
Abstract
Background
The IL-17 family cytokines are potent drivers of colorectal cancer (CRC) development. We and others have shown that IL-17 mainly signals to tumor cells to promote CRC, but the underlying mechanism remains unclear. IL-17 also dampens Th1-armed anti-tumor immunity, in part by attracting myeloid cells to tumor. Whether IL-17 controls the activity of adaptive immune cells in a more direct manner, however, is unknown.
Methods
Using mouse models of sporadic or inducible colorectal cancers, we ablated IL-17RA in the whole body or specifically in colorectal tumor cells. We also performed adoptive bone marrow reconstitution to knockout CXCR3 in hematopoietic cells. Histological and immunological experimental methods were used to reveal the link among IL-17, chemokine production, and CRC development.
Results
Loss of IL-17 signaling in mouse CRC resulted in marked increase in the recruitment of CD8+ cytotoxic T lymphocytes (CTLs) and regulatory T cells (Tregs), starting from early stage CRC lesions. This is accompanied by the increased expression of anti-inflammatory cytokines IL-10 and TGF-β. IL-17 signaling also inhibits the production of T cell attracting chemokines CXCL9 and CXCL10 by tumor cells. Conversely, the inability of hematopoietic cells to respond to CXCL9/10 resulted in decreased tumor infiltration by CTLs and Tregs, decreased levels of IL-10 and TGF-β, and increased numbers of tumor lesions. Blockade of IL-17 signaling resulted in increased expression of immune checkpoint markers. On the other hand, treatment of mouse CRC with anti-CTLA-4 antibody led to increased expression of pro-tumor IL-17.
Conclusion
IL-17 signals to colorectal tumor cells and inhibits their production of CXCL9/10 chemokines. By doing so, IL-17 inhibits the infiltration of CD8+ CTLs and Tregs to CRC, thus promoting CRC development. Cancer immunotherapy may be benefited by the use of anti-IL-17 agents as adjuvant therapies, which serve to block both IL-17-mediated tumor promotion and T cell exclusion.
Similar content being viewed by others


Background
The IL-17 family cytokines promote tumor development in multiple organs. Using mouse models of sporadic and inducible colorectal cancers (CRC), we and others have shown that IL-17 signals to transformed colorectal epithelial cells to drive tumor development [1, 2]. This IL-17-tumor cell signaling is necessary for the survival and outgrowth of early CRC lesions, and ablation of IL-17RA, the common receptor of IL-17 family cytokines, resulted in marked reduction in tumor numbers in mouse colon [1, 3]. IL-17 also activates production of chemokines, such as CXCL1 and CXCL2 that attract myeloid cells to sites of inflammation [4, 5]. Colorectal tumor cells exhibit defective epithelial barrier function. As a result, gut commensal bacteria and their degradative products invade tumor stroma, engage tumor-infiltrating myeloid cells, and activate the production of IL-23 and its downstream cytokine IL-17 [3]. Thus, the IL-17-myeloid cell pathway forms a self-enhancing loop that results in chronic tumor-associated inflammation. IL-17 is also known to block the effect of cytotoxic and anti-angiogenic chemotherapies against colorectal cancers [1, 6]. This correlates with the observation that loss of IL-17 signaling resulted in enhanced recruitment of CD8+ cytotoxic T lymphocytes (CTL) [1, 3, 7]. To date, it is unclear if IL-17 regulates the recruitment of adaptive immune cells to the site of CRC, and if so, what the underlying mechanism is.
The chemokine CXCL9 signals through CXCR3 and mediates migration of T cells to sites of inflammation [8]. In mouse models of transplanted tumors, CXCR3 signaling promotes CD8+ T cell infiltration that controls tumor growth [9,10,11]. The role of CXCL9 and its family members in sporadic CRC is unknown. Chemokine signaling through CXCR3 also mediates the recruitment of CD4+ T cells. Among them, Th17 cells promote CRC development by secreting IL-17 and IL-22 [1, 3, 12, 13], while Th1 cells have long known to inhibit tumor development [14]. Perhaps most intriguingly, regulatory T cells (Tregs) inhibit CRC development by dampening tumor-promoting inflammation [15]. Ablation of Treg-related cytokines IL-10 and TGF-β resulted in increased intestinal tumor burden [16, 17]. A high “Treg signature” in human CRC also indicates better prognosis [18]. The function of the CXCR3 cascade in CRC thus depends on the immune cell types that they recruit. The unique Treg-CRC relationship also complicates the use of Treg-targeting strategies, such as anti-CTLA4 for CRC treatment [19].
Here we show that IL-17 signals to transformed epithelial (tumor) cells to suppress the expression of CXCL9 and CXCL10 chemokines. Signaling of CXCL9/10 through CXCR3 is required for the recruitment of CD8+ CTLs and Tregs, but not Th1 or Th17 cells, to colorectal tumors. CXCR3 signaling to hematopoietic cells is required for the expression of IL-10 and TGF-β in tumors, and for the control of CRC development. Overall, IL-17 promotes CRC development by suppressing cells responsible for anti-cancer immunity, and fostering tumor-promoting gut inflammation. This novel mechanism pinpoints gut inflammation during cancer as a barrier for tumor control through the diverting action of IL-17 on the adaptive immune system.
Methods
Animal models
Il17ra−/− mice were from Amgen [20]. C57BL/6, ApcF/F [21], Cd8a−/− [22], B2m−/− [23], Cdx2-Cre [24], Cdx2-Cre-ERT2 [25], and Cxcr3−/− [26] mice were obtained from the Jackson Laboratory. Il17raF/F mice [1] were obtained from Dr. Michael Karin’s laboratory at University of California, San Diego.
