
Abstract
Chimeric Antigen Receptor (CAR) T cell therapies – adoptive T cell therapies that have been genetically engineered for a new antigen-specificity - have displayed significant success in treating patients with hematologic malignancies, leading to three recent US Food and Drug Administration approvals. Based on the promise generated from these successes, the field is rapidly evolving to include new disease indications and CAR designs, while simultaneously reviewing and optimizing toxicity and management protocols. As such, this review provides expert perspective on the significance and clinical considerations of CAR T cell therapies in order to provide timely information to clinicians about this revolutionary new therapeutic class.
Similar content being viewed by others


Background
Adoptive cell therapies (ACT) involve collection of immune cells from a patient or a donor, often followed by ex vivo manipulation and/or expansion and reinfusion, have been investigated for decades and are now a cornerstone of cancer immunotherapy. ACTs can be designed to overcome cancer immune evasion mechanisms by directly targeting cancer, thus activating a powerful and (ideally) specific immune response to the tumor. Multiple iterations of ACT are in development for the treatment of cancers, including dendritic cells, natural killer cells, and tumor-infiltrating lymphocytes [1,2,3]. Initial evidence of responses to ACT using patient-specific tumor infiltrating lymphocytes (TIL) described in the treatment of patients with advanced ovarian cancer, and has since been used to treat patients with melanoma, prostate cancer, and renal cancer, among others [4,5,6].
Successful ACT depends on adequate numbers of effector cells in the patient, which in turn requires precursors with natural anti-tumor recognition, or engineering of the T cells to provide this recognition. As such, researchers have developed strategies to increase tumor recognition of adoptively stimulated cells outside of T cell receptor (TCR)/major histocompatibility complex (MHC)/peptide recognition. Genetic engineering of novel receptors – now known as Chimeric Antigen Receptors (CAR) – can both recognize cancer-associated antigens and provide T cell activation, proliferation, and memory. CAR constructs are hybrid molecules that can replace major functions of the TCR, including surface antigen recognition as well as T cell activation and costimulation by incorporation of the intracellular domains, most commonly comprising CD3ζ, CD28, and/or 4-1BB [7]. Initial reports of CARs demonstrated successful CAR expression and antigen-specific cytotoxicity in T cells engineered with anti-2,4,6-trinitrophenyl (TNP) CAR constructs [8].
Clinical trials have shown high response rates after anti-CD19 CAR infusion in patients with B cell malignancies, including diffuse large B cell lymphoma (DLBCL) and B cell-precursor acute lymphoblastic leukemia (ALL), resulting in two FDA approved therapies [9, 10]. Studies have also observed specific toxicities – including cytokine release syndrome (CRS) and CAR T cell-related neurotoxicity (NTX) [11]. Based on these data, ongoing pre-clinical and clinical research programs are optimizing CAR T cell strategies in order to increase durability of response, target other cancers, and to control and prevent major toxicities. Our purpose here is to discuss the history of CAR T therapies, review the current toxicity management protocols, and provide an overview of in-development technologies for clinicians who will be treating patients with or referring patients for these groundbreaking therapies.
Main Text
CAR design across generations
CARs consist of three major domains: an ectodomain, transmembrane domain, and an endodomain. The ectodomain is the extracellular portion of the receptor that includes the antigen-recognition domain as well as a signal peptide for direction to the endoplasmic reticulum. The transmembrane domain primarily supports CAR stability. The intracellular endodomain facilitates signal transduction to activate T cells during antigen recognition [7].
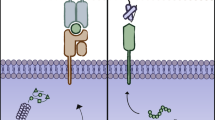
The endogenous TCR complex is expressed on the surface of T cells and can recognize antigens bound to MHC molecules on antigen-presenting cells to activate the T cell [12]. First-generation CARs were designed similarly to the endogenous TCR complex by incorporating a CD3ζ- chain or FcεRIγ intracellular domain, but instead incorporated extracellular antigen-recognition domains that allowed for direct antigen-recognition on the surface of tumor cells, allowing for MHC-independent T cell activation (Fig. 1). Importantly, these first generation designs did not include costimulatory domains to provide the second signal for full T cell activation. First-generation CAR T cells are more susceptible to apoptosis and have limited in vivo expansion and cytotoxicity [7].

Chimeric Antigen Receptor (CAR) design across generations. First generation Chimeric Antigen Receptors (CARs) include an extracellular antigen-binding domain and an intracellular T cell activation domain, commonly CD3ζ or FcεRIγ. Second generation CARs built upon first generation constructs by including an intracellular costimulatory domain, commonly 4-1BB or CD28, incorporated into the FDA approved CAR T therapies tisagenlecleucel and axicabtagene ciloleucel, respectively. Costimulatory domains help enhance CAR T cell cytotoxicity and proliferation compared to first generation designs. Third generation CARs include multiple costimulatory domains, primarily to increase CAR T cell proliferation and persistence
In distinction, second-generation CAR constructs incorporate both signal 1 and signal 2 for full T cell activation. Where signal 1 comes from CD3ζ and signal 2 comes from a costimulatory domain to promote interleukin-2 (IL-2) secretion, which promotes T cell activation and prevents apoptosis [7] Multiple costimulatory domains can be utilized to significantly change CAR T cell cytokine secretion profiles to increase cytotoxicity and T cell persistence, as FDA approved CARs incorporate either CD28 or 4-1BB (CD137) [13, 14]. The benefits of a specific costimulatory domain remain under investigation.
Third-generation CAR constructs, which combine multiple intracellular signaling domains, including combinations like CD3ζ-CD28-OX40 and CD3ζ-CD28-41BB are being investigated preclinically [15, 16]. Combination costimulatory domains further support increased cytokine production and could increase efficacy, but there is little clinical data available to assess this.
