
Abstract
Multiple myeloma (MM) is a hematological cancer characterized by the abnormal proliferation of plasma cells. Initial treatments often include immunomodulatory drugs (IMiDs), proteasome inhibitors (PIs), and monoclonal antibodies (mAbs). Despite salient progress in diagnosis and treatment, most MM patients typically have a median life expectancy of only four to five years after starting treatment. In recent developments, the success of chimeric antigen receptor (CAR) T-cells in treating B-cell malignancies exemplifies a new paradigm shift in advanced immunotherapy techniques with promising therapeutic outcomes. Ide-cel and cilta-cel stand as the only two FDA-approved BCMA-targeted CAR T-cells for MM patients, a recognition achieved despite extensive preclinical and clinical research efforts in this domain. Challenges remain regarding certain aspects of CAR T-cell manufacturing and administration processes, including the lack of accessibility and durability due to T-cell characteristics, along with expensive and time-consuming processes limiting health plan coverage. Moreover, MM features, such as tumor antigen heterogeneity, antigen presentation alterations, complex tumor microenvironments, and challenges in CAR-T trafficking, contribute to CAR T-cell exhaustion and subsequent therapy relapse or refractory status. Additionally, the occurrence of adverse events such as cytokine release syndrome, neurotoxicity, and on-target, off-tumor toxicities present obstacles to CAR T-cell therapies. Consequently, ongoing CAR T-cell trials are diligently addressing these challenges and barriers. In this review, we provide an overview of the effectiveness of currently available CAR T-cell treatments for MM, explore the primary resistance mechanisms to these treatments, suggest strategies for improving long-lasting remissions, and investigate the potential for combination therapies involving CAR T-cells.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is still the second most common hematological cancer globally, accounting for about 35,000 diagnoses in the United States and 590,000 worldwide each year, despite years of research and development into better understanding cancer mechanisms and treatment approaches [1, 2]. This aggressive bone marrow (BM) cancer arises from abnormal plasma cells (PCs), which are differentiated B cells [3]. Finding innovative treatments for MM patients who do not respond to standard therapies like IMiDs, PIs, and anti-CD38 monoclonal antibodies is a significant challenge. While the treatment of relapsed or refractory (R/R) MM is evolving to overcome tumor immune evasion mechanisms to improve patient survival, CAR T-cell therapies have shown clinical benefits for blood cancer patients, with six CAR-T products approved by the US FDA since 2017 [4,5,6,7,8,9]. Notably, Idecabtagene Vicleucel (ide-cel, bb2121, ABECMA) and Ciltacabtagene autoleucel (Cilta-cel, LCAR-B38M, JNJ-68284528, JNJ-4528, CARVYKTI), both approved BCMA-directed CAR-Ts, have demonstrated favorable responses in R/R MM patients. However, despite the prolonged responses of ide-cel and cilta-cel in some patients, a significant number of them eventually face relapse, typically with a response duration of approximately 8.6 months and 34.9 months, respectively [8,9,10,11,12,13].
This recurrence may be caused by factors such as inherent limitations in T-cell availability and persistence, alterations in T-cell features during treatment, and the nature of the disease, including changes in disease characteristics during treatment [14]. To tackle these issues, CAR-T manufacturing for MM patients has evolved through novel approaches aimed at optimizing and accelerating CAR-T production, reducing costs, and producing cells with a less differentiated phenotypethan conventional CAR T-cells. Additionally, engineering allogeneic T cells as a universal source to express CARs and utilizing CAR expression on other immune cells, such as natural killer (NK) cells, have been explored in various clinical trials [14, 15]. Furthermore, addressing disease features such as variability or shedding of antigen expression on myeloma cells, the presence of an immunosuppressive tumor microenvironment, and difficulties in CAR-T penetration into tumor cells are important issues to improve CAR T-cell efficacy [16, 17]. So, research has explored new designs of CARs, such as adopting dual or multi-specific antigen-targeting receptors, integrating diverse costimulatory domains, and making genetic alterations to co-express cytokines or inhibit intracellular exhaustion signals [18, 19].
Moreover, CAR T-cell treatments often come with undesirable side effects, including cytokine release syndrome (CRS), neurotoxicity, and cytopenia. Approaches to alleviate these effects include combining immunosuppressive drugs or developing innovative direct control systems. Additionally, there are ongoing explorations into creating CAR T-cells utilizing fully humanized monoclonal antibodies (mAbs) to minimize immunogenicity [20, 21].
Consequently, a profound understanding of the therapeutic potential of CAR T-cells, recognition of MM pathogenesis, and development of supplementary therapeutic agents can enhance the effectiveness of transplanted CAR T-cell therapy for MM patients. This article will elucidate various tactics utilized by myeloma cells to counteract the efficacy of CAR-T therapies and delve into advanced approaches to confront these challenges.
Efficacy and challenges of CAR T-cell therapy in MM clinical trials
CAR T-cell trials for hematological malignancies are more numerous than those for solid tumors [22]. MM is the third most common target for CAR T-cell therapy after B-NHL and ALL [22]. Most clinical trials included both genders, with an average age of 18 and 72, focusing on evaluating anti-tumor efficacy, persistence, and safety profile [22].
Several factors affect the effectiveness of CAR T-cell therapy for MM, which can be classified into three main categories: CAR T-cell manufacturing and administration processes, disease characteristics, and clinical considerations before or after CAR T-cell treatment (Fig. 1). The subsequent sections investigate in extensive detail the clinical trials conducted thus far on CAR T-cell therapy in the realm of MM disease.

Overview of Effectiveness and Challenges in CAR T-cell Therapy for Multiple Myeloma. A Manufacturing and Administration of CAR T-cells: This section highlights strategies for optimizing production and functionality, innovations in genetic modification, allogeneic CAR T-cell development, and early therapy implementation to ease healthcare system burdens. B Multiple myeloma Characteristics in CAR T-cell Therapy: It addresses the challenges posed by the heterogeneous nature of the disease and the immunosuppressive tumor microenvironment. Solutions include expanding antigen targets, using gamma-secretase inhibitors, surmounting immune checkpoints, and improving CAR T-cell trafficking. C Clinical Considerations and Safety Management: This part emphasizes the importance of managing CAR T-cell toxicity and enhancing safety features. It suggests combining pharmacological agents and supplementary treatments to improve overall therapy outcomes
Manufacturing and administration of CAR T-cells
Autologous CAR T-cell manufacturing
The standard process of making autologous CAR T-cells entails activating T cells from patients, genetically modifying them with CARs using retroviruses or lentiviruses, and expanding these cells ex vivo for at least two weeks before reintroducing them into the patient [23, 24]. CARs have an extracellular single-chain variable fragment (ScFv) for tumor antigen recognition without relying on major histocompatibility complex (MHC) restricted antigen presentation. Most CAR constructs incorporate at least one intracytoplasmic costimulatory domain, often called “second-generation,” such as CD28, 4-1BB, ICOS, or OX40, along with CD3ζ. Specific CAR designs incorporate additional costimulatory domains, cytokine receptors, or safety switches into the CAR structure, commonly termed “third- or fourth-generation CARs [25, 26]. Ide-cel and cilta-cel are both second-generation autologous CAR T-cell products. They employ the 4-1BB costimulatory domain, which has demonstrated effectiveness in the proliferation and persistence of CAR-Ts compared to other options. Both products utilize a third-generation lentiviral vector for CAR gene transfer and follow a similar approach to CAR T-cell expansion [9, 10].
Enhancing CAR design and CAR T-cell production to optimize persistence and functionality
During the initial phases of CAR T-cell therapy development, there was a belief that the sheer quantity of CAR T-cells administered would be a crucial factor in the therapy’s effectiveness. However, it has become apparent that beyond a certain point, the absolute number of infused CAR-Ts doesn’t directly correspond to their persistence and therapeutic success [27]. Consequently, researchers are exploring new methods in CAR T-cell production and administration to improve their longevity and efficacy.
In the case of MM patients, many are older and have lower T-cell counts and an altered CD4/CD8 ratio before undergoing CAR T-cell therapy, which can affect their T cells’ fitness. To address this issue, several clinical trials adjusted the CD4/CD8 ratio to 1:1 in the case of BCMA-directed CAR-Ts, such as orvacabtagene autoleucel (Orva-cel; JCARH125) (NCT03430011), or after gene transfer in the case of FCARH143 (NCT03338972) [28, 29] (Table 1).
Furthermore, a significant limitation of CAR T-cell therapy stems from the restricted lifespan of the transplanted T cells, a characteristic mainly linked to their inherent properties and the absence of memory attributes [24]. Current research indicates that augmenting CAR T-cell cultures with recombinant interleukins such as IL-5, IL-7, IL-8, IL-12, IL-15, IL-21, IL-23, and IL-36γ can enhance their persistence and promote a higher ratio of memory cells compared to effector cells [40]. In a preliminary clinical trial, a fourth-generation CAR T-cell targeting BCMA, which expresses IL-7 and CCL19 (known as 7 × 19 CAR T-cells), demonstrated superiority over traditional second-generation CAR T-cells in terms of expansion, accumulation of memory cells, migration, and cytotoxicity (NCT03778346) [41].
Recently, a phase I open-label trial (NCT04136756) investigated NKTR-255 in patients with R/R MM who had progressed after prior CAR-T therapy. NKTR-255 is an experimental IL-15 receptor agonist designed to boost Natural Killer (NK) cells and memory CD8 + T cells [42]. Accordingly, some clinical trials, including anti-BCMA CAR-Ts like Descartes-08 (NCT03448978, NCT04816526) and Descartes-11 (NCT03994705, NCT04436029), employed CD8 + cells, while P-BCMA-101 (NCT03288493, NCT03741127) and P-BCMA-ALLO1 (NCT04960579) utilized the T stem cell memory (Tscm) phenotype in their approaches [36, 37, 43]. Bb21217 also used the same CAR construct as ide-cel (bb2121); however, a PI3K inhibitor (bb007) was added to the ex vivo culturing process to enhance the memory-like T cell subpopulations (NCT03274219) [44]. The clinical pursuit of bb21217 showed an overall response rate (ORR) of 55%, which led to the completion of its development [30] (Table 1).
Transforming CAR T-cell production with fast-track innovations for patients with progressive diseases
The duration required for CAR T-cell production and the type of protocol used in ex vivo settings directly impact T cells’ vital outcomes such as cytotoxicity, cytokine production against target cells, persistence, and the prevalence of more resilient phenotypes like naïve, memory, and stem cell memory in T cells [23]. Leveraging the unique ability of lentiviral vectors to integrate into non-dividing cells, researchers have developed a highly efficient method to accelerate the production of CAR T-cells. This groundbreaking approach has slashed the manufacturing timeline from weeks to just one to two days, resulting in rapid CAR T-cell production with improved growth and reduced exhaustion [45, 46].
GC012F, a dual-targeting CAR-T product for BCMA/CD19 enabled by FasTCAR technology, is currently undergoing investigation in a Phase I trial conducted across multiple centers for MM in China (NCT04236011, NCT04182581, NCT04617704, NCT04935580, ChiCTR2100047061) [47]. Phase Ib/II trials for GC012F are also underway in the US and China (NCT05850234) [48, 49].
Additionally, CC-98633/BMS-986354, an enhanced version of orva-cel, a lentiviral CAR T-cell product that targets BCMA, employs the NEX-T approach to shorten the manufacturing period to 5 to 6 days. It is presently under evaluation in an ongoing phase I clinical trial (NCT04394650). Clinical studies have demonstrated that BMS-986354 is a less mature CAR T-cell product with enhanced effectiveness compared to Orva-cel, making CAR T-cell therapy more accessible and suitable for patients with progressive diseases [35].
PHE885, a fully human BCMA CAR T-cell product, is also rapidly manufactured with the T-Charge™ platform (< 2 days). It is currently undergoing Phase I trial evaluation (NCT04318327) to assess CAR T-cell expansion within the patient’s body [34, 50] (Table 1).
Advancements in virus-free genetic modification methods: enhancing safety and cost efficiency
Due to the complexity, extended duration, and high costs associated with viral vectors, there is a concerted effort to explore virus-free genetic modification methods in CAR T-cell manufacturing. These methods include transposon systems like PiggyBac (PB) and Sleeping Beauty (SB), as well as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), short/small hairpin RNA (shRNA/Hairpin) vectors, and clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 9 (CRISPR/Cas9). Additional approaches include ARCUS, a specific variant of the CRISPR/Cas9 gene editing system that serves as a fully synthetic enzyme derived from the natural homing endonuclease I-CreI, and Cas-CLOVER, which utilizes a dual-guided system to create double-strand cuts upon dimerization with nuclease Clo051. All of these methods have demonstrated gene edition efficiency in CAR T-cell manufacturing [51, 52].