To generate the mouse model of sporadic CRC, Cdx2-Cre and ApcF/F mice were crossed to generate Cdx2-Cre+/ ApcF/WT mice. These mice were sacrificed around 5 months of age for tumor analyses. Mouse colon was dissected, and colorectal tumors were excised with a scissor. Tumor-adjacent colon tissues were harvested and analyzed as “normal colon tissue” for comparison.
For tamoxifen-inducible tumorigenesis, Cdx2-Cre-ERT2+/ApcF/F mice were given 75 mg/kg body weight tamoxifen (Sigma, dissolved in 5% ethanol, 95% corn oil) i.p. on a daily basis for 3 consecutive days. Mice were sacrificed 4 to 5 weeks after the last dose of tamoxifen for tumor statistics and analysis. Mouse colon was dissected, and visible colorectal tumors (typically 1–2 mm in diameter) were excised with a scissor.
All mice were maintained in filter-topped cages on autoclaved food and water at UConn Health. All experiments used co-housed, gender matched littermates to ensure consistency of common microflora. Both male and female mice were used for all experiments.
Bone marrow transplantation
Six- to eight-week-old recipient mice were irradiated twice during 1 day to achieve a lethal dose (2 × 600 rad) and intravenously injected with single-cell suspension of 107 donor bone marrow cells. Recipients were co-housed littermates, which were transplanted with both gene-deficient and wild-type bone marrow for comparison. After transplantation the recipients were placed on sulphamethoxazole and trimethoprime in drinking water for 2 weeks, followed by regular water. Mice were sacrificed and analyzed for tumor development 4–5 months after transplantation.
Antibody treatment in mice
For sporadic CRC model (Cdx2-Cre+/ApcF/WT mice), IL-17A, CTLA-4, and PD-1 neutralizing antibodies or isotype control antibodies (Bio X Cell) were i.p. injected at a dose of 100 μg per mouse every 3 days until sacrifice.
For the tamoxifen inducible model of tumorigenesis, antibodies (100 μg per mouse, every 3 days) were injected 1 day after the dose of tamoxifen until sacrifice.
Immunofluorescent staining and ELISA
Immunofluorescent staining was performed on cryosectioned colorectal tumors with antibody against CD8α (Thermo Fisher), followed with Alexa-488-conjugated secondary antibody (Life Technology). Sections were further stained with DAPI and imaged under a confocal microscopy. For ELISA analysis of CXCL9 (Biolegend) and CXCL10 (R&D Systems), colonic tumors were cultured in opti-MEM containing 1% Antibiotic-Antimycotic (Life Technologies) for 24 h. Tissue culture supernatant was analyzed by ELISA. Concentrations of chemokines were normalized to the weight of tumors in each well.
Cell culture and cytokine treatment
Primary CRC tumor sphere culture was previously described [1]. Briefly, tumor cells were isolated from colorectal tumors of Cdx2-Cre-ERT2+/ApcF/F mice 4 weeks after tamoxifen injection. Cells were plated in Matrigel (BD Bioscience) and maintained in DMEM/F12 media (Life Technologies) containing B27 and N2 supplements (Life Technologies), 1.25 mM N-acetyl L-cysteine (Sigma), 100 ng/ml noggin (Peprotech), 50 ng/ml mEGF (Biosource), and 10% Rspo1-Fc-conditioned medium. To study IL-17 signaling in vitro, tumor spheres were replenished with serum and growth factor free medium for 24 h, and treated with 100 ng/ml recombinant human IL-17A, C or F for another 24 h.
Flow cytometry and cell sorting
Colorectal tumors were minced with scissors and digested with 1 mg/kg collagenase IV (Sigma Aldrich) for 20 min. Cells were filtered with 70-μm cell sieve, and stained with Live/Dead fixable exclusion dye (Tonbo Bioscience), followed by fluorochrome-conjugated antibodies in PBS with 2% fetal bovine serum (FBS) and 1 mM EDTA. Anti-CD3 (Cat # 100206), anti-CD4 (Cat # 100536), anti-CD45 (Cat # 103138), anti-CD19 (Cat # 152408), anti-CD11b (Cat # 101224), anti-F4/80 (Cat # 123108), anti-Gr-1 (Cat # 108428), anti-Ly6C (Cat # 128016), anti-Ly6G (Cat # 127641), anti-PD-1 (Cat # 135216), anti-Ep-CAM (Cat # 118216), anti-IL-10 (Cat # 505008), anti-IL-17A (Cat # 506904), anti-IFNγ (Cat # 505806), and anti-TNF-α (Cat # 506306) antibodies were from Biolegend. Anti-CD44 (Cat # 12–0441-82), anti-CD62L (Cat # 47–0629-42), anti-Foxp3 (Cat # 11–5773-82), and anti-Ki-67 (Cat # 11–5698-82) antibodies were from eBioscience. Anti-CD25 (Cat # 20–0251) and anti-CD3 (Cat # 20–0032) antibodies were from Tonbo Biosciences. Anti-CD8α antibody (Cat # 558106) was from BD Bioscience. For intracellular cytokine staining, cells were stimulated with Cell Stimulation Cocktail (eBioscience) for 4 h, followed with fixation and staining with Foxp3/transcription factor staining buffer set (eBioscience). Flow cytometry analyses were performed on a BD LSRII flow cytometer. Cell sorting was performed on a BD FACS ARIA II high speed cell sorter. Data was analyzed with FlowJo software.
Q-RT–PCR analysis
Total RNA was extracted with RNeasy Plus kit (Qiagen) and reverse transcribed using an IScript kit (Biorad). Q-RT-PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Biorad) on a Biorad CFX96 machine. Expression data were normalized to RPL32 mRNA levels. The data were calculated as 2(Ct(RPL32–gene of interest)) to compare experimental groups to controls, and are presented in arbitrary units. Primer sequences are listed in Additional file 1: Table S1. Whenever possible, primers were intron-spanning, such that amplification is feasible on complementary DNA.