CAR T cell production processes
Time from CAR T collection to patient infusion currently takes ~ 3 weeks. Peripheral blood mononuclear cells are collected from the patient using a large-volume leukapheresis procedure. The cells are then transferred to a GMP manufacturing facility for T cell engineering and expansion. Patient T cells are then incubated with CAR-encoding viral vectors, which enter the T cells and introduce CAR gene RNA. CAR RNA is then reverse-transcribed into DNA, which recombines into the T cell genome, resulting in permanent CAR gene incorporation. As this process is not 100% efficient, transformed T cells undergo ex vivo expansion for multiple days, resulting in a product that is ~ 90% CD3+ T cells, but variable fractions of which are CAR+. The cells are transferred back to the center for infusion, which typically happens as a single infusion for FDA approved therapies [13, 14, 17]. CAR T cell infusion recommendations vary across centers, sponsors, and products. Existing protocols include both inpatient and outpatient infusions, and dosage dependent on the total CAR+ population in the infusion product, as well as patient characteristics including age and weight [9, 10]. Of note, recent studies of a responding patient identified a single CAR T cell clone that made up for 94% of the total CAR T cell population [18]. These data suggest that infusion of a single CAR+ T cell can promote therapeutic response.
Initial CAR T therapy clinical testing
Initial clinical reports on CAR T cell therapeutic efficacy and safety began with a 2010 case report describing a partial response of B cell follicular lymphoma and ablation of blood and bone marrow B cells in a patient treated with a second-generation CD19 CAR T utilizing CD28 [19]. The patient underwent leukapheresis, lymphodepletion with fludarabine and cyclophosphamide (flu/cy), and the patient received the CAR product (infused over 2 days) followed by high-dose IL-2. The patient remained in partial remission for 32 weeks after treatment until discovery of progressive CD19+ lymphoma in lymph nodes. The patient then received chemotherapy plus CAR T cells/IL-2, and remains progression-free at the time of this report (personal communication, [20]). Further early development showed that this anti-CD19 CAR T cell product was highly effective against diffuse large B cell lymphoma, and this product went on to become the FDA approved therapy axicabtagene ciloleucel [9, 13, 21,22,23].
A report in 2011 described using a second generation, 4-1BB CD19 CAR for chronic lymphocytic leukemia (CLL), demonstrating complete responses (CR) [24]. This product became the first FDA approved CAR T cell therapy, tisagenlecleucel [10, 14]. Three patients with advanced, chemotherapy-resistant CLL (two p53-deficient) received autologous CD19 CAR T cells, with no exogenously administered cytokines. Two CRs (11+ mos and 10+ mos) and one partial response (7 mos) were observed. Toxicities occurred between days 7 and 21 in all patients, but all cases were reversible and treatable. Importantly, researchers noted that one patient had measurable CAR-expressing T cells up to 6 months post-treatment, and in vitro characterization confirmed that CAR T cell effector function was retained [24, 25]. Follow-up of these two patients show them to be in continued CR for > 5 years, with continued B cell aplasia suggesting continued functional persistence of the CAR T cells (personal communication).
Following pre-clinical studies showing efficacy of CD19 CAR T cells against ALL, the first data showing efficacy in relapsed/refractory ALL patients were published, with CRs observed responses in two children and five adults in separate studies, including patients with chemorefractory disease. Of the 5 adult patients who completed treatment, all were minimal residual disease (MRD)-negative and four underwent subsequent allogenic stem cell transplant (SCT). One patient died in CR post allo-BMT due to a pulmonary embolus [26]. The pediatric report demonstrated the role of IL-6 blockade in controlling CRS, and also showed that a mechanism of relapse is CD19 escape with CR functional persistence [27]. Pediatric patient 1 experienced a durable CR that corresponded with CAR T cell expansion, had severe CRS relieved by the IL-6 receptor blocking agent tocilizumab, and remains in remission in B cell aplasia without SCT 6 years later (personal communication). Unfortunately patient 2, who had a CR one month after CD19 CAR infusion after failure to respond to blinatumomab, relapsed after two months with CD19- ALL [27].
Current FDA CAR T approvals
To date, two CAR T therapies have been granted three total FDA approvals for the treatment of patients with hematologic malignancies. The first approval - granted on August 30th, 2017 – was awarded to 4-1BB-based CD19 CAR T cell therapy tisagenlecleucel (CTL019, Kymriah, Novartis, Basel, Switzerland) for the treatment of patients up to 25 years of age with ALL that is refractory or in second or later relapse [10]. Patients enrolled in the phase II global ELIANA registration trial (NCT02435849) were between the ages of three and 23 (n = 75) and had not received prior anti-CD19 therapy. All patients who were not leukopenic (72/75 patients) received lymphodepletion regimen before receiving a median dose of 3.1 × 106 cells/kg CAR T cells. Data supporting FDA approval is listed in Table 1. At publication of trial results, the overall remission rate in patients who received infusions of CAR T cells was 81% (95% CI: 71–89), and the median duration of remission had not yet been reached [17].
On October 18th, 2017, the FDA granted a second CAR T therapy approval to the CD28-based CD19 CAR T cell product axicabtagene ciloleucel (Axi-cel, Yescarta, Kite Pharma/Gilead, Los Angeles, CA) for the treatment of patients with DLBCL who have not responded or have relapsed after two prior treatment regimens [9]. 101 patients (77 DLBCL, 24 other) in the phase I/II ZUMA-1 trial (NCT02348216) received autologous CAR T cells (2 × 106 cells/kg) after flu/cy lymphodepletion. Data supporting FDA approval is listed in Table 2. At the time of publication, an objective response rate (ORR) of 82% (95% CI: 73–89) was observed, median time to response was 1 month (95% CI: 0.8–6), and the median duration of response was 8.1 months (95% CI: 3.3 – no estimate). 18-month overall survival (OS) was 52% [13].
On May 1st, 2018, tisagenlecleucel gained a second FDA approval, this time for the treatment of adult patients with relapsed/refractory DLBCL [10]. This approval was based on results from the phase II JULIET clinical trial (NCT02445248) where 81 evaluable patients (age 22–76 years, median = 56 years) were treated with tisagenlecleucel (1.0 × 107–6.0 × 108 CAR T cells) after lymphodepletion chemotherapy or prior leukopenia. Data supporting FDA approval is listed in Table 2. At time of publication, patient population ORR was 53.1% (CR = 39.5%, PR = 13.6, 95% CI: 42–64, p < 0.0001), and 6-month OS was 64.5% (95% CI: 51.5–74.8). Anti-CD19 CAR T cells were detected in responder blood for up to 367 days post-infusion. In 99 evaluable patients, CRS incidence was 58% (23% grade 3–4), and NTX incidence was 21% (12% grade 3–4). No deaths were reported due to therapy [14].