In the pursuit of enhancing CAR T-cell production quality, a novel category of CAR-Ts known as Descartes-08, derived from anti-BCMA mRNA, has been developed to reduce excessive in vitro proliferation time. The therapy has been evaluated in two clinical trials, identified as NCT03448978 and NCT04816526. The former has completed a phase I/II trial, while the latter has terminated a recent phase II trial.. Further, Descartes-11, an optimized and humanized version of Descartes-08, is under clinical trials (NCT03994705, NCT04436029) [43].
Additionally, P-BCMA-101 is a fully humanized anti-BCMA CAR T-cell employing a novel anti-BCMA VCAR with a non-immunoglobulin Centyrin scaffold produced using the transposon system. These VCARs, smaller than immunoglobulin-based CARs, offer advantages such as enhanced binding affinity, improved stability, reduced immunogenicity, and cost-effective production. P-BCMA-101 terminated a phase I trial (NCT03288493) in heavily treated R/R MM patients, achieving a 57% ORR [36]. A 15-year follow-up study (NCT03741127) has been initiated to delve deeper into the product’s efficacy and safety. Leveraging P-BCMA-101 data, an allogeneic CAR-T, P-BCMA-ALLO, using a similar PB construct was created and demonstrated excellent clinical safety and effectiveness in a phase I trial (NCT04960579) [36, 37, 52] (Table 1).
Innovations in allogeneic CAR T-cell development: safety enhancements and accessible off-the-shelf solutions
A group of ongoing research is dedicated to developing allogeneic CAR T-cell products that can be readily accessible “off the shelf” to patients with relapsed or refractory conditions. Such patients often confront rapid disease progression during the production of CAR-Ts or have diminished T-cell counts, rendering the receipt of autologous CAR T-cell therapy both time-consuming and financially burdensome. These limitations may result in missed opportunities for CAR T-cell treatment. To mitigate the risk of graft-versus-host disease (GvHD) and to enhance the cost-effective and expedited worldwide production and prescription procedures, cutting-edge genetic editing techniques are employed. These techniques either deactivate specific genes, such as the T cell receptor (TCR) αβ or γδ chains and MHC in T cells derived from healthy, universal donors, or introduce specific genes like CARs [51,52,53].
UCARTCS1A, the pioneering allogeneic CAR-T cell therapy in clinical development for MM, has eliminated both the SLAMF-7 antigen and TCR from T-cell surfaces by utilizing TALEN technology. This process also involved the integration of the SLAMF-7 CAR construct, and phase I trials for this therapy have been terminated (NCT04142619) [54]. Other treatments, ALLO-715 (NCT04093596) and ALLO-605 (NCT05000450), which are modified allogeneic anti-BCMA CAR T-cells, use TALEN technology to deactivate the T-cell receptor alpha constant gene (TRAC) and CD52. They are administered with ALLO-647, an anti-CD52 mAb, allowing for selective and temporary lymphodepletion before CAR-T therapy [51, 55, 56].
Moreover, P-BCMA-ALLO1 utilized Cas-CLOVER to disrupt β2 microglobulin (B2M), a component of MHC-I, as well as the TCRβ chain in T cells (NCT04960579) [37, 52]. In a terminated trial, PBCAR269A employed ARCUS technology to introduce TCR knock-outs and CAR T knock-ins for allogeneic BCMA-CAR T cells (NCT04171843).
BCMA-UCART utilized CRISPR/Cas9 (NCT03752541), while CB-011 employs CRISPR-Cas12a (NCT05722418) to target TRAC and MHC-I [57, 58]. In a similar vein, CTX120 is generated using the CRISPR/Cas9 system to remove TCR and MHC-I, followed by the specific insertion of the CAR at the TRAC locus using an AAV vector (NCT04244656) [59]. Furthermore, CYAD-211 is developed using shRNA/Hairpin and is currently under evaluation in the ongoing Phase I exploratory trial IMMUNICY-1 (NCT04613557) [39, 52] (Table 1).
Besides, double-negative T cells (DNT) are mature T cells that lack the CD4 and CD8 co-receptors on their surface. DNTs, recognized for their suppressive capabilities and non-specific antigen recognition, have demonstrated the ability to suppress GVHD independent of MHC limitations, indicating their potential role in allogeneic transplantation scenarios [60]. These cells have been studied in the context of allogeneic transplantation and cancer therapy, and they have been shown to persist in recipients for at least four weeks alongside conventional T cells without eliciting cytotoxic responses [61, 62]. Allogeneic double-negative CAR-T cells have demonstrated robust efficacy in inhibiting tumor growth in mouse models of blood cancers, suggesting their potential as a novel source for “off-the-shelf” CAR-T cell therapy [60].
Natural killer (NK) cells also possess inherent properties that make them suitable candidates for allogeneic cell therapy applications. Their ability to recognize and eliminate stressed or transformed cells without prior sensitization, coupled with a reduced risk of GVHD, positions them as promising targets for developing off-the-shelf allogeneic immunotherapies [63]. Several clinical trials are currently underway to evaluate BCMA-targeted CAR-NK cells for the treatment of multiple myeloma, such as NCT05652530, NCT05008536, NCT03940833, NCT06045091, and NCT05182073 [64, 65] (Table 1).
Earlier employment CAR T-cell therapy in the treatment continuum to alleviate healthcare delivery system burden
In the most recent clinical trials, CAR T-cell therapy is typically administered after multiple rounds of prior treatments for patients with R/R MM. However, the efficacy of successive therapies tends to diminish, with decreasing response rate, shallower depth of response, and shorter times to disease progression. Several factors contribute to this decline, including the development of resistance mechanisms, accumulation of treatment-related toxicities, selection of treatment-resistant cancer cell populations, and compromised T cell function due to prior therapies [23]. These phenomena pose challenges for implementing CAR-T treatment in later stages.
Cilta-Cel and Ide-cel are approved for myeloma patients undergoing disease progression after standard treatment with PIs, IMiDs, and anti-CD38 mAb. These approvals are based on the requirement that patients must have been exposed to triple-class therapy and received three or four prior lines of treatment in the EU and US, respectively [66]. On April 4, 2024, a significant modification to the approved Risk Evaluation and Mitigation Strategy (REMS) for Ide-cel was approved to reduce the burden on the healthcare delivery system. This modification applies to the treatment of adult patients with RR/MM who have undergone at least two prior therapies.
Additionally, the Phase I (THINK) study (NCT03018405) is currently evaluating the effectiveness of CYAD-01 without prior chemotherapy or lymphodepletion in MM patients, aiming for earlier employment of CAR T-cells [31, 67]. Similarly, phase II clinical trials for Descartes-08 (NCT04816526) and Descartes-11 (NCT04436029), along with completed safety assessments in the KarMMa-4 trial for ide-cel (NCT04196491), have been undertaken to evaluate the effectiveness of these interventions for individuals with newly diagnosed or high-risk multiple myeloma [65] (Table 1).
Multiple myeloma characteristics in CAR T-cell therapy
Heterogeneous nature and ability of BCMA to escape
Some particular antigens expressed on plasma cells can efficiently recruit specific CAR-Ts for MM. BCMA (CD269/TNFRSF17), with near-universal expression in MM, is the most intensively studied target for MM CAR T-cell treatments. One of the barriers to the success of CAR-T therapy is antigen heterogeneity, which interferes with T lymphocyte recognition and reduces the efficacy of the treatment [68]. The variable distribution of BCMA on myeloma cells and BCMA’s ability to escape immune cells is associated with resistance to current anti-BCMA CAR T-cell therapies. However, the underlying mechanisms of this phenomenon are still poorly understood [69].
Inherently, BCMA cleaved by gamma-secretase (a membrane-bound protease) can be released into the circulatory system or transferred spontaneously from tumors to T cells by a process known as trogocytosis, which causes T-cell fratricide (self-cytotoxicity) [70] (Fig. 2). This phenomenon may increase the risk of disease recurrence by making it more difficult for anti-BCMA CAR-Ts to find malignant cells [68, 71]. Consequently, the BCMA expression thresholds are currently being debated, and various results have been published regarding the use of BCMA expression thresholds as inclusion criteria for anti-BCMA CAR trials (NCT02546167, NCT03338972) [70, 72]

BCMA Transfer Mechanism in CAR T Cell Therapy. A Direct Contact: CAR T cells engineered to target BCMA form an immunological synapse with tumor cells, facilitating molecular exchange. B Trogocytosis: Membrane fragments containing BCMA are transferred from tumor cells to CAR T cells during synapse formation via trogocytosis, enhancing T cell activation. C BCMA Recognition: Transferred BCMA molecules are recognized by CARs on T cell surfaces, initiating T cell activation and tumor cell destruction. D Implications: While BCMA transfer boosts T cell killing, trogocytosis may inadvertently lead to fratricide, reducing therapy efficacy
Furthermore, one primary process of MM recurrence and refractoriness is the reduction of antigens or their disappearance under therapeutic pressure. While analyzing ide-cel, researchers discovered new resistance mechanisms of BCMA escape. They observed the downregulation of BCMA through non-truncating missense mutations or in-frame deletions in the extracellular domain of BCMA, in addition to tumor intrinsic resistance mechanisms that occurred at relapse after anti-BCMA CAR T therapy [73]. While both alleles were present before CAR T therapy, the majority of the MM cells displayed heterozygous deletion of the BCMA locus-containing region of chr16p together with nonsense mutation of the other allele, resulting in a biallelic suppression of BCMA [74]. So, defining tumor phenotypes before and after treatment may correlate with different clinical observations in the future [73]. Upon progression, BCMA loss might play a role in preventing the reapplication of the same CAR-Ts. Accordingly, cilta-cel’s unique structural design, with two tandemly arranged BCMA-targeting VHH (Variable heavy chain of heavy chain only antibody found in camelids) domains recognizing two different epitopes of antigens, exhibited a considerable therapeutic impact in R/R MM patients [10, 75, 76].
In the pivotal phase 2 KarMMa trial (NCT03361748), participants who received ide-cel infusion demonstrated an ORR of 73%, a median PFS of 8.6 months, and a median overall survival (OS) of 24.8 months [8, 77]. Meanwhile, the phase I/II CARTITUDE-1 trial (NCT03548207) demonstrated that cilta-cel delivered an impressive 98% ORR with a median PFS of 34.9 months [8, 11, 78]. In phase 3 trials, specifically CARTITUDE-4 (NCT04181827) and KarMMa-3 (NCT03651128), updated data was used to evaluate ide-cel and cilta-cel with a control arm, although these trials had different designs and patient populations. The results once again confirmed the effectiveness of ide-cel and cilta-cel treatments, with a hazard ratio for PFS of 0.49 for ide-cel [95% CI, 0.38–0.65; P < 0.001], and 0.26 for cilta-cel [95% CI, 0.18–0.38; P < 0.001] [12, 13, 79].
Expanding antigen targets to address heterogeneity and overcome BCMA evasion
Due to the limitations associated with targeting BCMA, numerous clinical trials are currently underway to investigate the incorporation of various intriguing antigens into CART cell therapy in MM therapy. These include some antigens such as signaling lymphocyte activation molecule family-7 (SLAMF-7) (expression in 95% MM), CD138 (expression in 90–100% MM), CD38 (expression in 80–100% MM), CD56 (expression in 70–90% MM), Kappa (κ) light chain (expression in 35% MM) as well as overexpressed antigens like natural-killer group 2 member D (NKG2D), G protein-coupled receptor class C group 5 member D (GPRC5D), integrin‑Beta7, MMG49, CD44v6, CD74, NY-ESO-1, TnMUC1, APRIL, TACI and CD19 [71, 80] (Table 2).
Moreover, the exploration of new targets, such as Fc receptor-homolog 5 (FcRH5) (also known as FcRL5, IRTA2, or CD307), can potentially expand the repertoire of CAR-T therapies for MM [92]. The following sections provide an overview of various myeloma antigens extensively investigated, either as single therapies or in combination as dual or multi-target strategies. These approaches are designed to address resistance issues observed in BCMA-targeted CAR T-cell therapies.
Targeting SLAMF-7
SLAMF-7, also known as CS1, is found in cancer-associated fibroblasts (CAFs), the predominant immunosuppressive components in the tumor microenvironment that promote MM cell proliferation [93]. Autologous SLAMF-7 targeting CAR-Ts (NCT04499339, NCT03958656, NCT03710421), allogeneic SLAMF-7 targeting CAR-Ts (NCT04142619), and BCMA/SLAMF-7 targeting CAR-Ts (NCT04662099, NCT04156269) have been engineered for R/R MM patients [54, 71, 84, 93].