Statistical analysis
Data are presented as averages +/− S.E.M. and were analyzed by the Students’ t test. P values less than 0.05 were considered significant.
Results
IL-17 inhibits the infiltration of tumor-associated CD4+ T cells and the production of IL-10 and TGF-β
Using mouse models of sporadic and inducible CRC, we set out to understand how IL-17 regulates adaptive immunity. The mouse model of sporadic CRC is based on allelic inactivation of one copy of the Apc tumor suppressor gene in colonic epithelial cells that is driven by a Cdx2-Cre transgene (Cdx2-Cre+/ApcF/+) [24, 27]. Subsequent Apc loss-of-heterozygosity (LOH) results in the development of large colonic adenomas and adenocarcinomas in the gut [24]. Tumors in this model are restricted to the colon and rectum and progress to adenocarcinomas, which makes this model more relevant to human CRC than the commonly used ApcMIN mice, where most of their tumors develop in the small intestine [28]. We also used a second model of synchronized colorectal tumorigenesis [25], which relies on tamoxifen-induced ablation of the Apc gene in Cdx2-Cre-ERT2+/ApcF/F mice permitting the study of early stage colorectal tumorigenesis [25]. Early CRC lesions can be detected histologically 1 week after tamoxifen injection. If undisrupted, these early lesions progress to visible colorectal tumors by 4 weeks. Using these tools, we found that IL-17 inhibits the production of IL-10 and TGF-β, both of which limit Th17 activity and inhibit CRC development [1]. IL-10 and TGF-β are produced by multiple immune and stromal cells within tumors, including Tregs [29]. Ablation of IL-17RA in the sporadic CRC model resulted in an increased level of Foxp3, a key marker for Tregs (Fig. 1a). Since our previous study showed that IL-17 is critical for the growth of early CRC lesions, we also examined the levels of IL-10 and TGF-β1 in early CRC tumors that were only 1 to 2 mm in diameter. Ablation of IL-17 signaling resulted in profoundly (more than 10-fold) increased levels of IL-10 and TGF-β1 mRNAs (Fig. 1b), and significantly induced the expression of Foxp3 in the tumor (Fig. 1b), suggesting a major role of IL-17 signaling in the suppression of anti-inflammatory cytokine production during early stage CRC. TGF-β1 is produced by multiple cell types within the tumor microenvironment, whereas IL-10 production appears to be restricted to CD4+ T cells (Fig. 1c). In early CRC lesions, ablation of IL-17 signaling resulted in increased recruitment of CD4+ T cells to the tumors and elevated numbers of IL-10+ CD4+ T cells that are either Foxp3+ (Tregs) or Foxp3− (Tr1 cells) (Fig. 1d, e). These data indicate that IL-17 inhibits the infiltration of Treg cells and the production of anti-inflammatory cytokines in early stage CRC.

IL-17 inhibits the infiltration of CD4+ T cells, and the production of TGF-β and IL-10 in CRC. a and b q-RT-PCR analyses of the indicated mRNAs in designated tissues of control and IL-17RA-deficient Cdx2-Cre+/ApcF/+ mice (a, n = 11), and Cdx2-Cre-ERT2+/ApcF/F mice (b, n = 5 for MLN, 11 for tumor). Mice in b received tamoxifen injection and were kept for 5 weeks for the development of early CRC tumors. Tumor-adjacent colonic tissues were used as “normal” control. c CD4+ T cells (CD45+ CD3+ CD4+), CD8+ T cells (CD45+ CD3+ CD8+), B cells (CD45+ CD19+), monocytes (CD45+ CD11b+ Ly-6CHigh), neutrophils (CD45+ CD11b+ Ly-6CLow, Ly-6G+), macrophages (CD45+ CD11b+, F4/80+), fibroblasts (CD45− EpCam−), and tumor cells (CD45− EpCam+) were FACS-sorted from pooled colonic tumors of 10 Il17ra+/+/Cdx2-Cre+/ApcF/+ mice. Purified cells were subjected to q-RT-PCR analysis, and the levels of each individual mRNA were shown as “1” in the cell type of highest expression. d Cdx2-Cre-ERT2+/ApcF/F mice that were Il17ra−/− or Il17ra+/− were given i.p. injection of tamoxifen (75 mg/kg body weight) daily for 3 consecutive days. Mice were sacrificed 5 weeks after tamoxifen-induced Apc ablation, and their mesenteric lymph nodes (MLN) and tumors were subjected to flow cytometry analysis. n = 5. Cells were isolated following collagenase digestion of the indicated tissues, followed by 4-h in vitro stimulation with PMA and ionomycin in the presence of Brefeldin A and monensin. e Representative flow cytometry plots for tumors samples from d. Data represent means ± S.E.M. *p < 0.05 in Students’ t test
IL-17 inhibits the infiltration of CTLs in early stage CRC
We have previously shown that IL-17 inhibits the expression of Th1/Tc1 signature genes [1]. This may result from IL-17-mediated inhibition on the infiltration of CD8+ CTLs to CRC. Indeed, immunostaining of cryosectioned colonic tumors showed that ablation of IL-17RA resulted in a marked increase in the number of CD8+ T cells in sporadic colorectal tumors (Fig. 2a, b). To test if this inhibition of CTL infiltration by IL-17 happens in early stage CRC, we performed flow cytometry analysis on tumors that developed following tamoxifen-induced deletion of Apc in colonic epithelium. Loss of IL-17RA in this model also resulted in marked elevation in the number of CD8+ CTLs in tumors (Fig. 2c), demonstrating an inhibitory role of IL-17 signaling in limiting CTL infiltration in early stage colon tumors. Early tumors that lost IL-17RA also exhibited elevated expression of IFN-γ and TNF-α (Fig. 2d). IL-17 signaling has no direct impact on the production of IFN-γ and TNF-α by T cells (Fig. 2e). Since CD8+ CTLs were long known to function in cancer immunesurveillance [30], the observed inhibition of CD8+ T cell infiltration by IL-17 is consistent with IL-17’s role in promoting early stage CRC development [1].