Lymphodepletion regimens prior to CAR T therapy
Studies indicate that patient lymphodepletion prior to CAR T therapy can improve clinical outcomes by increasing CAR T cell expansion and persistence. Tisagenlecleucel and axi-cel clinical trials included lymphodepletion regimens prior to patient CAR T infusion [13, 14, 17]. By far the most commonly used lymphodepletion regimens include flu/cy, although at a variety of doses. The cyclophosphamide dose commonly used ranges from 900 mg/m2 to 3600/m2 total dose. While the ELIANA trial testing tisagenlecleucel efficacy in patients with ALL used flu/cy in almost all patients, the JULIET trial of the same product in DLBCL utilized individualized lymphodepletion strategies based on patient therapeutic response history, blood cell counts, and organ function [14, 17]. Patient response to tisagenlecleucel was consistent across the overall patient population. Concerning axi-cel, patients enrolled in the ZUMA-1 trial received a fixed dose of fludarabine (30 mg/m2) and cyclophosphamide (500 mg/m2) on days 5, 4, and 3 pre-infusion [13]. Of note, some trials forgo lymphodepletion in patients who are already leukopenic with no evidence of detriment.
CAR T therapy toxicities
Toxicities observed in patients who receive CAR T therapies usually present within days of first infusion. The two most common toxicities are CRS and NTX, either of which can be lethal [28].
Cytokine release syndrome (CRS)
Overview
CRS is a condition hallmarked by increased cytokine production and associated inflammation. CRS generally correlates with increased IFNƴ,GM-CSF, IL-10, and IL-6, as well as CAR T cell expansion [29]. CRS clinical presentation includes fever, nausea, anorexia, tachycardia and/or hypotension, cardiac dysfunction, renal impairment, and hepatic failure among other conditions [29, 30]. Higher disease burdens correlate with greater CRS severity, especially in ALL [11]. Anti-tumor activity of CAR T therapies, however, is not predicated by disease severity, as patients who do not experience CRS may still respond to therapy [11].
CRS grading
Multiple grading scales for CAR T-related CRS have been developed. Among the most widely used is a system that reflects the potential severity of CRS in some CAR T cell patients (Fig. X) [29]. Using this scale, grade 1 CRS symptoms include non-life threatening events including fever and nausea. Grade 2 symptoms include oxygen requirement < 40%, hypotension that responds to fluids or low dose vasopressor, and grade 2 organ toxicity. Grade 3 CRS symptoms include an oxygen requirement ≥40%, hypotension requiring high-dose or multiple vasopressors, grade 3 organ toxicity, and/or grade 4 transaminitis. Grade 4 CRS include life-threatening symptoms that require ventilator support or grade 4 organ toxicity, excluding transaminitis [29].
CRS treatment
CRS treatment algorithms may utilize CRS grading scales or clinical factors, and work is underway to try to harmonize both CRS grading and treatment approaches. Grade 1 CRS often requires supportive care. Treatment of grade 1 CRS with corticosteroids is not recommended due to potential inhibitory activity upon CAR T cells. Nonsteroidal anti-inflammatory drugs for fever are also not recommended due to potential risk of hemorrhage in thrombocytopenic and/or coagulopathic patients [29]. Some patients with Grade 2 CRS and essentially all patients with Grade 3/4 CRS are treated with the IL-6 receptor agonist tocilizumab. Clinical trial results from the University of Pennsylvania/ Children’s Hospital of Philadelphia, as well as the ELIANA trial indicate that CRS severity specifically correlated with patient IL-6 levels [17]. As a result, the FDA concurrently approved tocilizumab with tisagenlecleucel for the treatment of CAR T-associated CRS. Subsequent studies have confirmed tocilizumab efficacy in reversing CRS symptoms, as well as having little to no inhibitory effect upon CAR T therapeutic efficacy [28, 31]. A stepwise CRS treatment algorithm uses tocilizumab first line. If severe CRS continues (especially hypotension), a short course of corticosteroids can be used [30]. Axi-cel specifically recommends 1 mg/kg methylprednisolone or equivalent dexamethasone for grade 3 CRS, and 1000 mg methylprednisolone at grade 4 [9].
Neurotoxicity (NTX)
Overview
NTX, which can present as CAR T-related encephalopathy (CRES), includes a variety of neurological symptoms - including confusion, delirium, expressive aphasia, obtundation, myoclonus, and seizure. White matter degradation has also been observed in some severe cases [11, 29, 30, 32]. To date, causality of CRES is unknown, but studies suggest that cytokine secretion and subsequent breakdown of the blood-brain barrier may play a role [33]. Of note, patients treated with blinatumomab – a bi-specific anti-CD19/CD3 BiTE antibody – also have increased incidence of neurological toxicities similar to those observed in patients treated with CAR T [34].
Grading
The University of Texas MD Anderson Cancer Center has developed a grading scale for CRES [32]. All CRES grades include a score from the CARTOX 10-point neurological assessment, where 1 point is assigned for successful completion of a basic neurological function (Fig. X). Grade 1 CRES is classified as a patient with CARTOX scores 7–9. Grade 2 CRES includes patients with CARTOX scores 3–6. Grade 3 CRES includes patients with CARTOX scores 0–2, stage 1/2 papilledema with < 20 mmHg CSF opening pressure, and/or partial seizure or non-convulsive seizures that respond to benzodiazepine. Grade 4 CRES is considered critical with a CARTOX score of 0, stage 3/4/5 papilledema, > 20 mmHg CSF opening pressure, and generalized seizures, convulsive or non-convulsive epilepticus, or motor weakness after CAR T infusion [32].
CRES treatment
The above grading scale also has corresponding treatment recommendations (Fig. X) [32]. Grade 1 CRES treatment includes supportive care and consistent neurological evaluation. Grade 2 CRES also requires a neurological evaluation, as well as tocilizumab administration if symptoms are associated with CRS. Unlike CRS where tocilizumab has a clear role, the efficacy of tocilizumab in reversing CRES is not well established. Grade 3/4 CRES requires ICU transfer, and some centers utilize tocilizumab administration if associated with CRS. Corticosteroids - including dexamethasone or methylprednisolone - are also recommended for grade 3/4 CRES [32].