Targeting GPRC5D
GPRC5D, a largely myeloma-specific orphan receptor with limited expression in other tissues, has been proposed as a valuable immunotherapeutic target for managing MM. Clinical trials are underway (NCT04674813, NCT05016778, NCT05739188, NCT05749133, NCT06084962, NCT05759793, NCT05219721) to assess autologous CAR T-cells targeting GPRC5D in R/R MM patients [88, 89, 94]. Additionally, there are CAR-T therapies targeting both BCMA and GPRC5D (NCT05431608, NCT06068400, NCT05325801, NCT05998928, and NCT05509530) [90, 95]. Moreover, MCARH109 (NCT04555551) has been administered to heavily treated MM patients, including those who relapsed after BCMA CAR T-cell therapy [90].
It is noteworthy that recent research has linked relapses in MM patients receiving anti-GPRC5D CAR T to focal biallelic deletions at the GPRC5D locus, while studies have observed the selective expansion of pre-existing subclones with biallelic GPRC5D loss, highlighting potential mechanisms of resistance to this targeted therapy [96, 97].
Targeting integrin β7
MM expresses high levels of integrin β7 receptor subfamily, contributing to adhesion, migration, homing, invasion, and drug resistance. CARs targeting the integrin β7 receptor are currently being evaluated in a multitarget setting (NCT03778346) [98].
Recently, a new generation of CARs has been developed targeting MMG49, an inducible epitope located within the N-terminal region of the integrin β7 chain in MM. Safety and effectiveness assessments for OPC-415 (a CART cell targeting MMG49) are also underway in another study (NCT04649073) [65].
Targeting NKG2D
Stress-induced ligands, including MHC-I chain-related proteins (MIC) like MICA and MICB, and structurally diverse unique long 16-binding proteins 1 to 6 (UL16-binding proteins or ULBPs), are typically upregulated in various hematological malignancies. When these ligands bind to NKG2D receptors on cytotoxic immune cells, such as NK or T cells, they form a hexameric structure with the adaptor protein DAP10 (DNAX-activating protein of 10 kDa). This interaction is crucial for eliminating tumor cells [65].
Consequently, CYAD-01, an autologous NKG2D-CAR T cell, was developed using a full-length natural NKG2D receptor, termed DAP10-NKG2D CAR, which is fused to the CD3ζ intracellular domain without any additional stimulation domains (NKG2D-CD3ζ CAR) [67, 91]. Encouraging outcomes observed in completed primary investigations (CM-CS1) involving CYAD-01 (NCT02203825) paved the way for a phase I clinical trial (THINK) (NCT03018405) [31].
Targeting TnMUC1
The membrane mucin 1 (MUC1) glycoprotein, primarily found on the luminal surface of the simple and glandular epithelium as well as on leukocytes, becomes altered in tumors. The TnMUC1 protein, marked by abnormal glycosylation, is often overexpressed in these tumors. CAR-TnMUC1, designed to target the Tn glycan epitope of MUC1, incorporates a transmembrane CD8α region along with two intracellular signaling domains, CD2 and CD3ζ. In a phase I study (NCT04025216), it was observed that CD2 signaling led to reduced exhaustion and prolonged persistence of T cells within the tumor microenvironment [99]. However, this trial has since been terminated.
Targeting BCMA/TACI
B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL) are recognized as pivotal signaling molecules contributing to MM resistance [100]. They bind to BCMA, BAFF-R, and transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) receptors, triggering expression of programmed cell death-ligand 1 (PD-L1) and exhausting T cells [101, 102]. The biological foundation of these molecules has spurred the creation of APRIL-CAR, a CAR that relies on a natural ligand instead of employing a scFv derived from an antibody. TACI and BCMA are targeted simultaneously in BCMA/TACI-based CARs to address heterogeneous antigens [101].
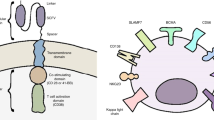
consequently, april car t-cells, including auto2 (nct03287804), APRIL (NCT04657861), and TriPRIL (NCT05020444), were developed to improve their binding capacity [103]. While AUTO2 was initially promising in preclinical and clinical trials, it was recently terminated due to concerns about durability and relapses in many patients [85]. On the other hand, TriPRIL, with its trimeric structure, has demonstrated superior binding capacity in initial studies compared to monomeric APRIL-based CAR-Ts [85, 103] (Fig. 3).

APRIL-based CAR T-cells. A APRIL and BAFF bind to BAFF-R, TACI, and BCMA receptors which leads to the expression of PDL-1 on the cancer cell and induces T cell exhaustion usingof PD-1 on the T-cell surface. B APRIL-base CARs can bind to the BCMA and TACI expressing multiple myeloma cells and inducing tumor shrinkage. The APRIL CAR should create a trimer protein to bind to the receptor, similar to APRIL binding to its target as a trimer protein. However, in TRiPIL CAR, an APRIL trimer protein serves as the binding domain so that oligomerization is not required to bind to receptors. TriPIL CAR is a superior choice for targeting MM cells
Multi-antigen targeting to address heterogeneity and overcome BCMA evasion
Prominent recent trials aiming to target dual or multiple antigens simultaneously on tumor cell surfaces typically utilize products combining a range of antigens. These combinations include BCMA, CD19, and CD138 (NCT03196414); BCMA, CD38, CD138, and CD56 (NCT03473496, NCT03271632); BCMA, CD38, CD138, SLAMF-7, and Integrin β7 (NCT03778346); and CD19, CD20, CD22, CD30, CD38, CD70, and CD123 (NCT03125577). These trials employ CAR-T therapies with bispecific targeting domains, tandem expression of both domains, or the co-infusion of distinct CAR-Ts targeting different antigens (Fig. 4) (Table 2). These innovative approaches are designed to address the heterogeneity of multiple myeloma and combat BCMA evasion or shedding by targeting a diverse array of surface antigens. [65, 104].

Managing tumor heterogeneity. A VHH-based CAR targeting two epitopes of the same antigen. B CAR T-cell targeting novel antigens like SLAMF7 C. Co-infusion of two distinct CAR T-cells targeting different antigens. D CAR T-cell expression two distinct CARs targeting different antigens. E Bispecific CAR T-cells targeting two different antigens. Recognition of each target antigen leads to T-cell activation and tumor shrinkage
After identifying a suitable target antigen, the next major challenge lies in enabling CAR T-cells to efficiently penetrate tumors characterized by dense barriers and a hostile microenvironment.
Utilizing gamma-secretase inhibitor to augment BCMA-Targeted CAR T-cell therapy
The high expression of Notch pathway receptors and ligands in MM cells and the bone marrow environment may explain their role in MM progression [105]. Notch signaling promotes cell proliferation and drug resistance through cell fate determination, a critical aspect of cell–cell communication. It acts through both homotypic activation, where MM cells interact via their own receptors and ligands, and heterotypic activation, involving interactions with bone marrow stromal cells, osteoclasts, and osteocytes [106].
Notch signaling regulates the CXCR4/SDF1α chemokine system and facilitates MM cell migration to the bone marrow [106]. This pathway also promotes MM cell proliferation by upregulating cyclin D1, hairy and enhancer of split-1 (HES1), and hairy/enhancer-of-split related with YRPW motif protein (HEY1) [105]. Notch receptor interacts with other pathways such as PI3K/AKT and NF-κB, further boosting MM cell survival and proliferation [105, 106].
Nirogacestat, a small-molecule gamma-secretase inhibitor (GSI), primarily inhibits Notch signaling and enhances the efficacy of BCMA-targeted therapies [107]. By blocking the proteolysis of Notch receptors, GSIs can potentially slow tumor growth and increase the susceptibility of MM cells to other treatments [108]. GSIs exhibit synergistic effects, enhancing the cytotoxic effects of other MM treatments, such as bortezomib [109]. Combining GSIs with traditional chemoradiotherapies can potentially overcome resistance mechanisms.
GSIs may induce differentiation and apoptosis in cancer stem-like cells, which are often responsible for treatment resistance and relapse in MM [110]. Additionally, by modulating the tumor microenvironment, GSIs can alter the interactions between MM cells and the bone marrow microenvironment, which is crucial for MM progression. [111, 112].
Moreover, GSIs increase the presence of BCMA on the cell surface and reduce the levels of soluble BCMA (sBCMA) by preventing the cleavage and shedding of BCMA from the surface of myeloma cells. GSIs can maintain the increased BCMA surface density for an extended period, potentially prolonging the effectiveness of CAR-T cell therapy with minimal impact on T cells [113].
Nirogacestat’s effectiveness has been assessed in several clinical trials involving “off-the-shelf” CAR T-cell products, as seen in studies such as NCT04093596 and NCT04171843 [38]. Another clinical trial utilized Crenigacestat (LY3039478; JSMD194) as a gamma-secretase inhibitor for MM patients with ide-cel in Phase I/II (KarMMa-7) (NCT04855136) and with anti-BCMA CAR T-cells (NCT03502577) [110]. (Table 4).
Immunosuppressive tumor microenvironment
Although multiple myeloma is often classified as blood cancer, uncontrolled plasma cell proliferation can lead to tumor formation, either within the bones (solitary plasmacytoma) or outside the bones (extramedullary plasmacytoma) [114]. A significant characteristic of MM is the creation of a favorable tumor microenvironment (TME), where local immunosuppression plays a critical role in continuously stimulating antigens [17].
The TME in MM is composed of a diverse array of cellular and acellular elements that contribute to the functional decline, exhaustion, senescence, and removal of CAR T-cells [115]. These include cellular components like regulatory T cells, bone marrow stromal cells (BMSCs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and tumor-associated neutrophils (TANs), in conjunction with various acellular elements like cytokines such as IL-6, transforming growth factor-β (TGF-β), IL-10, tumor-derived granulocyte–macrophage colony-stimulating factor (GM-CSF) and vascular endothelial growth factor (VEGF), proteins like programmed death-ligand 1 (PD-L1), indoleamine 2,3-dioxygenase (IDO), and CD38, and chemicals like prostaglandin E2 (PGE2) and Adenosine [16, 115]. Additionally, environmental conditions such as low extracellular pH (acidosis), limited nutritional resources, and low oxygen levels (hypoxia) further exacerbate the challenges faced by CAR T-cells within this microenvironment [116].
Enhancing our understanding of tumor-induced CAR T-cell suppression is critical for advancing immunotherapy, and various strategies are available to bolster CAR T-cells in this challenging microenvironment.
Surmounting immune checkpoints in CAR T-cell therapy
GM-CSF in the TME leads to MDSCs expansion, triggering signal transducer and activator of transcription 3.(STAT3)-mediated programmed death-ligand 1 (PD-L1) induction in them. PD-L1 directly interacts with PD-1 on CAR T-cells, leading to their suppression [117]. Moreover, the hypoxic conditions in the TME induce T cell anergy and exhaustion by increasing the expression of immune checkpoint molecules such as PD-1, cytotoxic T-lymphocyte associated protein 4 (CTLA4), lymphocyte activation gene 3 (LAG3), T-cell immunoglobulin mucin-3 (TIM-3), B and T lymphocyte attenuator (BTLA), and T cell immunoreceptor with Ig and ITIM domains (TIGIT), hindering their activation and cytotoxicity [118]. Furthermore, a significant increase in immunosuppressive regulatory T cells plays a pivotal role in maintaining immune tolerance by releasing immunosuppressive cytokines like TGF-β and IL-10 and expressing immune-inhibitory molecules such as PD1, TIM3, and CD38 [16, 118].
Approved mAbs targeting immune checkpoints can enhance CAR T-cell therapy. These include Ipilimumab for CTLA-4; Dostarlimab, Pembrolizumab, Cemiplimab, and Nivolumab for PD-1; and Atezolizumab, Durvalumab, and Avelumab for PD-L1 [119,120,121,122,123]. While current research provides valuable insights into the mechanisms and potential of these monoclonal antibodies targeting immune checkpoints, a select few have progressed to late-phase studies or limited clinical use in specific MM patient populations. Integrating these agents with CAR T-cell therapy represents a promising frontier in MM treatment, though their optimal application remains an active area of research. (Table 2).
However, researchers have discovered that directly disrupting the genes encoding these checkpoints through genomic editing technologies yields comparable improvements in CAR T-cell growth and cytotoxicity within the TME. CAR T-cells with genetic modifications, disrupting PD-1, TGF-β, CTLA-4, LAG3, or TIGIT genes, demonstrate an impressive ability to suppress target cells while retaining their essential T-cell characteristics [123,124,125,126]. Likewise, by knocking out the GM-CSF gene through techniques like TALEN or CRISPR/Cas9, CAR T-cells not only experience an enhancement in their cytotoxic activity but also a suppression in the secretion of CRS related biomarkers like IL-6 and IL-8 [127, 128].