IL-17 blocks the accumulation of CD8+ T lymphocytes starting from early stage CRC. a Immunostaining of colon tumors from 5-month-old Cdx2-Cre+/ApcF/+ mice that were heterozygous (+/−) or null (−/−) for Il17ra. Scale bar = 100 μm. b Statistics for the percentages of CD8+ cells shown in a. n = 8. c-e: Cdx2-Cre-ERT2+/ApcF/F mice that were Il17ra+/− or Il17ra−/− were sacrificed 5 weeks after tamoxifen-induced Apc ablation, and their MLN (c and d) and tumors (c, d, and e) were subjected to flow cytometry (c and e, n = 4 for Il17ra+/−, 10 for Il17ra−/−) and q-RT-PCR (d, n = 5 for MLN, 11 for tumor) analyses. Data represent means ± S.E.M. *p < 0.05 in Students’ t test
IL-17 suppresses the expression of CXCL9, 10, and 11
IL-17 is known to promote chemokine production and attraction of MDSCs to tumors [1, 7, 31, 32]. Ablation of IL-17RA in mice resulted in reduced intratumoral levels of CXCL1 and 2 [1], consistent with the known role of IL-17 in promoting CXCL1/2 production and myeloid cell recruitment [33, 34]. Whether IL-17 regulates T cell recruitment is unknown. We found that loss of IL-17 signaling resulted in increased levels of the T cell-attracting chemokines CXCL9, 10, and 11 in colonic tumors (Fig. 3a, b). Loss of IL-17 signaling also increased the expression of CXCR3, the cognate receptor for CXCL9/10/11 chemokines, likely reflecting an increased recruitment of CXCR3-expressing lymphocytes to the tumor (Fig. 3a). Upregulation of the CXCL9 family of chemokines was also seen in 5-month-old CRC tumors of mice receiving i.p. injections of anti-IL-17A neutralizing antibody, demonstrating the effect of acute IL-17A blockade in chemokine production (Fig. 3c). Consistent with the notion that IL-17 inhibits T cell infiltration during early stage CRC, ablation of its receptor in the mouse model of induced colorectal tumorigenesis resulted in elevated CXCL9 family chemokines in early CRC tumors (Fig. 3d).

IL-17 inhibits the production of CXCL9 family chemokines. a q-RT-PCR analyses of the indicated mRNAs in normal colon and colorectal tumor tissues of 5-month-old control (Il17ra+/−) and IL-17RA whole body knockout (Il17ra−/−) mice that also harbor Cdx2-Cre+/ApcF/+ genotypes (n = 12). b Colonic tumors from control (Il17ra+/−) and IL-17RA-deficient (Il17ra−/−) Cdx2-Cre+/ApcF/+ mice were cultured in Opti-MEM medium for 24 h. Concentrations of chemokines were tested using a bead-based immunoassay (Biolegend, for CXCL9), or plate based ELISA (R&D systems, for CXCL10). Data are shown as pg/ml chemokine per mg tumor in culture (n = 13). c 5-month-old Cdx2-Cre+/ApcF/+ mice were treated with i.p. injection of isotype or anti-IL-17A antibodies (100 μg per injection, every 3 days) for two weeks. Colonic tumors were harvested at the end of the study, and analyzed by q-RT-PCR for indicated mRNAs. n = 13. d Cdx2-Cre-ERT2+/ApcF/F mice (that were Il17ra−/− or Il17ra+/−) were sacrificed 5 weeks after tamoxifen-induced Apc ablation. Mouse MLN and tumors were subjected to q-RT-PCR analysis (n = 5 for MLN, 11 for tumor). Data represent means ± S.E.M. *p < 0.05 in Students’ t test
IL-17 signals to transformed colonic epithelial cells to suppress the production of CXCL9, 10, and 11
We have previously shown that IL-17 mainly signals to transformed colonic epithelial cells (tumor cells) to promote CRC development [1]. It is possible that the same signaling pathway also controls CXCL9 family chemokine production. To this end, we analyzed the expression of CXCL9 family chemokines in Cdx2-Cre+/ApcF/WT mice that harbor colon epithelial cell-specific deletion of IL-17RA. Loss of IL-17 signaling to colonic epithelial cells and their transformed counterparts resulted in elevated expression of CXCL9 family chemokines (Fig. 4a). To confirm a direct inhibitory effect of IL-17 on the production of chemokines, we isolated tumor cells from Cdx2-Cre-ERT+/ApcF/F mice, and cultured these cells in Matrigel to form tumor spheres. These tumor spheres were treated with recombinant IL-17A, C or F, and then analyzed for chemokine expression by q-RT-PCR. Consistent with the previously known role of IL-17 in promoting the production of myeloid cell attracting chemokines, treatment with recombinant IL-17 resulted in elevated levels of CXCL1 and 2 (Fig. 4b). However, in primary tumor spheres IL-17 stimulation resulted in reduced levels of CXCL9 and 10 mRNAs (Fig. 4b), thus confirming a direct inhibitory role of IL-17 on CXCL9/10 production.

