation regarding water maze analysis.
Funding
This work was supported by grants from the National Institutes of Health National Institute of Neurological Disorders and Stroke (NS072302 and NS077889) and the Kentucky Spinal Cord and Head Injury Research Trust (KSCHIRT 14-12A).
Author information
Authors and Affiliations
Contributions
EL and KES designed the experiments. EL and DS performed the surgeries. EL performed the immunostaining, imaging and image analysis and generated all figures. DS performed the RAWM testing. EL and KES analyzed and interpreted the data. EL and KES wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures involving animals were approved by the University of Kentucky’s Institutional Animal Care and Use Committee (approval # 2013–1156).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Littlejohn, E.L., Scott, D. & Saatman, K.E. Insulin-like growth factor-1 overexpression increases long-term survival of posttrauma-born hippocampal neurons while inhibiting ectopic migration following traumatic brain injury. acta neuropathol commun 8, 46 (2020). https://doi.org/10.1186/s40478-020-00925-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-020-00925-6