Other CAR T-associated adverse events
B cell aplasia
CD19 CAR T therapies can result in short or long-term patient B cell aplasia, which is also a marker of functional persistence of the CAR T cells [28]. Short-term B cell aplasia may not require treatment, while longer term B cell aplasia may require immunoglobulin replacement, especially in children.
Graft vs host disease (GVHD)
Patients who have had prior stem-cell transplant often retain full donor chimerism in the T cell compartment, so the “autologous” T cells obtained from the patient are usually of donor origin. CAR T clinical trials that supported FDA approvals did not enroll patients with active GVHD requiring systemic therapy [13, 14, 17]. In the absence of active GVHD, the donor origin cells obtained from the patient appear to be tolerated, and GVHD is either not seen or extremely rare.
Off-target toxicities
CAR constructs are designed to recognize specific markers, and healthy tissue presenting selected targets may be affected. Targets are optimized through pre-clinical research and early phase clinical trials.
Other clinical considerations for CAR T therapies
Treatment scheduling around stem cell transplant
Many studies have reported patients receiving hematopoietic stem cell transplant (HSCT) post-CAR T therapy to prevent relapse. Across these studies, durability of response between patients who went on to receive allo-HSCT vs those who did not are not statistically significant. These data indicate that CAR T therapy may be beneficial regardless of allo-HSCT consolidation [28, 31, 35, 36]. Whether a patient has MRD- status, however, may suggest a benefit for receiving allo-HSCT after CAR T therapy. One study assessed risk-of relapse post HSCT in patients with relapsed/refractory B-ALL who achieved MRD- after either anti-CD19 or anti-CD22 CAR T therapy. 24-month cumulative incidence of post-HSCT relapse of 25 evaluable patients was 13.5% (95% CI: 3.2–32.1), while 80% of patients who did not receive HSCT (n = 20) relapsed post-CAR T infusion [37]. Considering safety, incidence of graft vs host disease in patients who had prior CAR T therapy was consistent with historical rates of HSCT alone [37]. Future studies will need to determine whether CAR T therapy enhances response to HSCT, and whether specific CAR T therapies may be more likely to require HSCT post-infusion to prevent relapse.
Patient access to CAR T therapies
CAR T therapies require a significant logistical pipeline incorporating collection of patients leukocytes, engineering of CAR T cells, transportation of both the original and engineered cells, and patient infusion [38]. As of now, only specific quaternary care centers across the world are capable or certified to offer CAR T cell therapies to patients, and even fewer GMP cell manufacturing laboratories are capable of generating CAR T products. Center limitation presents two challenges. Time until CAR T therapy administration could become problematic as new FDA indications are granted or if, disregarding therapeutic cost, clinicians start to use therapies off-label in earlier disease settings.
There are very few treatment options available for relapsed/refractory pediatric ALL. Tisagenlecleucel is currently approved by the FDA for treating patients up to 25 years of age, and thus represents a critical option for this patient population [10]. Immune checkpoint inhibitors, for example, are usually not an option for pediatric patients, as they often require PD-L1 positivity or increased tumor mutational burden for on-label administration and efficacy. Checkpoint blockade, thus far, has shown limited efficacy in treating pediatric cancers [39].
While not fully within the scope of this manuscript, it is worth mentioning that price of CAR T therapies could become limiting, especially if clinicians start to recommend off-label treatments in earlier disease settings. One mechanism in place to relieve cost concerns is attached to tisagenlecleucel, in which the parent company Novartis will not charge patients who did not have evidence of response to treatment within one month post-infusion [40]. Because of rapid advancements in this field, it will also be imperative for clinicians to standardize treatment scheduling of CAR T therapies and inform insurance companies of new standard-of-care therapies to avoid incorrect patient charges.
Future advances for CAR T therapies
Improvements upon FDA approved CAR T strategies
Despite the recent success of CAR T therapies in the clinic, FDA approved therapies can be improved upon concerning increased efficacy and CAR T proliferation/persistence, lowering the potential for acquired resistance, and reducing the risk for severe toxicities. Current therapeutic strategies have demonstrated that CD19 is a viable target for elimination of various hematologic malignancies, but improvements concerning T cell phenotypic abundance, use of alternative costimulatory domains, and combining anti-CD19 CAR T cells with other therapies are currently being tested in numerous ongoing clinical trials. Among the most advanced CAR T therapy currently in development is lisocabtagene maraleucel (liso-cel, Juno Therapeutics/Celgene, Seattle, WA) – formerly known as JCAR017 – that is being evaluated for safety and efficacy in the phase 1 TRANSCEND study (NCT02631044) for the treatment of patients with relapsed/refractory DLBCL. Liso-cel utilizes an anti-CD19 second generation CAR construct with a 4-1BB costimulatory domain. In December 2017, 91 total patients (ECOG PS 0–2, no previous autologous stem cell transplant) were treated with liso-cel. The ‘core’ group of patients – patients with DLBCL and ECOG PS 0–1 – were separated into three dosage cohorts. Patients within the DL2 cohort were treated with 100 million cells (n = 29), patients within the DL1 cohort were treated with 50 million cells (n = 34), and 4 patients were treated with 50 million cells twice, with 2 weeks between infusions (n = 4). Across doses within the core group, 6 month ORR was 92%, with 80% of patients with CR at 3 months remaining in remission at 6 months. Across the trial population, CRS was observed in 35% of patients (1% grade 3–4) and 19% experienced NTX (12% grade 3–4) [41].
New disease settings for CAR T therapies
Multiple ongoing clinical trials are evaluating CAR T therapies in disease settings beyond B-ALL and DLBCL. Among the most advanced data to date concern CAR T therapies for the treatment of patients with multiple myeloma and chronic lymphocytic leukemia.