IL-17 signals to transformed epithelial cells to suppress CXCL9 family chemokine production. a Cdx2-Cre+/ApcF/+ mice were crossed to Il17ra-flox mice to generate a conditional ablation of Il17ra gene in colorectal epithelial cells and tumor cells. These mice carry Cdx2-Cre+/ApcF/+/Il17raF/− genotypes and are labeled as “Il17raF/−”. Cdx2-Cre+/ApcF/+/Il17raF/+ mice (labeled as Il17raF/+) were used as controls. Both groups of mice were sacrificed at 5 months of age. Colorectal tumors and adjacent normal colon tissues were harvested for q-RT-PCR analysis. n = 6. b Tumor cells were isolated from colorectal tumors of Cdx2-Cre-ERT2+/ApcF/F mice 4 weeks after tamoxifen injection. Cells were then cultured a 3-D system to allow their development into primary tumor spheres. Tumor spheres were subsequently treated with vehicle control (PBS with 0.1% BSA) or 100 ng/ml recombinant human IL-17A, C, or F for 24 h, followed by q-RT-PCR analysis (n = 3, and data represent one of three consistent tests). Data represent means ± S.E.M. *p < 0.05 in Students’ t test
CXCR3 signaling attracts CTLs and Treg cells to inhibit CRC development
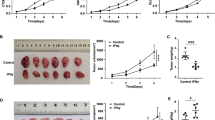
CXCL9 and 10 are expressed by tumor cells and tumor-infiltrating myeloid cells, and their receptor CXCR3 is restricted to T lymphocytes (Fig. 5a). We reasoned that this chemokine pathway may be responsible for IL-17’s inhibitory role in T cell migration to CRC. Indeed, ablation of CXCR3 in all blood cells by means of bone marrow reconstitution resulted in reduced recruitment of CD8+ T cells, and to a lesser extent, Tregs to colorectal tumors (Fig. 5b, c). Ablation of CXCR3 in blood cells also reduced the levels of IL-10 and TGF-β in tumors (Fig. 5d), both of which have been shown to inhibit CRC development by dampening tumor-promoting inflammation [15, 17, 35, 36]. CXCR3 ablation also resulted in a marked decrease in the level of Foxp3 in tumors (Fig. 5d), suggesting a reduced Treg recruitment upon loss of CXCR3. CXCR3 signaling in hematopoietic cells is dispensable for the recruitment of Th1, Th17, and myeloid cells to colorectal tumors (Additional file 1: Figure S1). Ablation of CXCR3 in bone marrow cells showed no impact on IL-17 expression in tumors, and resulted in elevated IFN-γ levels (Fig. 5d). Loss of CXCR3 also did not impact the activation and expansion of tumor-infiltrating T cells (Additional file 1: Figure S2). Consistent with the known role of CD8+ CTLs in limiting cancer development, loss of these cells in mice that lack Cd8α or Beta-2-Microglobulin (a subunit of MHC I complex) both resulted in elevated tumor development in the large intestine (Additional file 1: Figure S3a, b). Given the role of CXCR3 in mediating CD8+ CTL and Treg recruitment, we reasoned that loss of CXCR3 should also result in accelerated colorectal tumorigenesis. Indeed, mice that lack CXCR3 in hematopoietic cells developed increased numbers of colorectal tumors, without changes in tumor sizes (Fig. 5e). The expression levels of CXCL9 family chemokines were not affected by the loss of CD8+ T cells in CRC-bearing mice (Additional file 1: Figure S3c), suggesting that these cells are not required for the production of CXCL9 family chemokines. Overall, these data show that CXCR3 signaling selectively attracts CD8+ CTLs and Tregs to CRC, and inhibits CRC development.

CXCR3 mediates the attraction of CD8+ CTLs and Treg cells, and inhibits the CRC development. a FACS-purified cells (as depicted Fig. 1c) from colonic tumors of Cdx2-Cre+/ApcF/WT mice were subjected to q-RT-PCR analysis. b-e Bone marrow cells were harvested from WT and Cxcr3−/− mice, and transferred into lethally irradiated 6–8-week-old Cdx2-Cre+/ApcF/WT mice. Recipient mice were sacrificed at 5 months of age, and their colorectal tumors were used for flow cytometry (b and c, n = 7), q-RT-PCR (d, n = 16), and tumor statistics (e, n = 9). Cells shown in c were gated as live/CD45+. Data represent means ± S.E.M. *p < 0.05 in Students’ t test
IL-17 blockade upregulates the expression of immune checkpoint markers
Immune checkpoint inhibitors, such as antibodies that block CTLA-4 and PD-1 signaling, are effective only in a fraction of colorectal cancers that show microsatellite instability (MSI) [37, 38]. Given the role of IL-17 in inhibiting the infiltration of CTLs and Treg cells to CRC, we next tested if its blockade would impact immune checkpoint signaling. Ablation of IL-17RA in the mouse sporadic tumor model resulted in elevated expression of CTLA-4 (Fig. 6a), a cell surface protein that is constitutively expressed in Tregs and mediates part of their immune suppressive function [39, 40]. In addition, IL-17RA-null tumors also exhibited elevated expression levels of PD-L1 and PD-L2 (Fig. 6a). Similar changes were observed in Cdx2-Cre+/ApcF/WT mice treated with neutralizing antibody against IL-17A (Fig. 6b). Upregulation of CTLA-4 and PD-1 pathway markers were also observed in the mouse model of early stage colorectal tumorigenesis (Fig. 6c), suggesting an antagonism of IL-17 and immune checkpoint pathways beginning at the early phase of CRC development. We have previously shown that ablation of IL-17RA in CRC led to an increased level of IFN-γ [1], which is known to upregulate PD-L1 expression [41]. IL-17 signaling does not impact proliferation or activation of tumor-infiltrating CD4+ and CD8+ T cells (Fig. 6d). Both tumor-infiltrating CD8+ and CD4+ T cells express PD-1, and the proportion of PD-1-positive T cells decreased modestly upon ablation of IL-17 signaling (Fig. 6d). Therefore, the increase in overall PD-1 expression in CRC likely reflects the substantial increase in T cell infiltration upon blockade of IL-17 signaling, and not increased PD-1 expression on a per cell basis. CTLA-4 immunotherapy has been tested in human cancers and showed variable efficacy [42, 43]. Consistent with the role of Tregs in limiting tumor-associated inflammation, blockade of CTLA-4 signaling by antibody increased the expression of IL-17A in tumors (Fig. 7a). In contrast, expression of IL-17 was not changed in mice that received blocking antibody for PD-1 (Fig. 7b). These results suggest that CTLA-4 blockade upregulates pro-tumor IL-17 in colorectal tumors. Taken together, our data show that IL-17 signals to tumor cells to downregulate the production of chemokines CXCL9/10, which are required for attracting CD8+ CTLs and Tregs to CRC. Inhibition of CXCL9/10 signaling by IL-17 thus reduces the activity of anti-cancer immunity, and fosters stronger tumor-promoting inflammation (Fig. 7c).