Armored CAR T-cells are also engineered to combat the immunosuppressive TME through various innovative strategies. Some armored CAR T-cells are designed to express cytokines or ligands like IL-7, IL-12, IL-15, IL-18, and CD40L, which boost T-cell proliferation, survival, and persistence within the tumor [129]. Others express anti-apoptotic factors from the BCL-2 family of proteins, like BCLxL (BCMA-BCL2L1), to enhance T-cell survival [130]. Additionally, some armored CAR T-cells are engineered to express enzymes such as heparanase, which degrades the extracellular matrix to improve tumor infiltration, thereby protecting against oxidative stress in the TME [131]. Specific therapeutic approaches such as pro-survival gene modifications, including AKT overexpression, have the potential to improve CAR T-cell function in nutrient-poor conditions [132, 133]. Metabolic enhancements, such as overexpressing phosphoenolpyruvate carboxykinase 1 (PCK1) or glutamine synthetase, can allow CAR T-cells to thrive in glucose- or glutamine-depleted environments [134].
Additionally, some armored CAR T-cells are engineered to secrete antibodies targeting PD-1 or PD-L1, effectively preventing the activation of this immunosuppressive pathway [135,136,137] An investigation is underway using anti-BCMA CAR T-cells to produce mutant PD-1 Fc fusion protein in R/R MM patients (NCT04162119) (Table 2).
Improving CAR T-cell trafficking
Recent research has explored utilizing the CXCL12-CXCR4 chemokine axis to improve the intratumoral trafficking of adoptive T cells [138]. In an effort to enhance the homing of CAR T-cells into the CXCL12-rich bone marrow, BCMA-targeted CAR T-cells were engineered to co-express CXCR4. A clinical trial for multiple myeloma (NCT04727008) demonstrated that this approach led to a reduction in tumor burden [139] (Table 2). Additionally, preclinical studies showed that concurrent CXCR4 and anti-BCMA CAR expression on NK cells effectively inhibited MM progression in mouse models [140].Ulocuplumab, the first mAb targeting CXCR4, has shown promise in a phase Ib/II study focused on R/RMM [141].
However, enhancing CAR T-cells’ effectiveness may lead to increased inflammatory signals and associated toxicities. Therefore, precise immunomodulation is crucial for the future application of CAR T-cells in multiple myeloma treatment, aiming to enhance control over the tumor microenvironment while minimizing potential adverse effects.
Clinical considerations and safety management in CAR T-cell therapy for multiple myeloma
Addressing cytotoxicity challenges in CAR T-cell therapy
While CAR-T therapies have demonstrated remarkable therapeutic outcomes in R/R MM, they can also lead to significant toxicities. [142, 143]. The two main complications are cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS), caused by the release of inflammatory cytokines such as TNF-α, IFN-γ, GM-CSF, IL-1, IL-2, IL-6, IL-8, and IL-10, as well as immune responses mediated by other innate immune cells, including immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome (IEC-HS) and macrophage activation cells (MAC). These processes often form a self-aggravating cycle [144,145,146].
CRS typically occurs within 1 to 7 days post-infusion of CAR T-cells, presenting with symptoms ranging from mild flu-like symptoms to severe organ failure, hypotension, hypoxia, and even cardiac arrest in extreme cases.Tumor lysis syndrome (TLS) can also develop in hematological malignancies, presenting symptoms that overlap with those of CRS [147]. The severity of CRS is typically graded on a scale from 1 to 5. Most CRS events are classified as mild(grade 1 or 2), while severe cases are designated as grade 3 or 4. Grade 5, representing the highest severity level, is rarely observed [148].
‘ICANS can occur concurrently with or independently of CRS and typically occurs 3 to 8 days after CAR T-cell infusion. Neurotoxicity can range from mild headaches or delirium to severe confusion, apraxia, or seizures [149].
For cilta-cel, CRS occurred in approximately 95% of patients, but only 4.1% of these cases were severe. ICANS was observed in about 16.5% of patients at any grade, with severe cases reported in 2.1% of patients [150]. In the case of ide-cel, the incidence of CRS was also about 84% of patients, with severe CRS occurring in approximately 5%. ICANS was observed in about 17% of patients at any grade, with severe cases in 3% of patients [151]. Although currently, studies are underway to reduce the mentioned side effects [152].
Although at lower levels, desired antigens like BCMA are also expressed by some normal cells. Consequently, CAR T-cell therapy targeting these antigens may lead to the elimination of healthy cells, a phenomenon known as “on-target off-tumor” toxicities.. This can result in complications such as B-cell aplasia, cytopenia, hypogammaglobulinemia, and immunosuppression, which may increase patients’ susceptibility to various types of infectious diseases [153].
Researchers have identified a correlation between the number of prior therapies, the occurrence of CRS, and the extent of bone marrow tumor burden. These factors were associated with prolonged hematological recovery periods. Patients experiencing severe CRS had lower neutrophil counts, hematocrit levels, and platelet nadirs, necessitating more platelet and red cell transfusions compared to those with mild CRS [20].
Additionally, in MM patients, research has shown that soluble BCMA (sBCMA) levels correlate with laboratory evaluations of tumor burden and prognosis. Elevated sBCMA levels are associated with worse outcomes and a higher incidence of adverse events, including CRS and ICANS) [154]. Currently, the measurement of sBCMA is not standard practice in clinical settings. Instead, most clinicians rely on other assessments of tumor burden, including bone marrow plasma cell percentage, β2 microglobulin levels, serum protein electrophoresis (SPEP)/M-spike, and values associated with light chain disease. However, sBCMA shows potential for patient stratification in clinical trials and real-time disease progression monitoring [155].
To enhance safety, recent studies have explored various CAR T-cell strategies, such as reducing the number of CAR T-cells administered to patients, combining small molecule drugs with CAR T-cells, or mitigating toxicity by engineering CAR T-cells with suicide genes (Table 3).
Combining pharmacological immunosuppressive agents
Numerous studies have investigated drugs aimed at neutralizing the toxicity associated with CAR T-cell therapy in multiple myeloma. Tocilizumab, a mAb against IL-6 receptors (IL-6RA), and corticosteroids are traditionally used to manage toxicities (NCT02215967, NCT02546167, NCT02658929, NCT03090659, NCT03274219, NCT03548207, NCT03361748, NCT03287804) [142]. For ICANS and more severe forms of CRS, corticosteroids are beneficial since Tocilizumab does not penetrate the blood–brain barrier effectively [170] (Table 3).
Clinical trials have revealed that while Tocilizumab effectively mitigated cytotoxicity without compromising CAR T-cell efficacy, approximately 30% of patients still experienced severe grade 3 or higher CRS/ICANS. Even the early administration of steroids failed to alleviate around 13% of the severe cases [171, 172]. Therefore, Tocilizumab-refractory CRS or steroid-refractory ICANS patients require novel treatments [173].
Anakinra, an FDA-approved recombinant IL-1 receptor antagonist (IL-1RA) protein typically used to treat auto-inflammatory diseases, has shown promise in managing CAR T-cell therapy-associated toxicities [172, 174, 175]. For instance, a Phase II study assessing the efficacy of Orva-cel (NCT03430011) included a group of patients given prophylactic Anakinra treatment, which substantially reduced CRS levels following infusion [29]. Recent consensus guidelines suggest Anakinra as a third-line agent for refractory CAR T-associated toxicities [172].
Dasatinib, an inhibitor of lymphocyte-specific protein tyrosine kinase (LCK), blocks the phosphorylation of CD3ζ and ZAP70 [176]. It protects CAR-Ts from lethal CRS by limiting cytolytic activity, cytokine production, and expansion, potentially serving as a pharmacologic on/off switch for CAR T-cells [18]. Dasatinib has been shown to ameliorate the exhausted phenotype of CAR T-cells by upregulating memory-associated genes (such as TCF7 and CCR7) and downregulating the immune checkpoint molecule like PD1, as well as exhaustion-related regulators (such as NR4A1, BATF3, ATF4, and FO) [177]. A Phase I clinical trial (NCT04603872) is recruiting R/R MM patients for CAR T-cells targeted at CD19/BCMA in combination with Dasatinib [80]. In addition, Siltuximab, an anti-IL-6 mAb, has been used in multiple myeloma CAR T-cell trials for severe cases of CRS/ICANS (NCT04975555) (Table 3).
The ZUMA-19 trial (NCT04314843) is investigating the use of Lenzilumab, an anti-human GM-CSF neutralizing mAb, for Large B-cell lymphoma to assess its efficacy and safety in preventing or minimizing CRS/ICANS in combination with CAR T-cell therapy. This aligns with the FDA guidance on prospective registration phase 3 studies for various CAR T-cell assessments [178].
Directly addressing CAR T-cell toxicity
Suicide genes are genetic elements that allow for the permanent removal of introduced cells through the use of pharmaceutical agents. This removal is achieved via metabolic processes that transform non-toxic substances into toxic agents, primarily when unexpected adverse reactions arise. Prominent instances of inducible suicide genes include herpes simplex virus thymidine kinase (HSV-TK) and recombinant human inducible caspase 9 (iCasp9/iC9). Additionally, the expression of truncated epidermal growth factor receptor (tEGFR/EGFRt) or CD20 molecular proteins are utilized to selectively eliminate CAR T-cells that express these genes when necessary [179, 180] (Fig. 5).

Approaches to mitigating the toxicity linked with CAR T-cell therapy directly. A The HSV-TK safety switch and its mode of action. Ganciclovir (GCV) administration leads to HSV-TK gene activation and its conversion to GCV-monophosphate (MP) and eventually further phosphorylated to convert to a toxic GCV-trisphosphate (TP). GCV-TP disrupts DNA synthesis, selectively eliminating CAR T cells. B The inducible caspase 9 (iCasp9) suicide switch and its mechanism. Dimerizing drug administration induces iCasp molecules (FK506) to form dimers, activating downstream apoptotic cascades, and resulting in CAR T cell death. C Examples of mAb-based safety switches and their functions. Co-expression of CAR and cell surface proteins such as EGFRt and CD20 facilitates selective CAR T cell elimination through pharmaceutical-grade mAbs like Cetuximab and Rituximab, which engage immune effector cells or complement fixation cells via mechanisms such as antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) upon specific mAb introduction
HSV-TK
In cases of severe toxicity, a non-toxic prodrug, ganciclovir (GCV), can be administered to selectively activate the HSV-TK gene within transplanted CAR T-cells. This is due to HSV-TK’s high affinity for GCV, which mammalian cell thymidine kinase lacks [181]. Upon HSV-TK phosphorylation of GCV, GCV-monophosphate (MP) eventually converts to a toxic GCV-trisphosphate (TP). This toxic form competitively inhibits guanosine trisphosphate (dGTP), resulting in the cessation of DNA chain synthesis and the demise of specific proliferating cells [182, 183]. The HSV-TK-GCV suicide switch system also activates caspases through ligand-independent aggregation of CD95 and the formation of Fas-associated death domain protein (FADD) and TNF-R-mediated apoptosis in CAR T-cells [184].
Numerous clinical trials have explored CAR-Ts containing the HSV-TK gene for hematological malignancies.A Phase I/II clinical trial investigated the HSV-TK Mut2 gene in anti-CD44v6 CAR T-cells in R/R MM patients (NCT04097301) [185]. However, this trial has since been terminated [181] (Table 3).
iCasp9
The other suicide gene, known as inducible caspase 9 (iCasp9), consists of a sequence from the human FK506-binding protein (modified FKBP12) with an F36V mutation linked to the gene encoding human caspase 9 (CASP9), which lacks its endogenous caspase activation and recruitment domain. FK506 (FKBP12-F36V) strongly binds to an inert small-molecule dimerizing agent, serving as a selectable marker. In the presence of this agent, the iCasp9 pro-molecule undergoes dimerization and activates the intrinsic apoptotic pathway, leading to the death of CAR T-cells expressing this fusion protein [186].
P-BCMA-101 (NCT03288493, NCT03741127) and P-BCMA-ALLO1 (NCT04960579) are autologous and allogeneic CAR-T therapies, respectively, that target BCMA with a second-generation CAR design (4-1BB-iCasp9), specifically incorporating this safety switch [36, 52, 104]. Additionally, a third-generation CAR-T targeting SLAMF-7 (CD28 or 4-1BB/CD3ζ-iCasp9) was investigated in a completed clinical trial (NCT03958656), and a fourth-generation CAR-T (CD28/CD27/CD3ζ-iCasp9) with multiple antigen targeting features is under evaluation in the clinical trial (NCT03125577). In instances of severe toxicity, the activation of inducible caspase 9 is initiated by administering Rimiducid as a safety switch activator [71] (Table 3).