Multiple myeloma
B cell maturation antigen (BCMA) has been identified as a CAR target for the treatment of multiple myeloma and were first tested in a single-center clinical trial [42,43,44]. A different anti-BCMA CAR T-cell product known as bb2121 (Bluebird Bio/Celgene) has progressed to a multicenter clinical trial, and on November 16th 2017, the FDA granted a breakthrough designation to bb2121 for the treatment of patients with relapsed/refractory multiple myeloma [45]. Bb2121 is a second-generation CAR construct that contains an anti-BCMA antigen-recognition domain and an intracellular 4-1BB costimulatory domain [45]. BCMA – a member of the tumor necrosis factor receptor superfamily – is required for long-term survival of plasma cells and is often present on multiple myeloma cells [46]. Preliminary results from the phase I CRB-401 study (NCT02658929) involving bb2121 were presented in June 2017. In all, 21 patients (3 prior treatments or double refractory, ≥50% BCMA expression) underwent leukapheresis and subsequent cyclophosphamide/fludaribine lymphodepletion, and then received between 5 × 107 and 1.2 × 109 anti-BCMA CAR T cells. ORR across all patients and doses was 89% (75% PR, 27% CR, 95% CI: 65–99). All patients with CR were MRD-. DOR was > 134 days (95% CI: 7–361). Anti-BCMA CAR T cell expansion was observed, and CAR T cells were measurable in patient blood for up to 24 weeks post-infusion. 15 total cases of CRS were observed, with two grade 3 cases. No grade 3/4 NTX were observed [47]. An update in December 2017 reported an ORR of 94% and a 56% CR rate. Additionally, median PFS was not yet reached after 40 weeks of follow up. The phase 2 KarMMa trial (NCT03361748) is ongoing and will serve as the basis for regulatory submission to the FDA [48].
Similar to bb2121, LCAR-B38M – an anti-BCMA CAR T therapy (Legend/GenScript Biotech, Nanjing, China) – has been evaluated for efficacy in patients with multiple myeloma. Data presented in June 2017, 33/35 patients achieved CR, with 14/19 evaluable patients with at least four months of follow up remaining in remission [49]. LCAR-B38M is currently in phase 1/2 clinical trials (NCT03090689). Additionally, data concerning LCAR-B38M has fueled the development of the anti-BCMA CAR T therapy JNJ-68284528 (Janssen/Johnson & Johnson, Beerse, Belgium) that is currently being evaluated in a phase 1b/2 clinical trial (NCT03548207) for the treatment of patients with relapsed/refractory multiple myeloma [50].
Chronic lymphocytic leukemia
Among the first studies investigating anti-CD19 CAR T therapeutic efficacy was a case report treating a patient with CLL. Studies involving tisagenlecleucel for the treatment of patients with CLL reveal a 57% ORR (8/14) with 4 CR. Responses have been durable, with patients with CR remaining disease-free 40 months post-infusion [51]. Since this initial trial, over 60 patients with relapsed/refractory CLL have been treated with anti-CD19 CAR T cells at the University of Pennsylvania and response continues to be significant (personal communication). Interestingly, clinical trial data suggests that combination ibrutinib - a small molecule targeting Bruton’s tyrosine kinase on B cells – may improve tisagenlecleucel efficacy in treating patients with relapsed/refractor CLL. CR was observed in 11 patients at follow up (3–12 months) who received combination therapy. Additionally, no detectable CLL was observed in 10/11 patients at 3 months [52].
Acute myeloid leukemia
Phase 1 clinical trials assessing efficacy of various CAR T strategies in patients with acute myeloid leukemia (AML) are also underway. As CD19+ AML is considered rare, alternative antigens – including CD33, CD38, CD56, CD117, CD123, Lewis-Y, Muc-1, and NKGDL – are being considered as targets for developmental CAR T strategies [53,54,55,56]. Patient response has been observed in early results. In one example, six adult patients with relapsed AML were treated with anti-CD123 CAR T cells at two doses: 50 × 106 or 100 × 106 cells. In all, one patient receiving 50 × 106 cells experienced MRD-level disease response, two patients receiving 100 × 106 cells achieved CR, and two patients receiving 100 × 106 cells achieved PR [57].
Solid tumors
CAR T therapies offer significant promise in treating a variety of solid tumors, but many challenges concerning optimal cellular targets, tumor immune resistance, and toxicities will need to be resolved. We recognize the potential of this ongoing research, but further discussion of these treatments is beyond the scope of this manuscript.
Expanded indications for FDA approved anti-CD19 CAR T therapies
Axi-cel is also being tested in alternative disease settings. The most advanced data is from the phase 1/2 ZUMA-3 trial (NCT02614066) testing efficacy and safety in the treatment of adult patients with relapsed/refractory ALL (n = 23). At median follow-up (2.7 months), CR rate was at 71%, and 17 patients were MRD-. Grade 3/4 CRS was observed in 22% of patients, and grade 3/4 NTX occurred in 12% of patients. Two deaths were reported due disease progression. This trial is ongoing [58].
New CAR designs
Next-generation CAR T constructs are being designed to help overcome a variety of potential issues including elimination of pre-infusion leukodepletion, overcoming inhibitory factors within the tumor microenvironment, and reducing the risk of relapse. Additionally, CAR T cells constructs are being developed to target antigens other than CD19 on hematologic malignancies.
Targeting alternative antigens
CAR constructs targeting antigens other than CD19 are currently being developed. Among the most heavily studied alternative targets for the treatment of hematologic malignancies are CD20, CD22, and CD30. Anti-CD20 CAR T therapies in a phase 1/2a clinical trial (NCT01735604) have thus far demonstrated similar response rates (ORR = 81.8%) in patients with non-Hodgkin lymphoma (n = 11) compared to anti-CD19 CAR T therapies after 5 years of follow up [59]. Phase I clinical trial results assessing efficacy of anti-CD22 CAR T cells in treating patients with B-ALL who are naïve or resistant to anti-CD19 CAR T therapy reported a 73% complete remission rate in evaluable patients (11/15) [60]. Anti-CD30 CAR T therapies have also induced complete responses in patients with Hodgkin lymphoma (2/7) or anaplastic large cell lymphoma (1/2) in phase I clinical trials [61].