Blockade of IL-17 signaling in CRC results in enhanced immune checkpoint signaling. a q-RT-PCR analysis of normal colon tissue and colorectal tumors from Cdx2-Cre+/ApcF/+ mice that harbor heterozygous or null alleles of Il17ra gene (n = 12). b 4-month-old Cdx2-Cre+/ApcF/+ mice received i.p. injection of 100 μg isotype or anti-IL-17A antibodies every 3 days for 1 month. Mice were sacrificed for q-RT-PCR analysis (n = 9). c Cdx2-Cre-ERT2+/ApcF/F mice that were Il17ra+/− or Il17ra−/− were sacrificed 5 weeks after tamoxifen-induced Apc ablation, and their MLN and tumors were subjected to q-RT-PCR analysis (n = 5 for MLN, 11 for tumor). d Cdx2-Cre-ERT2+/ApcF/F mice that were Il17ra−/− or Il17ra+/− were sacrificed 5 weeks after tamoxifen-induced Apc ablation, and their MLN and tumors were subjected to flow cytometry analysis. n = 5. Data represent means ± S.E.M. *p < 0.05 in Students’ t test

Blockade of CTLA-4 induced the expression of IL-17 in CRC. a, b 5-month-old Cdx2-Cre+/ApcF/+ mice received i.p. injection of 100 μg isotype or blocking antibodies against CTLA-4 (a, n = 24) or PD-1 (b, n = 8) every 3 days for 2 weeks, and were sacrificed for q-RT-PCR analysis. Data represent means ± S.E.M. *p < 0.05 in Students’ t test. c: IL-17 signals directly to tumor cells in CRC to inhibit the production of CXCL9 and CXCL10. These two chemokines are required for the recruitment of CD8+ CTLs and Tregs, which inhibits CRC development by targeting cancer cells and dampening tumor-promoting inflammation, respectively
Discussion
IL-17 is known to promote neutrophil infiltration by activating the production of their attracting chemokines. In mouse model of CRC, ablation of IL-17 resulted in reduced levels of CXCL1 and CXCL2, which correlates with decreased numbers of tumor infiltrating myeloid cells [1, 4, 5, 44]. We also showed that these tumor-infiltrating myeloid cells respond to bacterial products that pass through defective surface barrier due to tumorigenesis, and produce IL-23 [3]. IL-23 in turn promotes the production of IL-17 by T cells and innate lymphoid cells [3]. In this way, IL-17 and tumor-infiltrating myeloid cells form an auto-enhancing loop to promote tumor-associated inflammation. Combined with our new finding that IL-17 inhibits T cell infiltration through the downregulation of CXCL9/10, it is now clear that IL-17 skews tumor immune environment towards an innate cell-dominant, tumor-promoting inflammation. In different settings, IL-17 has also been shown to promote the infiltration and develop of myeloid-derived suppressor cells (MDSC), which inhibit the activity of CTLs and thus promotes tumor development [31, 45]. The contribution of MDSC to T cell inactivation in sporadic CRC is unknown, but may represent an alternative pathway by which IL-17 indirectly inhibits CD8+ CTL activity. It is therefore possible that tumor-infiltrating myeloid cells play dual roles in CRC: 1) these cells respond to commensal bacteria and promote tumor-associated inflammation (such as the production of IL-23 and IL-17), which subsequently leads to reduced CXCL9/10 production and T cell attraction; 2) these cells may serve as suppressors of anti-tumor immunity. Additional research is required to dissect the inflammation-promoting v.s. T cell inactivating roles of myeloid cells in tumors. For example, one may employ myeloid-specific ablation of effector molecules (such as arginase [45]) to examine the effect of MDSC in sporadic CRC.
Chemokine signaling through CXCR3 has been shown to inhibit tumor growth in several transplantable tumor models [10, 11, 46]. This anti-tumor function of CXCR3 and its cognate ligands were attributed to the recruitment of CD8+ CTLs into tumors. Consistently, in human CRC, a high CXCL10 level correlates with CD8+ T cell infiltration [47]. In our study, we also observed reduced CTL number in colorectal tumors upon ablation of CXCR3 in hematopoietic cells. In contrast, CXCR3 signaling was dispensable for Th1 and Th17 cell infiltration. Intriguingly, we found that CXCR3 functions to recruit Treg cells to CRC tumors, and CXCR3 loss results in marked decreases in the levels of IL-10 and TGF-β. Given the anti-tumor roles of IL-10 and TGF-β in early stage colon cancer development, we concluded that CXCR3 inhibits early stage colorectal tumorigenesis by attracting both CTLs and Treg cells. This conclusion was supported by the observation that loss of CXCR3 in blood cells resulted in increased tumor incidence in mouse colon, but no changes in tumor size. It is also in agreement with the known role of IL-17 in promoting early stage CRC development [1].