EGFRt or CD20
One approach to eliminating toxic CAR T-cells is the selective targeting of these cells with antibodies [187]. This can be achieved by co-expressing cell surface proteins that are known to have established commercial mAbs against them, providing a suitable method for the specific reduction of CAR-Ts. This approach facilitates the selective elimination of CAR T-cells through mechanisms like antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) upon introducing a specific mAb [187, 188].
In MM research, the clinical application of mAbs like Cetuximab, which targets EGFRt, and Rituximab, designed for CD20, shows promise for their integration into CAR T-cell therapy [189]. Clinical trials have explored the use of second-generation CAR T-cells targeting BCMA (4-1BB-EGFRt) (NCT03070327, NCT03093168, NCT03502577), as well as third-generation CAR T-cells targeting both BCMA and CD19 (OX40/CD28-EGFRt) (NCT03455972), in MM patients. These trials focused on evaluating the efficacy of these CAR-T therapies, especially when administered alongside supplementary adjuvant treatments [71, 110, 190, 191]. Furthermore, it has been demonstrated that NKTR-255 therapy enhances ADCC with Rituximab and Cetuximab in patients who previously received CAR-T therapy [192]. However, the true efficacy of Cetuximab or Rituximab in MM patients remains a subject of ongoing investigation (Table 3).
Enhancing safety features of antibody binding domains in CAR T-cells
Multiple clinical trials have validated CAR T-cell therapies using human or humanized anti-BCMA scFv binding domains for R/R MM. These include trials for various products such as NCT03093168, NCT03070327, NCT03288493, NCT03741127, NCT04960579, NCT03338972, NCT04309981, NCT02546167, NCT04194931, NCT05201118, NCT03815383, NCT06045091, NCT04670055, NCT03975907, NCT03915184, NCT04003168 and NCT05698303. Additionally, ddBCMA, featuring a smaller-sized scFv consisting of 73 amino acids (NCT04155749), anti-BCMA VHH antigen binding domain (NCT03664661, NCT03661554), and anti-BCMA VH (engineered human antibody fragment, consisting of the variable domain of the heavy chain) (NCT03602612) have demonstrated the potential to decrease immunogenicity and achieve high response rates in patients with R/R MM [65, 193] (Table 3).
Supplementary treatments preceding or following CAR-T therapy in multiple myeloma
Commonly used PIs such as Bortezomib, Ixazomib, and Carfilzomib; IMiDs including Lenalidomide, Iberdomide, and Pomalidomide; and monoclonal antibodies such as anti-CD38 mAbs like Daratumumab, as well as anti-SLAMF7 mAbs like Elotuzumab, are often combined with chemotherapeutic drugs such as fludarabine and cyclophosphamide in MM CAR T-cell therapies [12, 194] (Table 4).
Isatuximab, another anti-CD38 mAb approved for adult patients with R/R MM, can potentially enhance CAR-T efficacy [71]. However, the decline in CD38 antigen presence and the emergence of CD38-negative plasma cells pose a significant challenge in R/R MM clinical trials. Research findings suggest that all-trans retinoic acid (ATRA) can enhance CD38 expression, leading to improved efficacy of CD38 monoclonal antibodies and anti-CD38 CAR T-cell therapy [198]. This could expand the repertoire of treatment options available for multiple myeloma.
Mounting evidence underscores the transformative role of various immunotherapies, such as novel monoclonal antibodies, bispecific T-cell engagers (BiTEs), and antibody–drug conjugate (ADC) therapies as innovative treatment modalities for R/R MM [199]. These approaches can potentially complement CAR T-cell regimens. Clinical trials are evaluating several BiTEs targeting BCMA, GPRC5D, and FcRH5 in heavily pretreated MM patients. BCMA-targeted BiTEs like Teclistamab and Elranatamab, GPRC5D-directed BiTE Talquetamab, and BCMA-targeted ADC Belantamab mafodotin have already gained approval for R/R MM [200,201,202,203]. Clinical trials are currently assessing the effectiveness of Belantamab mafodotin (NCT05117008, NCT05789303) and Talquetamab (NCT06066346) within the context of MM CAR T-cell therapies (Table 4). Several BiTEs are currently in clinical trials for MM treatment, showing promise for FDA approval and potential integration with CAR T-cell therapies. Linvoseltamab and F182112 target BCMA, while Cevostamab targets FcRH5. [204]. Early results look promising, with Linvoseltamab showing a 64% response rate and F182112 showing a 43.8% response rate [205, 206].
In a study focusing on a fully human BCMA-targeting CAR T-cell therapy (CT103A), researchers have incorporated Selinexor, a selective inhibitor of nuclear export. Selinexor functions by disrupting the activity of exportin-1 (XPO1), a protein crucial for transporting various proteins and ribonucleoproteins from the nucleus to the cytoplasm. This inhibition prevents the nuclear export of tumor suppressor proteins and cell cycle regulators, leading to their accumulation in the nucleus [207, 208]. The potential of Selinexor in combination with CAR T-cell treatment strategies is being investigated for managing R/R MM (NCT05201118) (Table 4).
sBuilding on this approach, additional research studies are currently underway to further enhance the treatment of multiple myeloma patients. These studies involve various strategies, including autologous hematopoietic stem cell (Auto-HSC) transplantation followed by CART-19/BCMA infusion (NCT03455972) or in combination with CAR T-BCMA therapy (NCT05887167, NCT05632380, NCT05257083), radiation therapy post CAR T-BCMA therapy (NCT05336383, NCT03070327, NCT02546167) [209,210,211], and CAR T-cell re-treatment following previous CAR T-cell therapy (NCT03672253) [65] (Table 4).
Perspectives on the future of MM-CAR T cells in clinical trials
For most MM patients with several previous treatments, allogeneic leukapheresis products from healthy donors offer more cost-effective and life-preserving opportunities [53]. Despite allogeneic CAR T-cells’ short in vivo lifespan, patient outcomes have been encouraging, necessitating further research [51]. Following the FDA’s grant of fast-track designation to ALLO-605 (NCT05000450) in June 2021 and its inclusion in the FDA orphan product development program in April 2022, the decision to advance from Phase II to Phase III is expected to be guided by insights from global data concerning the success rate of transitioning between these clinical trial phases.
Furthermore, MM’s autologous CAR T-cell therapy field has seen significant advancements, with several notable achievements. BCMA CAR T-cell therapy NXC-201 (formerly HBI0101) (NCT04720313) demonstrated a 90% ORR in relapsed/refractory MM patients and a 100% ORR in heavily pre-treated relapsed/refractory amyloid light chain (AL) amyloidosis in the phase Ia/Ib NEXICART-1 trial [212, 213]. Additionally, BCMA/CD19 CAR T-cell (GC012F) trials (NCT04236011, NCT04182581, NCT04617704, NCT04935580, NCT05850234, ChiCTR2100047061) achieved a higher 90% ORR [32, 48, 49, 214]. The autologous GPRC5D CAR T-cell therapy OriCAR-017 showed nearly a 100% ORR in its phase I POLARIS trial (NCT05016778) [87, 215]. These promising results have led to orphan drug designations from the FDA for MM treatment.
Conclusion
Although initial clinical trials demonstrated noteworthy potential for traditional CAR T-cells in the treatment of multiple myeloma, the limited accessibility or persistence, considerable costs, and time involved in manufacturing, coupled with challenges such as patient relapse or side effects following anti-BCMA CAR-T therapy, emphasize the necessity for enhanced efficacy. This situation presents a complex and demanding challenge in the field. Consequently, clinical and basic science have taken on a crucial role in three key objectives: enhancing CAR T-cell manufacturing and persistence, developing insights into disease features, and addressing supplementary therapeutic aspects both prior to and following CAR T-cell therapy in the context of MM.
In this article, we begin by examining strategies employed to enhance the persistence of CAR T-cells through structural and administration optimization. This entails the consideration of immunophenotype characterization factors, including achieving a specific CD4/CD8 ratio, utilizing CD8 + or T stem cell memory phenotype, enhancing CAR T-cell cultures, or equipping them with pro-inflammatory cytokines or ligands to promote a memory CAR T-cell-enriched phenotype [28, 36, 42, 43]. Additionally, it involves the exploration of rapid CAR-T manufacturing methods through novel genetic modifications to reduce transplant timing and costs while maintaining lower-differentiated T cell phenotypes [47, 48]. Initiating CAR T-cell therapy at earlier stages of treatment and the development of universal allogeneic CAR T-cells or even NK-CAR cells introduce new opportunities for patients with high-risk or relapsed/refractory multiple myeloma (R/R MM) [51,52,53, 64] (Table 1).
Regarding the second aspect, the article delves into the intricacies associated with the inherent factors of MM. These complexities include the heterogeneous nature of the cancer and the potential for BCMA evasion or shedding, which can be tackled through innovative CAR designs, such as dual or multitarget CAR T-cells [71, 104] (Table 2). The use of gamma-secretase inhibitors has also been explored to address these challenges [70, 105, 110] (Table 4). Moreover, strategies to counteract the presence of an immunosuppressive tumor microenvironment have been discussed. This involves inhibiting intracellular exhaustion signals through genetic modifications or employing inhibitors as part of the treatment [135, 137, 138, 141] (Table 2).
The final category encompasses clinical aspects associated with CAR T-cell transplantation. This involves addressing unwanted side effects that may occur after CAR T-cell transplantation, such as cytokine release syndrome, neurotoxicity, or cytopenia. These side effects can be managed by utilizing pharmacological immunosuppressive agents in combination or suicide genes that induce direct interactions leading to CAR T-cell death through spatial drugs. Further, the immunogenicity of CAR T-cells can be mitigated by designing humanized scFv or VHH for their recognition sites [172, 216] (Table 3). It also includes the incorporation of additional treatments like proteasome inhibitors, immunomodulators, monoclonal antibodies, bispecific T-cell engagers, antibody–drug conjugates, chemotherapy, corticosteroid therapy, radiation therapy, and autologous hematopoietic stem cell transplantation in combination with CAR-T therapy to enhance its efficacy [66, 194, 201, 202] (Table 4).
Availability of data and materials
Not applicable.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- MM:
-
Multiple myeloma
- CAR:
-
Chimeric antigen receptor
- BCMA:
-
B-cell maturation antigen
- IMiD:
-
Immunomodulatory drugs
- PI:
-
Proteasome inhibitor
- mAb:
-
Monoclonal antibody
- PCs:
-
Plasma cells
- BM:
-
Bone marrow
- R/R MM:
-
Relapsed/ refractory multiple myeloma
- CRS:
-
Cytokine release syndrome
- ICANS:
-
Immune effector cell-associated neurotoxicity syndrome
- TME:
-
Tumor microenvironment
- ALL:
-
Acute Lymphoblastic Leukemia
- DLBCL:
-
Diffuse Large B cell Lymphoma
- FL:
-
Follicular lymphoma
- ScFv:
-
Single-chain variable fragment
- MHC-I:
-
Major histocompatibility complex class I
- MIC:
-
MHC class I chain-related protein
- NKG2D:
-
Natural-killer group 2 member D
- GPRC5D:
-
G protein-coupled receptor class C group 5 member D
- BAFF:
-
B-cell activating factor
- APRIL:
-
A proliferation-inducing ligand
- TRIPRIL:
-
Trimeric APRIL-based CAR
- CAF:
-
Cancer-associated fibroblast
- SLAMF-7:
-
Signaling lymphocyte activation molecule family-7
- FAP:
-
Fibroblast activation protein
- ULBP:
-
Unique long 16-binding protein
- TnMUC1:
-
Tn antigen on membrane mucin 1
- c-Met:
-
Cellular-mesenchymal-epithelial transition
- NY-ESO-1:
-
New York Esophageal Squamous Cell Carcinoma-1
- ddBCMA:
-
D-domain-based antigen binder B-Cell Maturation Antigen
- MSLN:
-
Mesothelin
- NK:
-
Natural Killer
- CB-NK:
-
Cord blood-derived natural killer
- IPSCs:
-
Induced Pluripotent Stem Cells
- MDSC:
-
Myeloid-derived suppressor cells
- GM-CSF:
-
Granulocyte–macrophage colony stimulating factor
- PD-1:
-
Programmed death protein 1
- CTLA4:
-
Cytotoxic T-lymphocyte associated protein 4
- LAG3:
-
Lymphocyte-activation gene 3
- TIM-3:
-
T-cell immunoglobulin mucin-3
- BTLA:
-
B and T-lymphocyte attenuator
- TGF-β:
-
Transforming Growth Factor-beta
- VEGF:
-
Vascular endothelial growth factor
- PGE2:
-
Prostaglandin E2
- TNF-α:
-
Tumor Necrosis Factor-alpha
- IFN-γ:
-
Interferon-gamma
- TIGIT:
-
T-cell immunoreceptor with Ig and ITIM domains
- GSI:
-
Gamma secretase inhibitor
- TLS:
-
Tumor lysis syndrome
- GvHD:
-
Graft-versus-host disease
- PI3K:
-
Phosphoinositide 3-kinase
- MAS:
-
Macrophage activation syndrome
- HLH:
-
Hemophagocytic lymphohistiocytosis
- IL-6RA:
-
IL-6 receptor antagonist
- IL-1RA:
-
IL-1 receptor antagonist
- HSV-TK:
-
Herpes simplex virus thymidine kinase
- tEGFR/EGFRt:
-
Truncated epidermal growth factor receptor
- LCK:
-
Lymphocyte-specific protein tyrosine kinase
- GCV:
-
Ganciclovir
- FADD:
-
Fas-associated death domain protein
- iCasp9:
-
Inducible caspase 9
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- CDC:
-
Complement-dependent cytotoxicity
- MDS:
-
Myelodysplastic syndrome
- SAMT:
-
Subsequent anti-myeloma therapy
- BiTE:
-
Bispecific T-cell engager
- ADC:
-
Antibody–drug conjugate
References
Sung H, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Cowan AJ, et al. Diagnosis and management of multiple myeloma: a review. JAMA. 2022;327(5):464–77.
van Beurden-Tan CH, et al. Systematic literature review and network meta-analysis of treatment outcomes in relapsed and/or refractory multiple myeloma. 2017;35(12):1312–9.