Bi-specific CAR T cells
While anti-CD19 CAR T therapies provide initial complete responses, a significant number of patients relapse, partially due to loss of the targeted antigen. As such, it has been hypothesized that CAR constructs that target more than one antigen may reduce the risk of relapse. Based on studies involving single targeted antigens, clinical trials are underway assessing efficacy of anti-CD19/CD22 (NCT03448393) and anti-CD19/CD20 bi-specific CAR T cells (NCT03271515) to overcome relapse due antigen loss, and to provide treatment options to patients with refractory disease to antigen-specific targeted therapies.
IL-12 secreting CAR T cells
Building upon second-generation CAR T constructs, it has been hypothesized that additional IL-12 – a cytokine normally generated from antigen-presenting cells that promotes CD8+ T cell activation through increased IFNƴ secretion – may further improve CAR T cell proliferation and anti-tumor activity, as well as assist in overcoming inhibitory factors associated with the tumor microenvironment [62]. Systemic IL-12 immunotherapies showed promising antitumor activity in pre-clinical models as a monotherapy and in combination with other agents. Clinical trials assessing antitumor efficacy of systemic IL-12, however, failed due to severe toxicities including hematologic and hepatic dysfunction [63,64,65]. As such, researchers constructed IL-12 secreting anti-CD19 CAR T cells – termed ‘armored’ CAR T cells – and showed enhanced proliferation and increased cytotoxicity compared to non-IL-12 secreting variants when exposed to cell-free ascites from human ovarian tumor-bearing mice [66].
Both pre-clinical and clinical data suggest that leukodepletion is important for effective CAR T cell antitumor activity. Studies are investigating whether the addition of an IL-12 secretion domain in CAR constructs may also eliminate the requirement for leukodepletion. In support, IL-12 secreting CAR T cells were able to clear lymphoma from lymphoreplete mice, while non-IL-12 secreting CAR T cells were unable to do so [67].
Targeting T cell metabolism
Towards the goal of improving efficacy and reducing toxicities of cancer immunotherapies, many research programs are working to better understand T cell metabolism and the impact it may have on differentiation, effector function, and interactions with cancerous cells in the tumor microenvironment [68, 69]. For example, one report notes that in vitro 4-1BB costimulation enhances T cell mitochondrial activity that promotes anti-PD-1 response [70]. As CAR T cells retain innate metabolic programming compared to their non-CAR variants, manipulation of CAR T cell metabolism is a potentially viable mechanism to increase effector function and promote proliferation/persistence. While studies are ongoing, preliminary in vitro experiments suggest that CAR T cells with a 4-1BB costimulatory domain generally rely upon oxidative metabolism and display enhanced persistence compared to CD28 CAR T cells that display glycolytic metabolic function [71].
Next-generation CAR T engineering
Using CRISPR in CAR T cell generation
Current CAR T cell engineering methods do not control for CAR gene localization within the T cell genome. Recent developments in Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) genetic techniques – derived from a bacterial anti-viral defense system – now allow for precise integration of genetic material into target cell genomes [72]. As such, studies are currently underway assessing efficacy of CRISPR-generated CAR T cells. One study notes that CRISPR/Cas9-driven CAR localization to the T cell receptor α constant (TRAC) genomic location allows for uniform CAR expression across all generated T cells, as well as increased cytotoxicity in a murine acute lymphoblastic leukemia model [73].
Safety switches
Multiple ‘safety switches’ are being tested in CAR constructs in order to reduce toxicities through more precise control of cell proliferation and activity. For example, one concept being tested incorporates caspase-9/human FK506-binding protein-hybrids that may promote CAR T cell apoptosis in the presence of a synthetic molecule that can be administered to patients after tumor eradication [74]. Other techniques include antibody-based control of CAR T cell activity, as well as the engineering of regulatory mechanisms to control CAR T cell gene expression via a secondary medication [75, 76].
Controlling CAR T product composition
Biomarkers are becoming increasingly important for evaluating patient prognosis and predicting immunotherapy success. As CAR T cells present biomarkers of their own, characterization of infusion product functionality and immune marker composition may help identify specific cellular mixes that allow for both anti-cancer activity as well as reduced toxicities. Liso-cel pre-infusion products, for example, are analyzed and subsequently balanced for CD4+ and CD8+ cells prior to CAR transfection [41].
Allogeneic ‘off the shelf’ CAR T cells
The development of universal, allogenic CAR T cells – cells engineered from a single donor source that can be used in multiple patients – could help expand CAR T cell therapy access, reduce time to first infusion, and provide product composition consistency towards reducing toxicity and increasing efficacy [77]. Multiple hurdles remain concerning this technology, most notably the ability to overcome potential graft vs host disease and rejection. Allogenic CAR T cells are currently being tested in phase I clinical trials.
Conclusion
CAR T cell therapies have offered significant promise in the treatment of patients with hematologic malignancies, providing a foundation for the development of treatment strategies for other cancers. FDA approvals for CAR T therapeutic development has moved from bench to bedside more quickly compared to other immunotherapies. While it is important to be able to offer these life-changing therapies to patients as soon as possible, many questions have been left unanswered for the field to discern along the way. Additionally, as toxicities are better understood and treatment scheduling and dosage is further optimized with clinical data, technologies will be continually refined and improved upon. It is clear that CAR T cell therapies are heavily entrenched in the future of cancer immunotherapy. With the unprecedented speed of this field, however, researchers and clinicians must remain vigilant in providing information to clinicians about the multiple caveats of this therapeutic class, allowing for the best patient outcomes possible.