In this study, we report a novel mechanism by which IL-17 inhibits the recruitment of CD8+ CTLs and Treg cells by downregulating the production of CXCL9/10 chemokines. Such knowledge will demonstrate the feasibility of interfering with IL-17-Treg interaction for CRC prevention and immunotherapy. For instance, blockade of IL-17 signaling may be useful for the prevention of CRC in genetically susceptible populations, such as FAP (familial adenomatous polyposis) patients that harbor germline mutations in the Apc tumor suppressor gene. Given the known role of IL-17 in promoting early stage CRC development [1], and its negative impact in the inhibition of CD8+ CTLs and Tregs, blocking IL-17 may suppress tumor-promoting inflammation, activate tumor immunosurveillance, and reduce the rate of tumorigenesis in this genetically predisposed population.
Immunotherapy against human CRC has shown limited success, as it is effective only in microsatellite instable (MSI) cases [37, 38]. For the 85% of microsatellite stable CRC, checkpoint inhibition largely does not work. Our mouse models of CRC are based on allelic inactivation of the Apc tumor suppressor gene [24, 25, 27], and do not carry MSI lesions. Yet, in both sporadic and early stage CRC models, ablation of IL-17 signaling resulted in increased recruitment of anti-tumor CD8+ CTLs via upregulation of CXCL9 family chemokines, without the requirement of MSI. It is possible that in human CRC that are microsatellite stable, blockade of IL-17 can also result in increased production of CXCL9 family chemokines and enhanced infiltration of CD8+ T cells to tumors, which is a desirable trait for cancer immunotherapy. Upregulation of IL-17 in mouse models of CRC stems from the loss of surface barrier function in the process of epithelium transformation. In this regard, it remains to be tested if IL-17 plays a similar role in limiting T cell infiltration in MSI tumors.
While IL-17 blockade may also increase the number of Tregs in human CRC, blockade of immune checkpoints should be sufficient to neutralize their inhibition on anti-cancer immunity. In this regard, neutralizing antibodies against IL-17A and IL-17RA, which have been tested safe and effective for the treatment of autoimmunity in humans [48], may be tested as adjuvant therapies that accompany current cancer immunotherapies. IL-17 production is restricted to the CRC tumor site, and its blockade should result in selective upregulation of CXCL9 family chemokines in tumors. In this perspective, IL-17 blockade should be effective in attracting T cells to tumors, and poses less risk of systemic immune activation.
Conclusions
Our data show a novel role of IL-17 in inhibiting the infiltration of CD8+ CTLs and Tregs to CRC. This is mediated by IL-17’s signaling to colorectal tumor cells, which leads to the reduced production of CXCL9/10 chemokines. CXCL9/10 chemokines, signaling through their cognate receptor CXCR3, recruit CD8+ CTLs and Tregs to CRC, but are dispensable for the recruitment or activation of other T cells and myeloid cells. By excluding Tregs and CTLs from CRC, IL-17 fosters the dominance of tumor-promoting inflammation. To this end, cancer immunotherapy may be benefited by the use of anti-IL-17 agents, as blockade of IL-17 reduces the rate of tumor growth and increases the infiltration of CTLs that are vital for effective cancer treatment.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- B2m:
-
Beta-2-microglobulin
- CRC:
-
Colorectal cancer
- CTL:
-
Cytotoxic T lymphocytes
- CTLA4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CXCL10:
-
C-X-C Motif Chemokine Ligand 10
- CXCL11:
-
C-X-C Motif Chemokine Ligand 11
- CXCL9:
-
C-X-C Motif Chemokine Ligand 9
- Foxp3:
-
Forkhead box P3
- IFN-γ:
-
Interferon gamma
- IL-10:
-
Interleukin-10
- IL-17:
-
Interleukin-17
- IL-17A:
-
Interleukin-17A
- IL-17C:
-
Interleukin-17C
- IL-17F:
-
Interleukin-17F
- IL-17RA:
-
Interleukin-17 receptor A
- MDSC:
-
Myeloid-derived suppressor cells
- MLN:
-
Mesenteric lymph node
- PD-1:
-
Programmed cell death-1
- PD-L1:
-
Programmed death-ligand 1
- PD-L2:
-
Programmed death-ligand 2
- Th1:
-
Type 1 T helper cells
- Th17:
-
T helper 17 cells
- TNF-a:
-
Tumor Necrosis Factor-α
- Treg:
-
Regulatory T cells
References
Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity. 2014;41(6):1052–63.
Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(12):5540–4.
Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491(7423):254–8.
Awane M, Andres PG, Li DJ, Reinecker HC. NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J Immunol. 1999;162(9):5337–44.
Datta S, Novotny M, Pavicic PG Jr, Zhao C, Herjan T, Hartupee J, et al. IL-17 regulates CXCL1 mRNA stability via an AUUUA/tristetraprolin-independent sequence. J Immunol. 2010;184(3):1484–91.
Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013;19(9):1114–23.
Thiele Orberg E, Fan H, Tam AJ, Dejea CM, Destefano Shields CE, Wu S, et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol. 2017;10(2):421–33.
Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - a target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–7.
Chheda ZS, Sharma RK, Jala VR, Luster AD, Haribabu B. Chemoattractant receptors BLT1 and CXCR3 regulate antitumor immunity by facilitating CD8+ T cell migration into tumors. J Immunol. 2016;197(5):2016–26.