O’Leary MC, et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin Cancer Res. 2019;25(4):1142–6.
Bouchkouj N, et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2019;25(6):1702–8.
Voelker R. CAR-T therapy is approved for mantle cell lymphoma. JAMA. 2020;324(9):832–832.
Mullard A. FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discovery. 2021;20(3):166–7.
Munshi NC, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–16.
Berdeja JG, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. The Lancet. 2021;398(10297):314–24.
Raje N, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–37.
Lin Y, et al. CARTITUDE-1 final results: phase 1b/2 study of ciltacabtagene autoleucel in heavily pretreated patients with relapsed/refractory multiple myeloma. Am Soc Clin Oncol. 2023;41(16_suppl).
Rodriguez-Otero P, et al. Ide-cel or standard regimens in relapsed and refractory multiple myeloma. N Engl J Med. 2023;388(11):1002–14.
San-Miguel J, et al. Cilta-cel or standard care in lenalidomide-refractory multiple myeloma. New England J Med. 2023;389:335–47.
Lv Z, Luo F, Chu Y. Strategies for overcoming bottlenecks in allogeneic CAR-T cell therapy. Front Immunol. 2023;14:1199145.
Zhang X, et al. CAR-T cell therapy in multiple myeloma: Current limitations and potential strategies. Front Immunol. 2023;14:1101495.
Manier S, et al. Bone marrow microenvironment in multiple myeloma progression. BioMed Res Int. 2012;2012(1):157496.
Holthof LC, Mutis T. Challenges for immunotherapy in multiple myeloma: bone marrow microenvironment-mediated immune suppression and immune resistance. Cancers. 2020;12(4):988.
Safarzadeh Kozani P, et al. Strategies for dodging the obstacles in CAR T cell therapy. Front Oncol. 2021;11:627549.
García-Guerrero E, Sierro-Martínez B, Pérez-Simón JA. Overcoming chimeric antigen receptor (CAR) modified T-cell therapy limitations in multiple myeloma. Front Immunol. 2020;11:1128.
Schubert M-L, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. 2021;32(1):34–48.
Asaadi Y, et al. A comprehensive comparison between camelid nanobodies and single chain variable fragments. Biomarker Research. 2021;9:1–20.
Barros LRC, et al. Systematic review of available CAR-T cell trials around the world. Cancers. 2022;14(11):2667.
Puertas B, Mateos M-V, González-Calle VJH. Anti-BCMA CAR T-cell therapy: changing the natural history of multiple myeloma. Hemasphere. 2022;6(3):e691.
Nasiri F, Muhammadnejad S, Rahbarizadeh F. Effects of polybrene and retronectin as transduction enhancers on the development and phenotypic characteristics of VHH-based CD19-redirected CAR T cells: a comparative investigation. Clin Exp Med. 2022;23(6):2535–49.
Stoiber S, et al. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. 2019;8(5):472.
Guedan S, et al. Engineering and design of chimeric antigen receptors. Mol Ther-Methods Clin Dev. 2019;12:145–56.
Vucinic V, et al. Production and application of CAR T cells: current and future role of Europe. 2021. p. 1343.
Green DJ, et al. Fully human Bcma targeted chimeric antigen receptor T cells administered in a defined composition demonstrate potency at low doses in advanced stage high risk multiple myeloma. Blood. 2018;132(Supplement 1):1011–1011.
Mailankody S, et al. Orvacabtagene autoleucel (orva-cel), a B-cell maturation antigen (BCMA)-directed CAR T cell therapy for patients (pts) with relapsed/refractory multiple myeloma (RRMM): update of the phase 1/2 EVOLVE study (NCT03430011). ASCO Annual Meeting. J Clin Oncol. 2020;38(15_suppl).
Raje NS, et al. Updated clinical and correlative results from the phase I CRB-402 study of the BCMA-targeted CAR T cell therapy bb21217 in patients with relapsed and refractory multiple myeloma. Blood. 2021;138:548.
Sallman DA, et al. CYAD-01, an autologous NKG2D-based CAR T-cell therapy, in relapsed or refractory acute myeloid leukaemia and myelodysplastic syndromes or multiple myeloma (THINK): haematological cohorts of the dose escalation segment of a phase 1 trial. Lancet Haematol. 2023;10(3):e191–202.
Du J, et al. Updated results of a phase I open-label single-arm study of dual Targeting BCMA and CD19 Fastcar-T cells (GC012F) as first-line therapy for transplant-eligible newly diagnosed high-risk multiple myeloma. Blood. 2023;142:1022.
Du J, et al. P869: updated results of a phase I, open-label study of BCMA/CD19 dual-targeting FASTCAR-T GC012F for patients with RELAPSED/REFRACTORY MULTIPLE MYELOMA (RRMM). HemaSphere. 2023;7(S3):e84060bf.
Sperling AS, et al. Updated phase I study results of PHE885, a T-Charge manufactured BCMA-directed CAR-T cell therapy, for patients (pts) with r/r multiple myeloma (RRMM). J Clin Oncol. 2023;41:8004.
Costa LJ, et al. Results from the first phase 1 clinical study of the B-Cell Maturation Antigen (BCMA) Nex T Chimeric Antigen Receptor (CAR) T cell therapy CC-98633/BMS-986354 in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM). Blood. 2022;140(Supplement 1):1360–2.
Costello CL, et al. Phase 1/2 study of the safety and response of P-BCMA-101 CAR-T cells in patients with relapsed/refractory (r/r) multiple myeloma (MM)(PRIME) with novel therapeutic strategies. Blood. 2020;136:29–30.
Kocoglu M, et al. 47P Phase I study to assess the safety and efficacy of P-BCMA-ALLO1: a fully allogeneic CAR-T therapy, in patients with relapsed/refractory multiple myeloma (RRMM). Immuno-Oncol Technol. 2022;16(Supplement 1):100152.
Mailankody S, et al. Universal updated phase 1 data highlights role of allogeneic anti-BCMA ALLO-715 therapy for relapsed/refractory multiple myeloma. Blood. 2022;140(Supplement 1):4620–2.
Al-Homsi A-S, et al. Immunicy-1: targeting BCMA with cyad-211 to establish proof of concept of an shRNA-based allogeneic CAR T cell therapy platform. Blood. 2021;138:2817.
Briukhovetska D, et al. Interleukins in cancer: from biology to therapy. Nat Rev Cancer. 2021;21(8):481–99.
Duan D, et al. The BCMA-targeted fourth-generation CAR-T cells secreting IL-7 and CCL19 for therapy of refractory/recurrent multiple myeloma. Front Immunol. 2021;12:609421.
Turtle CJ, et al. Pharmacodynamic analysis of CAR-T cell persistence in patients with hematologic malignancies treated with NKTR-255, an IL-15 receptor agonist that enhances CD8+ T-cells: preliminary results from a phase 1 study. Blood. 2021;138:2815.
Lin L, et al. Preclinical evaluation of CD8+ anti-BCMA mRNA CAR T cells for treatment of multiple myeloma. Leukemia. 2021;35(3):752–63.
Finney OC, et al. Molecular and phenotypic profiling of drug product and post-infusion samples from CRB-402, an ongoing: phase I clinical study of bb21217 a BCMA-directed CAR T cell therapy. Blood. 2020;136:3–4.
Ghassemi S, et al. Rapid manufacturing of non-activated potent CAR T cells. 2022;6(2):118–28.
Manceur AP, et al. Scalable lentiviral vector production using stable HEK293SF producer cell lines. Hum Gene Ther Methods. 2017;28(6):330–9.
Jiang H, et al., Long-term follow-up results of a multicenter first-in-human study of the dual BCMA/CD19 Targeted FasT CAR-T GC012F for patients with relapsed/refractory multiple myeloma. 2021, Wolters Kluwer Health.
Chen X, et al. P1218: First-in-human study of CD19/BCMA dual-targeting FAST CAR-T GC012F for patients with relapsed/refractory B-CELL NON-non-Hodgking’s lymphoma. HemaSphere. 2022;6:1104–5.
Wu JF, Dhakal B. BCMA-targeted CAR-T cell therapies in relapsed and/or refractory multiple myeloma: latest updates from 2023 ASCO annual meeting. J Hematol Oncol. 2023;16(1):86.
Sperling A, et al. P1446: phase I study data update of PHE885, a fully human BCMA-directed CAR-T cell therapy manufactured using the T-ChargeTM platform for patients with relapsed/refractory (R/R) multiple myeloma (MM). Hemasphere. 2022;6:1329–30.
Du Z, et al. Non-conventional allogeneic anti-BCMA chimeric antigen receptor-based immune cell therapies for multiple myeloma treatment. Cancers. 2023;15(3):567.
Costello C, et al. Clinical trials of BCMA-targeted CAR-T cells utilizing a novel non-viral transposon system. Blood. 2021;138:3858.
Caldwell KJ, Gottschalk S, Talleur AC. Allogeneic CAR cell therapy—more than a pipe dream. Front Immunol. 2021;11: 618427.
Mathur R, et al. Universal SLAMF7-specific CAR T-cells as treatment for multiple myeloma. Blood. 2017;130:502.
Sommer C, et al. Preclinical evaluation of ALLO-605, an allogeneic BCMA turbocar TTM cell therapy for the treatment of multiple myeloma. Blood. 2020;136:8.
Mailankody S, et al. Universal: an allogeneic first-in-human study of the anti-BCMA ALLO-715 and the anti-CD52 ALLO-647 in relapsed/refractory multiple myeloma. Blood. 2020;136:24–5.
Akram F, et al. CRISPR/Cas9: A revolutionary genome editing tool for human cancers treatment. Technol Cancer Res Treat. 2022;21:15330338221132078.
Berdeja J, et al. A first-in-human phase 1, multicenter, open-label study of CB-011, a next-generation CRISPR-genome edited allogeneic anti-BCMA immune-cloaked CAR-T cell therapy, in patients with relapsed/refractory multiple myeloma (CAMMOUFLAGE trial). Am Soc Clin Oncol. 2023;41(16_suppl).
Dar H, et al. Preclinical development of CTX120, an allogeneic CAR-T cell targeting Bcma. Blood. 2018;132(Supplement 1):1921–1921.
Vasic D, et al. Allogeneic double-negative CAR-T cells inhibit tumor growth without off-tumor toxicities. Sci Immunol. 2022;7(70):eabl3642.
Lee JB, et al. Developing allogeneic double-negative T cells as a novel off-the-shelf adoptive cellular therapy for cancer. Clin Cancer Res. 2019;25(7):2241–53.
Tang B, et al. Allogeneic double-negative T cell therapy for relapsed acute myeloid leukemia patients post allogeneic hematopoietic stem cell transplantation: a first-in-human phase I study. Am J Hematol. 2022;97(7):E264–7.
Biederstädt A, Rezvani K. Engineering the next generation of CAR-NK immunotherapies. Int J Hematol. 2021;114(5):554–71.
Lin X, et al. IPSC-derived CAR-NK cells for cancer immunotherapy. Biomed Pharmacother. 2023;165: 115123.
Kegyes D, et al. Patient selection for CAR T or BiTE therapy in multiple myeloma: which treatment for each patient?. J Hematol Oncol. 2022;15(1):78.
Ferreri CJ, et al. Real-world experience of patients with multiple myeloma receiving ide-cel after a prior BCMA-targeted therapy. Blood Cancer J. 2023;13(1):117.