Abbreviations
- ACT:
-
Adoptive cell therapy
- ALL:
-
Acute lymphoblastic leukemia
- BCMA:
-
B cell maturation antigen
- CAR:
-
Chimeric antigen receptor
- CI:
-
Confidence interval
- CLL:
-
Chronic lymphocytic leukemia
- CR:
-
Complete response
- CRES:
-
CAR T cell-related encephalopathy syndrome
- CRISPR:
-
Clustered Regularly Interspaced Short Palindromic Repeats
- CRS:
-
Cytokine release syndrome
- DLBCL:
-
Diffuse large B cell lymphoma
- FDA:
-
U.S. Food and Drug Administration
- Flu/cy:
-
Fludarabine/cyclophosphamide
- GM-CSF:
-
Granulocyte macrophage colony stimulating factor
- GMP:
-
Good manufacturing practice
- GVHD:
-
Graft vs host disease
- HSCT:
-
Hematopoietic stem cell transplant
- IFN:
-
Interferon
- IL:
-
Interleukin
- MHC:
-
Major histocompatibility complex
- MRD:
-
Minimal residual disease
- NTX:
-
Neurotoxicity
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PR:
-
Partial response
- SCT:
-
Stem cell transplant
- TCR:
-
T cell receptor
- TIL:
-
Tumor infiltrating lymphocytes
- TNP:
-
Trinitrophenyl
References
Perica K, Varela JC, Oelke M, Schneck J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med J. 2015;6(1):e0004.
Baggio L, Laureano AM, Silla L, Lee DA. Natural killer cell adoptive immunotherapy: coming of age. Clin Immunol. 2017;177:3–11.
Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15(7):e257–67.
Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–80.
Tang X, Liu T, Zang X, Liu H, Wang D, Chen H, Zhang B. Adoptive cellular immunotherapy in metastatic renal cell carcinoma: a systematic review and meta-analysis. PLoS One. 2013;8(5):e62847.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22.
Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. 2017;5:22.
Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–8.
YESCARTA Perscribing information. In.
KYMRIAH Perscribing information. In.
Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011.
Attaf M, Legut M, Cole DK, Sewell AK. The T cell antigen receptor: the Swiss army knife of the immune system. Clin Exp Immunol. 2015;181(1):1–18.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44.
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–54.
Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12(5):933–41.
Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18(2):413–20.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48.
Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O'Connor RS, Hwang WT, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563–71.
Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–102.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–9.
Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, Xue A, Goff SL, Yang JC, Sherry RM, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum Interleukin-15 levels. J Clin Oncol. 2017;35(16):1803–13.
Kochenderfer JN, Somerville RPT, Lu T, Yang JC, Sherry RM, Feldman SA, McIntyre L, Bot A, Rossi J, Lam N, et al. Long-duration complete remissions of diffuse large B cell lymphoma after anti-CD19 chimeric antigen receptor T cell therapy. Mol Ther. 2017;25(10):2245–53.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73.
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra138.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95.
Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–30.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28.
Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62.
Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7(12):1404–19.
Topp MS, Gokbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, Dombret H, Fielding AK, Heffner L, Larson RA, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224)):224ra225.
Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–38.
Shalabi H DC, Stetler-Stevenson M, et al.: Chimeric antigen receptor t-cell (CAR-T) therapy can render patients with all into PCR-negative remission and can be an effective bridge to transplant (HCT). . Presented at: the 2018 ASPHO conference; may 2-5, 2018; Pittsburgh, PA Poster 1017.
Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92–101.
Park JA, Cheung NV. Limitations and opportunities for immune checkpoint inhibitors in pediatric malignancies. Cancer Treat Rev. 2017;58:22–33.
Novartis: Novartis receives first ever FDA approval for a CAR-T cell therapy, Kymriah(TM) (CTL019), for children and young adults with B-cell ALL that is refractory or has relapsed at least twice. In.; 2017.
Abramson JS, Siddiqi T, Palomba ML, Gordon LI, Lunning MA, Arnason JE, Wang M, Forero-Torres A, Albertson T, Dehner C, et al. High durable CR rates and preliminary safety profile for JCAR017 in R/R aggressive B-NHL (TRANSCEND NHL 001 study): a defined composition CD19-directed CAR T cell product with potential for outpatient administration. Biol Blood Marrow Transplant. 2018;24(3):S25.
Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, Brudno JN, Stetler-Stevenson M, Feldman SA, Hansen BG, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128(13):1688–700.
Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, Stetler-Stevenson M, Salem D, Yuan C, Pavletic S et al: T Cells Genetically Modified to Express an Anti–B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol , 0(0):JCO.2018.2077.8084.
Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, Gress RE, Hakim FT, Kochenderfer JN. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048–60.
Berdeja JG LY, Raje N, et al. : Durable Clinical Responses in Heavily Pretreated Patients with Relapsed/Refractory Multiple Myeloma: Updated Results from a Multicenter Study of bb2121 Anti-Bcma CAR T Cell Therapy. . Presented at: ASH Annual Meeting and Exposition; Dec 9–12, 2017; Atlanta Abstract 740.
Tai YT, Anderson KC. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy. 2015;7(11):1187–99.
Berdeja JG, Lin Y, Raje NS, Siegel DSD, Munshi NC, Liedtke M, Jagannath S, Maus MV, Turka A, Lam LP, et al. First-in-human multicenter study of bb2121 anti-BCMA CAR T-cell therapy for relapsed/refractory multiple myeloma: Updated results. J Clin Oncol. 2017;35(15_suppl):3010 3010.
Raje NS BJ, Lin Y, et al.: bb2121 anti-BCMA CAR T-cell therapy in patients with relapsed/refractory multiple myeloma: Updated results from a multicenter phase I study. J Clin Oncol 2018;36 (suppl; abstr 8007).
Fan F, Zhao W, Liu J, He A, Chen Y, Cao X, Yang N, Wang B, Zhang P, Zhang Y et al: Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol 2017, 35(15_suppl):LBA3001-LBA3001.
Johnson J: Janssen Announces Initiation of Phase 1b/2 Clinical Development Program Evaluating JNJ-68284528 CAR-T Cells for the Treatment of Multiple Myeloma. In.; 2018.
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139.
Gill S, Frey NV, Hexner EO, Lacey SF, Melenhorst JJ, Byrd JC, Metzger S, Marcus T, Gladney W, Marcucci K et al: CD19 CAR-T cells combined with ibrutinib to induce complete remission in CLL. J Clin Oncol 2017, 35(15_suppl):7509–7509.
Tasian SK. Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: how far up the road have we traveled? Ther Adv Hematol. 2018;9(6):135–48.
Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol. 2012;1(1):36.
Tsuchiya H, ElSonbaty SS, Nagano K, Watanabe M, Migita M, Mitsubuchi H, Kaneko Y, Matsuda I. Acute myeloblastic leukemia (ANLL-M2) with t(8;21)(q22;q22) variant expressing lymphoid but not myeloid surface antigens with a high number of G-CSF receptors. Leuk Res. 1993;17(4):375–7.