Gorbachev AV, Kobayashi H, Kudo D, Tannenbaum CS, Finke JH, Shu S, et al. CXC chemokine ligand 9/monokine induced by IFN-gamma production by tumor cells is critical for T cell-mediated suppression of cutaneous tumors. J Immunol. 2007;178(4):2278–86.
Yang X, Chu Y, Wang Y, Zhang R, Xiong S. Targeted in vivo expression of IFN-gamma-inducible protein 10 induces specific antitumor activity. J Leukoc Biol. 2006;80(6):1434–44.
Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity. 2014;40(5):772–84.
Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491(7423):259–63.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30.
Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, et al. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res. 2005;65(10):3998–4004.
Dennis KL, Wang Y, Blatner NR, Wang S, Saadalla A, Trudeau E, et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 2013;73(19):5905–13.
Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21(4):491–501.
Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011;71(4):1263–71.
Passardi A, Canale M, Valgiusti M, Ulivi P. Immune Checkpoints as a Target for Colorectal Cancer Treatment. Int J Mol Sci. 2017;18(6):1–12.
Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194(4):519–27.
Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278(5335):120–3.
Fung-Leung WP, Schilham MW, Rahemtulla A, Kundig TM, Vollenweider M, Potter J, et al. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell. 1991;65(3):443–9.
Koller BH, Marrack P, Kappler JW, Smithies O. Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+ T cells. Science. 1990;248(4960):1227–30.
Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67(20):9721–30.
Feng Y, Sentani K, Wiese A, Sands E, Green M, Bommer GT, et al. Sox9 induction, ectopic Paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. Am J Pathol. 2013;183(2):493–503.
Cuenca AG, Wynn JL, Kelly-Scumpia KM, Scumpia PO, Vila L, Delano MJ, et al. Critical role for CXC ligand 10/CXC receptor 3 signaling in the murine neonatal response to sepsis. Infect Immun. 2011;79(7):2746–54.
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–67.
Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from min mice. Cancer Res. 1994;54(22):5947–52.
Wang K, Vella AT. Regulatory T cells and Cancer: a two-sided story. Immunol Investig. 2016;45(8):797–812.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–8.
Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8.
Novitskiy SV, Pickup MW, Gorska AE, Owens P, Chytil A, Aakre M, et al. TGF-beta receptor II loss promotes mammary carcinoma progression by Th17 dependent mechanisms. Cancer Discov. 2011;1(5):430–41.
Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat Immunol. 2011;12(9):853–60.
Iyoda M, Shibata T, Kawaguchi M, Hizawa N, Yamaoka T, Kokubu F, et al. IL-17A and IL-17F stimulate chemokines via MAPK pathways (ERK1/2 and p38 but not JNK) in mouse cultured mesangial cells: synergy with TNF-alpha and IL-1beta. Am J Physiol Renal Physiol. 2010;298(3):F779–87.
Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B, et al. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am J Pathol. 2003;162(2):691–702.
Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, et al. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 2003;63(18):6042–50.
Arora SP, Mahalingam D. Immunotherapy in colorectal cancer: for the select few or all? J Gastrointest Oncol. 2018;9(1):170–9.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–5.
Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–65.
Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64(3):1140–5.
Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–14.
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–61.
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6(11):1133–41.
He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, et al. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol. 2010;184(5):2281–8.
Hensbergen PJ, Wijnands PG, Schreurs MW, Scheper RJ, Willemze R, Tensen CP. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J Immunother. 2005;28(4):343–51.
Zumwalt TJ, Arnold M, Goel A, Boland CR. Active secretion of CXCL10 and CCL5 from colorectal cancer microenvironments associates with GranzymeB+ CD8+ T-cell infiltration. Oncotarget. 2015;6(5):2981–91.
Kurschus FC, Moos S. IL-17 for therapy. J Dermatol Sci. 2017;87(3):221–7.
Acknowledgements
We thank Dr. Michael Karin at University of California, San Diego for the Il17raF/F mice, and Ms. Li Zhu at the UConn Health Flow Cytometry Core Facility for technical assistance.
Funding
All work was conducted at UConn Health and supported by institutional startup funds to K.W. Salary support for J.C. was provided by UConn Health; Salary support for X.Y. was provided by UConn Health and Shenzhen PKU-HKUST Medical Center; Salary support for E.P. was provided by NIH T90 training grant (T90-DE022526) under the support of Dr. Mina Mina, and UConn Health; Salary support for M.L. was provided by UConn Health, Beijing University of Chinese Medicine, and the China Scholarship Council. J.W. was supported by the National Natural Science Foundation of China (81571043), Natural Science Foundation of Guangdong Province (2016A030312016), and Shenzhen Basic Research Grants (JCYJ20170306161450254 and JCYJ20170815153617033).
Author information
Authors and Affiliations
Contributions
JC and KW conceived the project. JC, XY, EP, ML, EJ, and KW performed the experiments and analyzed the data. JW, AA and AV provided conceptual advice. JC and KW wrote the manuscript, with all authors contributing to the writing, editing, and providing key advice. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All animal experiments were approved by the IACUC of UConn Health, and performed in accordance with the university and NIH guidelines and regulations.
Consent for publication
The work does not contain data from any individual person, and therefore consent for publication is not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Primer Sequences for q-RT-PCR. Figure S1. CXCR3 signaling is dispensable for the recruitment of Th1, Th17 and myeloid cells. Figure S2. CXCR3 signaling is dispensable for the activation of T cells. Figure S3. CD8+ T cells inhibit the development of sporadic CRC.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, J., Ye, X., Pitmon, E. et al. IL-17 inhibits CXCL9/10-mediated recruitment of CD8+ cytotoxic T cells and regulatory T cells to colorectal tumors. j. immunotherapy cancer 7, 324 (2019). https://doi.org/10.1186/s40425-019-0757-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40425-019-0757-z