Sallman DA, et al. Results from the completed dose-escalation of the hematological arm of the phase I think study evaluating multiple infusions of NKG2D-based CAR T-cells as standalone therapy in relapse/refractory acute myeloid leukemia and myelodysplastic syndrome patients. Blood. 2019;134:3826.
Reguera-Ortega JL, García-Guerrero E, Pérez-Simón JAJH. Current status of CAR-T Cell therapy in multiple myeloma. Hemato. 2021;2(4):660–71.
Wang B, et al. Chimeric antigen receptor T cell therapy in the relapsed or refractory multiple myeloma with extramedullary disease–a single institution observation in China. Blood. 2020;136:6.
Green DJ, et al. Response to Bcma CAR-T cells correlates with pretreatment target antigen density and is improved by small molecule inhibition of gamma secretase. Blood. 2019;134:1856.
Teoh PJ, Chng WJ. CAR T-cell therapy in multiple myeloma: more room for improvement. Blood Cancer J. 2021;11(4):84.
Cohen AD, et al. B cell maturation antigen–specific CAR T cells are clinically active in multiple myeloma. J Clin Investig. 2019;129(6):2210–21.
Samur MK, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun. 2021;12(1):868.
Da Vià MC, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med. 2021;27(4):616–9.
Qian Y, et al. Successful treatment of relapsed/refractory extramedullary multiple myeloma with anti-BCMA CAR-T cell therapy followed by haploidentical hematopoietic stem cell transplantation: a case report and a review of the contemporary literature. 2021. p. 483.
Zhao W-H, et al. Four-year follow-up of LCAR-B38M in relapsed or refractory multiple myeloma: a phase 1, single-arm, open-label, multicenter study in China (LEGEND-2). J Hematol Oncol. 2022;15(1):1–12.
Hillengass J, et al. The phase 2 CARTITUDE-2 trial: updated efficacy and safety of ciltacabtagene autoleucel in patients with multiple myeloma and 1–3 prior lines of therapy (cohort A) and with early relapse after first line treatment (cohort B). Blood. 2023;142:1021.
Anderson J, Larry D, et al. Idecabtagene vicleucel (ide-cel, bb2121), a BCMA-directed CAR T cell therapy, in relapsed and refractory multiple myeloma: updated KarMMa results. In 18th International Myeloma Society Workshop. 2021.
Dhakal B, et al. First phase 3 results from CARTITUDE-4: Cilta-cel versus standard of care (PVd or DPd) in lenalidomide-refractory multiple myeloma. Am Soc Clin Oncol. 2023;41(17_suppl):LBA106.
Atrash S, Moyo TK. A review of chimeric antigen receptor T-Cell therapy for myeloma and lymphoma. Onco Targets Ther. 2021;14:2185–201.
Lee LSH, et al. Development of a phase 1 study evaluating the activity of modular CAR T for multiple myeloma (MCARTY) targeting BCMA and CD19 for improved persistence. Blood. 2023;142:350.
Shi X, et al. Anti-CD19 and anti-BCMA CAR T cell therapy followed by lenalidomide maintenance after autologous stem-cell transplantation for high-risk newly diagnosed multiple myeloma. Am J Hematol. 2022;97(5):537–47.
Wudhikarn, K., S. Mailankody, and E.L. Smith, Future of CAR T cells in multiple myeloma. Hematology 2014, the American Society of Hematology Education Program Book, 2020. 2020(1): p. 272–279.
Li C, et al. Bispecific CS1-BCMA CAR-T cells are clinically active in relapsed or refractory multiple myeloma. Leukemia. 2023;38(1):149–59.
Lee, L., et al., Limited efficacy of APRIL CAR in patients with multiple myeloma indicate challenges in the use of natural ligands for CAR T-cell therapy. J Immunother Cancer. 2023;11(6).
Yan L, et al. Sequential CD19 and BCMA-specific CAR T-cell treatment elicits sustained remission of relapsed and/or refractory myeloma. Cancer Med. 2021;10(2):563–74.
Huang H, et al. OriCAR-017, a novel GPRC5D-targeting CAR-T, in patients with relapsed/refractory multiple myeloma: long term follow-up results of phase 1 study (POLARIS). J Clin Oncol. 2024;42(16_suppl):7511.
Li S, et al. Safety and efficacy of GPRC5D CAR T cell therapy in relapsed/refractory multiple myeloma patients. Blood. 2023;142(Supplement 1):3472–3472.
Bal S, et al. S193: BMS-986393 (CC-95266), AG protein–coupled receptor class C group 5 member D (GPRC5D)–targeted CAR T-cell therapy for relapsed/refractory multiple myeloma (RRMM): results from a phase 1 study. Hemasphere. 2023;7(S3):e9863287.
Mailankody S, et al. GPRC5D-targeted CAR T cells for myeloma. N Engl J Med. 2022;387(13):1196–206.
Baumeister SH, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. 2019;7(1):100–12.
Jiang D, et al. Chimeric antigen receptor T cells targeting FcRH5 provide robust tumour-specific responses in murine xenograft models of multiple myeloma. Nat Commun. 2023;14(1):3642.
Sakemura R, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood, J Am Soc Hematol. 2022;139(26):3708–21.
Xia J, Li Z, Xu K. Immunotherapies targeting GPRC5D in relapsed or refractory multiple myeloma: latest updates from 2022 ASH annual meeting. J Hematol Oncol. 2023;16(1):60.
Smith EL, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci transl Med. 2019;11(485):eaau7746.
Mi X, et al. Genetic basis of relapse after GPRC5D-targeted CAR T cells. Blood. 2023;142:336.
Lee H, et al. Mechanisms of antigen escape from BCMA-or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med. 2023;29(9):2295–306.
Hosen N, et al. The activated conformation of integrin β7 is a novel multiple myeloma–specific target for CAR T cell therapy. Nat Med. 2017;23(12):1436–43.
Gutierrez R, et al. Phase I experience with first in class TnMUC1 targeted chimeric antigen receptor T-cells in patients with advanced TnMUC1 positive solid tumors. J Clin Oncol. 2021;39(15_suppl):e14513.
Tai Y-T, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood. 2016;127(25):3225–36.
Wong DP, et al. A BAFF ligand-based CAR-T cell targeting three receptors and multiple B cell cancers. Nat Commun. 2022;13(1):1–17.
Lee L, et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood. 2018;131(7):746–58.
Schmidts A, et al. Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv. 2019;3(21):3248–60.
Jiao C, et al. 4SCAR2. 0: a multi-CAR-T therapy regimen for the treatment of relapsed/refractory B cell lymphomas. Blood Cancer J. 2021;11(3):59.
Sabol, H.M. and J. Delgado-Calle, The multifunctional role of Notch signaling in multiple myeloma. J Cancer Metastasis Treat. 2021;7.
Colombo M, et al. Notch signaling deregulation in multiple myeloma: a rational molecular target. Oncotarget. 2015;6(29):26826–40.
Chen H, et al. γ-secretase inhibitors augment efficacy of BCMA-targeting bispecific antibodies against multiple myeloma cells without impairing T-cell activation and differentiation. Blood Cancer J. 2022;12(8):118.
McCaw TR, et al. Gamma secretase inhibitors in cancer: a current perspective on clinical performance. Oncologist. 2021;26(4):e608–21.
Chen F, et al. Gamma-secretase inhibitor enhances the cytotoxic effect of bortezomib in multiple myeloma. Cell Oncol. 2011;34:545–51.
Cowan AJ, et al. γ-Secretase inhibitor in combination with BCMA chimeric antigen receptor T-cell immunotherapy for individuals with relapsed or refractory multiple myeloma: a phase 1, first-in-human trial. Lancet Oncol. 2023;24(7):811–22.
Majumder S, et al. Targeting Notch in oncology: the path forward. Nat Rev Drug Discovery. 2021;20(2):125–44.
Song C, et al. The critical role of γ-secretase and its inhibitors in cancer and cancer therapeutics. Int J Biol Sci. 2023;19(16):5089.
Pont MJ, et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood, J Am Soc Hematol. 2019;134(19):1585–97.
Alrasheed N, et al. Marrow-infiltrating regulatory T cells correlate with the presence of dysfunctional CD4+ PD-1+ cells and inferior survival in patients with newly diagnosed multiple myeloma. Clin Cancer Res. 2020;26(13):3443–54.
Fonkoua LAK, et al. CAR T cell therapy and the tumor microenvironment: current challenges and opportunities. Mol Ther-Oncolytics. 2022;25:69–77.
Lopes R, et al. The immune microenvironment in multiple myeloma: friend or foe? Cancers. 2021;13(4):625.
Groth C, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120(1):16–25.
Swamydas M, et al. Deciphering mechanisms of immune escape to inform immunotherapeutic strategies in multiple myeloma. J Hematol Oncol. 2022;15(1):17.
Savoia P, Astrua C, Fava P. Ipilimumab (Anti-Ctla-4 Mab) in the treatment of metastatic melanoma: Effectiveness and toxicity management. Hum Vaccin Immunother. 2016;12(5):1092–101.
Schachter J, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. 2017;390(10105):1853–62.
Dang TO, et al. Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert Rev Anticancer Ther. 2016;16(1):13–20.
Herbst RS, et al. Atezolizumab for first-line treatment of PD-L1–selected patients with NSCLC. N Engl J Med. 2020;383(14):1328–39.
Tang N, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5(4):e133977.
Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. 2022;21(1):1–13.
Rupp LJ, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):1–10.
Zhang P, et al. Targeting TIGIT for cancer immunotherapy: recent advances and future directions. Biomark Res. 2024;12(1):7.
Sterner RM, et al. Using CRISPR/Cas9 to knock out GM-CSF in CAR-T cells. JoVE (Journal of Visualized Experiments). 2019;149:e59629.
Sterner RM, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood J Am Soc Hematol. 2019;133(7):697–709.
Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–54.
Poorebrahim M, et al. Bclxl Prevents Progressive Exhaustion in BCMA CAR T Cells with Maintenance of High TCF1 Expressing T Cells. Blood. 2023;142:454.
Caruana I, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–9.
Guedan S, et al. Enhancing CAR T cell persistence through ICOS and 4–1BB costimulation. JCI Insight. 2018;3(1):e96976.
Kawalekar OU, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–90.
Ho P-C, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162(6):1217–28.
Suarez ER, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7(23):34341.
Wang Y, et al. Immune-restoring CAR-T cells display antitumor activity and reverse immunosuppressive TME in a humanized ccRCC mouse model. Iscience. 2024;27(2):108879.
Rafiq S, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36(9):847–56.
Dewan M, et al. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed Pharmacother. 2006;60(6):273–6.
Bianchi ME, Mezzapelle R. The chemokine receptor CXCR4 in cell proliferation and tissue regeneration. Front Immunol. 2020;11:2109.
Ng YY, et al. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther. 2022;29(5):475–83.
Ghobrial IM, et al. A phase Ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin Cancer Res. 2020;26(2):344–53.
Zhou X, et al. Toxicities of chimeric antigen receptor T cell therapy in multiple myeloma: an overview of experience from clinical trials, pathophysiology, and management strategies. Front Immunol. 2020;11:620312.
Roex G, et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J Hematol Oncol. 2020;13:1–14.
Lindo L, Wilkinson LH, Hay KA. Befriending the hostile tumor microenvironment in CAR T-cell therapy. Front Immunol. 2021;11:618387.
Mehta P, et al. Silencing the cytokine storm: the use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. 2020;2(6):e358–67.
Hines MR, et al. Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome (IEC-HS). Transplant Cell Ther. 2023;29(7):438.e1–438.e16.
Rahmani B, et al. Current understanding of tumor lysis syndrome. Hematol Oncol. 2019;37(5):537–47.
van de Donk NW, et al. Biological correlative analyses and updated clinical data of ciltacabtagene autoleucel (cilta-cel), a BCMA-directed CAR-T cell therapy, in patients with multiple myeloma (MM) and early relapse after initial therapy: CARTITUDE-2, cohort B. Am Clin Oncol. 2022;40(16_suppl):8029.
Cohen AD, et al. Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. 2022;12(2):1–9.
Martin T, et al. Ciltacabtagene autoleucel, an anti–B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J Clin Oncol. 2023;41(6):1265–74.
Hansen DK, et al. Idecabtagene vicleucel for relapsed/refractory multiple myeloma: real-world experience from the myeloma CAR T consortium. J Clin Oncol. 2023;41(11):2087–97.
Cohen A, et al. Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. Blood Cancer J. 2022;12:32 Case series on patients with neurocognitive and movement toxicity after treatment with anti-BCMA CAR-T and strategies implemented for mitigation of neurotoxicity Article PubMed PubMed Central.