Khalil SH, Jackson JM, Qari MH, Pyle H. Acute myeloblastic leukemia (AML-M2) expressing CD19 B-cell lymphoid antigen without myeloid surface antigens. Leuk Res. 1994;18(2):145.
Budde L, Song JY, Kim Y, Blanchard S, Wagner J, Stein AS, Weng L, Del Real M, Hernandez R, Marcucci E, et al. Remissions of acute myeloid leukemia and Blastic Plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial. Blood. 2017;130(Suppl 1):811 811.
Shah BD OO, Baer MR, et al.: Outcomes of patients (pts) treated with prior blinatumomab (Blin) in ZUMA-3: A study of KTE-C19, an anti-CD19 chimeric antigen receptor (CAR) t cell therapy, in adult pts with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). Abstract #7006 Presented at the 2018 ASCO annual meeting, June 2, 2018; Chicago, IL.
Zhang WY, Wang Y, Guo YL, Dai HR, Yang QM, Zhang YJ, Zhang Y, Chen MX, Wang CM, Feng KC, et al. Treatment of CD20-directed chimeric antigen receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an early phase IIa trial report. Signal Transduct Target Ther. 2016;1:16002.
Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, Wolters P, Martin S, Delbrook C, Yates B, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–8.
Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, Bilgi M, Wu MF, Liu H, Grilley B, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest. 2017;127(9):3462–71.
Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. OncoImmunology. 2015;4(3):e994446.
Lenzi R, Edwards R, June C, Seiden MV, Garcia ME, Rosenblum M, Freedman RS. Phase II study of intraperitoneal recombinant interleukin-12 (rhIL-12) in patients with peritoneal carcinomatosis (residual disease < 1 cm) associated with ovarian cancer or primary peritoneal carcinoma. J Transl Med. 2007;5:66.
Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, Palmer DC, Reger RN, Borman ZA, Zhang L, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70(17):6725–34.
Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, Nahvi AV, Ngo LT, Sherry RM, Phan GQ, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21(10):2278–88.
Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep. 2017;7(1):10541.
Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully Lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics. 2018;8:41–51.
Le Bourgeois T, Strauss L, Aksoylar HI, Daneshmandi S, Seth P, Patsoukis N, Boussiotis VA. Targeting T cell metabolism for improvement of Cancer immunotherapy. Front Oncol. 2018;8:237.
Herbel C, Patsoukis N, Bardhan K, Seth P, Weaver JD, Boussiotis VA. Clinical significance of T cell metabolic reprogramming in cancer. Clin Transl Med. 2016;5(1):29.
Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, Watkins SC, Delgoffe GM. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. 2018.
Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, Patel PR, Guedan S, Scholler J, Keith B, et al. Distinct signaling of Coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–90.
Chira S, Gulei D, Hajitou A, Zimta AA, Cordelier P, Berindan-Neagoe I. CRISPR/Cas9: transcending the reality of genome editing. Mol Ther Nucleic Acids. 2017;7:211–22.
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gonen M, Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–7.
Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, Heslop HE, Spencer DM, Rooney CM. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105(11):4247–54.
Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113(4):E459–68.
Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, Durett AG, Dakhova O, Savoldo B, Di Stasi A, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood. 2015;125(26):4103–13.
Ruella M, Kenderian SS. Next-generation chimeric antigen receptor T-cell therapy: going off the shelf. BioDrugs. 2017;31(6):473–81.
Acknowledgements
The authors acknowledge Peter J. Intile, PhD at the Society for Immunotherapy of Cancer for medical writing support and development of this manuscript. The authors also thank SITC staff for administrative support.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
MMB, MVD, RJB, JNK, SSN, MVM, DLP, DGM, SAG, CLM, CHJ, and MRB participated in drafting, writing, and reviewing the manuscript. MMB and MRB participated in figure selection. All authors approved the final version of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
JNK has received research funding from Kite Pharma and Bluebird Bio. He has also received royalties from intellectual property interests through Kite Pharma, Bluebird Bio, and Novartis. SSN has received research funding from Kite Pharma, Merck, Bristol-Myers Squibb, Cellectis, Poseida, Acerta, Karus. He has also served as a consultant for Kite Pharma, Novartis, Unum Therapeutics, Celgene, and Merck. MVM has received consulting fees from Novartis, Juno, and Kite Pharma, and has intellectual property interests through the University of Pennsylvania. DLP has received research funding from Novartis, and has served on advisory boards for Kite Pharma and Bellicum. In addition, Dr. Porter reports intellectual property interests through the University of Pennsylvania, and that his spouse is employed by Genentech. DGM has received research funding from Juno Therapeutics and Kite Pharma, and has received personal fees for serving on the scientific advisory board from Celgene, BioLink RX, Eureka, Roche/Genentech, Kite Pharma/Gilead, Novartis, Bristol-Myers Squibb, Immunogen, and Seattle Genetics. In addition, he has a pending patents related to cellular therapies. SAG has served on the SCC for Novartis and on the scientific advisory board for TCR2 Therapeutics, Eureka Therapeutics, Adaptimmune, and Cellectis/Servier. CLM has received personal fees for serving on the scientific advisory board for Unum Therapeutics, Glaxo-Smith Kline, Adaptimmune, Servier/Pfizer, Vor Pharmaceuticals, Apricity Health, and Allogene. In addition, she has intellectual property interests through Juno Therapeutics, as well as multiple pending patents related to CAR T therapies. CHJ has received research funding and has intellectual property interests through Novartis. He is also a scientific founder of Tmunity Therapeutics and reports stock holdings. MRB has served as a clinical investigator and advisory board member for Novartis and Juno Therapeutics. He has also served as a clinical investigator, advisory board member, and on the Speakers Bureau with honoraria for Kite Pharma. MMB, MVD, and RJB declare no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Boyiadzis, M.M., Dhodapkar, M.V., Brentjens, R.J. et al. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. j. immunotherapy cancer 6, 137 (2018). https://doi.org/10.1186/s40425-018-0460-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40425-018-0460-5