Chohan KL, Siegler EL, Kenderian SS. CAR-T cell therapy: the efficacy and toxicity balance. Curr Hematol Malig Rep. 2023;18(2):9–18.
Ghermezi M, et al. Serum B-cell maturation antigen: a novel biomarker to predict outcomes for multiple myeloma patients. Haematologica. 2017;102(4):785.
Visram A, et al. Serum BCMA levels predict outcomes in MGUS and smoldering myeloma patients. Blood Cancer J. 2021;11(6):120.
Lin Y, et al. Idecabtagene vicleucel for relapsed and refractory multiple myeloma: post hoc 18-month follow-up of a phase 1 trial. Nat Med. 2023;29(9):2286–94.
Xu J, et al. Long-term remission and survival in patients with relapsed or refractory multiple myeloma after treatment with LCAR-B38M CAR T cells: 5-year follow-up of the LEGEND-2 trial. J Hematol Oncol. 2024;17(1):23.
Brudno JN, et al. T cells genetically modified to express an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267.
Mailankody S, et al. Clinical responses and pharmacokinetics of MCARH171, a human-derived Bcma targeted CAR T cell therapy in relapsed/refractory multiple myeloma: final results of a phase I clinical trial. DC: American Society of Hematology Washington; 2018.
Du J, et al. CAR-T cell therapy targeting B cell maturation antigen is effective for relapsed/refractory multiple myeloma, including cases with poor performance status. Am J Hematol. 2022;97(7):933–41.
Fernandez de Larrea C, et al. Long-term follow-up of ARI0002h (cesnicabtagene autoleucel), an academic point-of-care B-cell maturation antigen (BCMA)-directed chimeric antigen receptor (CAR) T-cell strategy: Activity and safety after fractionated initial therapy and booster dose in 60 patients with relapsed/refractory multiple myeloma (RRMM). J Clin Oncol. 2024;42(16_suppl).
Li C, et al. CT103A, a novel fully human BCMA-targeting CAR-T cells, in patients with relapsed/refractory multiple myeloma: updated results of phase 1b/2 study (FUMANBA-1). Am Soc Clin Oncol. 2023;41(16_suppl):8025.
Fu C, et al. Three-year follow-up on efficacy and safety results from phase 1 lummicar study 1 of Zevorcabtagene Autoleucel in chinese patients with relapsed or refractory multiple myeloma. Blood. 2023;142(Supplement 1):4845–4845.
Kumar SK, et al. Results from Lummicar-2: a phase 1b/2 study of fully human B-cell maturation antigen-specific CAR T cells (CT053) in patients with relapsed and/or refractory multiple myeloma. Blood. 2020;136:28–9.
Qu X, et al. Phase 1 study of C-CAR088, a novel humanized anti-BCMA CAR T-cell therapy in relapsed/refractory multiple myeloma. J Immunoth Cancer. 2022;10(9):e005145.
Wang Q, et al. An alternative fully human anti-BCMA CAR-T shows response for relapsed or refractory multiple myeloma with anti-BCMA CAR-T exposures previously. Cancer Gene Ther. 2024;31(3):420–6.
Frigault MJ, et al. Phase 1 study of CART-ddBCMA for the treatment of subjects with relapsed and refractory multiple myeloma. Blood Adv. 2023;7(5):768–77.
Mikkilineni L, et al. T cells expressing an anti-B-cell maturation antigen (BCMA) chimeric antigen receptor with a fully-human heavy-chain-only antigen recognition domain induce remissions in patients with relapsed multiple myeloma. Blood. 2019;134:3230.
Han, L., et al., The clinical study of anti-BCMA CAR-T with single-domain antibody as antigen binding domain. Wolters Kluwer Health 2021.
Santomasso BD, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell Therapy in patients with B-cell acute lymphoblastic LeukemiaBiomarkers of neurotoxicity in CD19 CAR T cell therapy. Cancer Discov. 2018;8(8):958–71.
Topp M, et al. Earlier steroid use with axicabtagene ciloleucel (Axi-Cel) in patients with relapsed/refractory large B cell lymphoma. Biol Blood Marrow Transplant. 2019;134:243.
Wehrli M, et al. Single-center experience using anakinra for steroid-refractory immune effector cell-associated neurotoxicity syndrome (ICANS). J Immunother Cancer. 2022;10(1):e003847.
Si S, Teachey DTJT, Management CR. Spotlight on tocilizumab in the treatment of CAR-T-cell-induced cytokine release syndrome: clinical evidence to date. 2020;16:705.
Mehta P, et al. Silencing the cytokine storm: the use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. 2020;2(6):e358–67.
Norelli M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–48.
Weber EW, et al. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019;3(5):711.
Zhang H, et al. Dasatinib enhances anti-leukemia efficacy of chimeric antigen receptor T cells by inhibiting cell differentiation and exhaustion. J Hematol Oncol. 2021;14:1–6.
Nizzoli MA, Bavieri A, Luminari S. CAR-T cell therapy. In: A New milestone in the treatment of B-cell lymphomas. 2022.
Moghanloo E, et al. Remote controlling of CAR-T cells and toxicity management: molecular switches and next generation CARs. Transl Oncol. 2021;14(6):101070.
Jones BS, et al. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol. 2014;5:254.
Carrabba MG, et al. Phase I-IIa clinical trial to assess safety and efficacy of MLM-CAR44. 1, a CD44v6 directed CAR-T in relapsed/refractory acute myeloid leukemia (AML) and multiple myeloma (MM). Blood. 2018;132:5790.
Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Can Res. 1986;46(10):5276–81.
Elion GB, et al. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci. 1977;74(12):5716–20.
Beltinger C, et al. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci. 1999;96(15):8699–704.
Stornaiuolo A, et al. Characterization and functional analysis of CD44v6. CAR T cells endowed with a new low-affinity nerve growth factor receptor-based spacer. Hum Gene Ther. 2021;32(13–14):744–60.
Di Stasi A, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–83.
Li H, Zhao Y. Increasing the safety and efficacy of chimeric antigen receptor T cell therapy. Protein Cell. 2017;8(8):573–89.
Kieback E, et al. A safeguard eliminates T cell receptor gene-modified autoreactive T cells after adoptive transfer. Proc Natl Acad Sci. 2008;105(2):623–8.
Introna M, et al. Genetic modification of human T cells with CD20: a strategy to purify and lyse transduced cells with anti-CD20 antibodies. Hum Gene Ther. 2000;11(4):611–20.
Jain T, et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic malignancies. Blood Adv. 2020;4(15):3776–87.
Liu Y, et al. Durable remission achieved from Bcma-directed CAR-T therapy against relapsed or refractory multiple myeloma. Blood. 2018;132:956.
Shah N, et al. Safety, tolerability, PK/PD and preliminary efficacy of NKTR-255, a novel IL-15 receptor agonist, in patients with relapsed/refractory hematologic malignancies. Blood. 2021;138:3134.
Frigault MJ, et al. Phase 1 Study of CART-ddBCMA, a CAR-T therapy utilizing a novel synthetic binding domain, for the treatment of subjects with relapsed and refractory multiple myeloma. Blood. 2020;136(Supplement 1):2.
Chekol Abebe E, et al. Ciltacabtagene autoleucel: The second anti-BCMA CAR T-cell therapeutic armamentarium of relapsed or refractory multiple myeloma. Front Immunol. 2022;13:991092.
Hillengass J, et al. The phase 2 cartitude-2 trial: updated efficacy and safety of ciltacabtagene autoleucel in patients with multiple myeloma and 1–3 prior lines of therapy (Cohort A) and with early relapse after first line treatment (Cohort B). Transplant Cell Ther Off Publ Am Soc Transplant Cell Ther. 2024;30(2):S36–7.
Usmani S, et al. KarMMa-2 cohort 2a: efficacy and safety of idecabtagene vicleucel in clinical high-risk multiple myeloma patients with early relapse after frontline autologous stem cell transplantation. Blood. 2022;140(Supplement 1):875–7.
Xu Y, et al. Promising Safety and Efficacy of Trovocabtagene Autoleucel (C-CAR088) Followed ASCT in Ultra High-Risk Multiple Myeloma (UHR-MM) Patients Who Failed or Had Suboptimal Response to Standard First Line Triplet Based Therapy. Blood. 2023;142(Supplement 1):6864–6864.
Peng Z, et al. All-trans retinoic acid improves NSD2-mediated RARα phase separation and efficacy of anti-CD38 CAR Tcell therapy in multiple myeloma. J Immunother Cancer. 2023;11(3):e006325.
Offidani M, et al. Novel experimental drugs for treatment of multiple myeloma. J Exp Pharmacol. 2021;13:245–64.
van de Donk NW, et al. Long-term follow-up from MajesTEC-1 of teclistamab, a B-cell maturation antigen (BCMA) x CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma (RRMM). Am Soc Clin Oncol. 2023;41(16_suppl):8011.
Mohty M, et al. Elranatamab, a B-cell maturation antigen (BCMA)-CD3 bispecific antibody, for patients (pts) with relapsed/refractory multiple myeloma (RRMM): extended follow up and biweekly administration from the MagnetisMM-3 study. Am Soc Clin Oncol. 2023;7(Suppl ):e1309654.
Schinke CD, et al. Pivotal phase 2 MonumenTAL-1 results of talquetamab (tal), a GPRC5DxCD3 bispecific antibody (BsAb), for relapsed/refractory multiple myeloma (RRMM). HemaSphere. 2023;7(S3):e5955094.
Baines AC, et al. FDA approval summary: belantamab mafodotin for patients with relapsed or refractory multiple myeloma. Clin Cancer Res. 2022;28(21):4629–33.
Zhao J, et al. Bispecific antibodies targeting BCMA, GPRC5D, and FcRH5 for multiple myeloma therapy: latest updates from ASCO 2023 Annual Meeting. J Hematol Oncol. 2023;16(1):92.
Lee HC, et al. LINKER-MM1 study: Linvoseltamab (REGN5458) in patients with relapsed/refractory multiple myeloma. Am Soc Clin Oncol. 2023;41(16_suppl).
Sun M, et al. Results from a first-in-human phase I study of F182112, a B-cell maturation antigen (BCMA)-CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma. Am Soc Clin Oncol. 2023;41(16_suppl).
Wang D, et al. A novel two-step administration of XPO-1 inhibitor may enhance the effect of anti-BCMA CAR-T in relapsed/refractory extramedullary multiple myeloma. J Transl Med. 2023;21(1):812.
Mo CC, et al. Selinexor: targeting a novel pathway in multiple myeloma. eJHaem. 2023;4:792–810.
Smith EL, et al. BCMA-targeted CAR T-cell therapy plus radiotherapy for the treatment of refractory myeloma reveals potential synergy. Cancer Immunol Res. 2019;7(7):1047–53.
Manjunath SH, et al. The safety of bridging radiation with anti-BCMA CAR T-cell therapy for multiple myeloma. Clin Cancer Res. 2021;27(23):6580–90.
Huan T, Li H, Tang B. Radiotherapy plus CAR-T cell therapy to date: a note for cautions optimism?. Front Immunol. 2022;13:1033512.
Lebel E, et al. Feasibility of a novel academic anti-BCMA chimeric antigen receptor T-cell (CART)(HBI0101) for the treatment of relapsed and refractory AL amyloidosis. Blood. 2023;142:538.
Lebel E, et al. Safety and efficacy of a locally produced novel anti-BCMA chimeric antigen receptor T-Cell (CART)(HBI0101) for the treatment of relapsed and refractory multiple myeloma. Blood. 2023;142:4852.
Du J, et al. Updated results of a phase I, open-label study of BCMA/CD19 dual-targeting fast CAR-T GC012F for patients with relapsed/refractory multiple myeloma (RRMM). J Clin Oncol. 2023;41(Supplement 16):8005.
Zhang M, et al. GPRC5D CAR T cells (OriCAR-017) in patients with relapsed or refractory multiple myeloma (POLARIS): a first-in-human, single-centre, single-arm, phase 1 trial. Lancet Haematol. 2023;10(2):e107–16.
Si S, Teachey DT. Spotlight on tocilizumab in the treatment of CAR-T-cell-induced cytokine release syndrome: clinical evidence to date. In: Therapeutics and clinical risk management. 2020. p. 705–14.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
F.N. wrote the manuscript with support from Y.A., F.M., S.M., S.P., S.A. and SM. Y.A. prepared the figures. All authors provided critical feedback and edited the original manuscript. F.R. supervised the project.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nasiri, F., Asaadi, Y., Mirzadeh, F. et al. Updates on CAR T cell therapy in multiple myeloma. Biomark Res 12, 102 (2024). https://doi.org/10.1186/s40364-024-00634-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-024-00634-5