
Abstract
TOX (thymocyte selection-associated HMG BOX) is a member of a family of transcriptional factors that contain the highly conserved high mobility group box (HMG-box) region. Increasing studies have shown that TOX is involved in maintaining tumors and promoting T cell exhaustion. In this review, we summarized the biological functions of TOX and its contribution as related to lymphocytic malignancies. We also discussed the potential role of TOX as an immune biomarker and target in immunotherapy for hematological malignancies.
Similar content being viewed by others


Background
TOX (thymocyte selection-associated HMG BOX), a transcription factor, belongs to the high mobility group box (HMG-box) superfamily. In 2002, Wilkinson et al. demonstrated for the first time that TOX plays an important role in the differentiation of CD4 + CD8+ double-positive thymocytes by gene microarray technology. Numerous studies have shown that TOX plays a vital role in the development and formation of various immune organs and cells such as CD4+ T cells and natural killer cells (NK cells). Recently, it has been reported that TOX may be involved in promoting CD8+ T cell exhaustion and the development of various malignant tumors [1, 2].
Structure and biological function of TOX
There are four human TOX genes: TOX (also known as TOX1), TOX2, TOX3, and TOX4. These genes are located on different chromosomes and have different functions (Fig. 1, Table 1) [16].

Schematic diagram of the TOX gene structure and location. The bars represent exons and the lines represent introns. The dotted box indicates the regions of exons that encode the HMG box. All exon lengths are drawn to scale with the scale bar indicating 400 nucleotides
TOX
TOX, also known as KIAA0808, is located on q12.1 of human chromosome 8 and has one transcript. The length of the TOX mRNA is 4076 bp, and it contains nine exons that translate into a protein consisting of 526 amino acids. TOX is expressed in many human tissues, including hematopoietic and immune tissues, such as the tonsils, thymus, spleen, and bone marrow, and non-hematopoietic tissues, such as the intestines, lung, kidney, ovary, pancreas, breast and testis [17, 18]. In addition, TOX expression is relatively different in different tissues. For example, TOX is highly expressed in the thymus but has low expression in the spleen [19]. Overall, TOX plays a vital role in the immune system [3]. TOX expression gradually increases as NK cells begin to differentiate and decreases as NK cells mature. After silencing TOX by RNA interference, it was found that decreased TOX expression leads to a reduction in T-bet, thereby inhibiting the differentiation of NK cells [20, 21]. TOX is related to the formation of lymph nodes and Peyer’s patches, and knockout of TOX in mice resulted in the loss of lymph nodes and Peyer’s patches [21]. It has been found that both TOX and TOX2 are related to CD8+ T cell exhaustion [4]. Recently, novel findings showed that higher TOX expression is associated with better prognosis in breast cancer. Thus, it is considered that the alteration of TOX may be a potential prognostic marker for breast cancer [18].
TOX2
TOX2, also known as C20orf100, is located on q13.12 of human chromosome 20 and has four transcripts. The TOX2 transcript encoding the longest isoform contains nine exons with an mRNA length of 2491 bp that is translated into a protein consisting of 506 amino acids. There are few studies on TOX2 at present. TOX2, similar to TOX, affects the development of NK cells by regulating the expression of T-bet. Interestingly, contrary to TOX, TOX2 is highly expressed in mature NK cells but not in the early stages of developing NK cells [7]. These data suggest that TOX and TOX2 may contribute to NK cell development at different time points.
TOX3
TOX3, also known as TNRC9, was initially found in the brain [22]. This gene is located on q12.1 of human chromosome 16 and has two transcripts. The TOX3 transcript encoding the longest isoform contains seven exons, and the length of its mRNA is 4959 bp, which translated into a protein containing 576 amino acids. In 2009, Yuan et al. found that TOX3 is an important calcium-dependent transactivator in neurons and has a protective effect in neurons [9]. In patients with spinal cord injury, up-regulation of TOX3 can increase oligodendrocyte survival after injury, thereby reducing damage due to secondary degeneration during early spinal cord injury [10]. Genome-wide association studies (GWAS) have indicated that the variable expression of TOX3 might play a different role in different tumors. For example, TOX3 is highly expressed in breast cancer (particularly luminal breast cancer) and is associated with poor prognosis. Interestingly, this finding is opposite to the finding of TOX in breast cancer [18]. While in lung cancer, high expression of TOX3 is associated with better prognosis [11,12,13].
TOX4
TOX4, also known as KIAA0737, is located on q11.2 of human chromosome 14 and has two transcripts. The transcript encoding the longest isoform contains nine exons that leads to an mRNA 4514 bp in length that is translated into a protein containing 621 amino acids. TOX4 is involved in the regulation of the cell cycle and fate. In HEK293 cells, TOX4 forms a complex with protein phosphatase 1 (PP1), which participates in regulation of the cell cycle, proliferation, apoptosis, and other biological processes [23]. TOX4 reprograms somatic cells to form induced pluripotent stem cells (iPSCs) that differentiate these cells into a neuronal fate. Moreover, TOX4 is an indispensable factor for maintaining and establishing the pluripotency of embryonic stem cells and epiblast stem cells [14, 15].
HMG-box of TOX
Although the number of exons in each TOX family member is different, the HMG-box region encoded by two or three exons in vertebrates is highly conserved among family members [24, 25]. The traditional HMG-box protein family can be divided into two classes. Class I HMG box group proteins are transcription factors containing a single HMG box that can bind DNA [25], and class II proteins have two DNA-binding motifs [26,27,28] that can bind DNA and alter chromatin structure [29,30,31]. TOX contains a single highly conserved HMG-box region and is a class I HMG-box protein. Most characteristics of TOX are similar to other HMG-box proteins; however, TOX lacks a key hydrophobic wedge residue that disables the induction of DNA bending [3]. In recent years, increasing studies have shown that TOX is a new class of HMG-Box proteins because TOX combines and recruits various proteins to form protein complexes that bind to DNA instead of directly binding DNA [5, 32]. Thus, it has been suggested TOX may function through a combination of different protein complexes on DNA to alter chromatin structure, which affects the expression of other genes.
The role of TOX in T cell immunity
TOX and T cell differentiation
TOX was demonstrated to be involved in the proliferation and differentiation of T cells in TOX knock-out mice. The expression of TOX transiently increased during mouse β selection and positive selection in the thymus, and it decreased with the maturation of CD4+ and CD8+ T cells [16, 33, 34]. During the positive selection process, TOX determines the fate of CD8+ T cells by upregulating runt-related transcription factor 3 (RUNX3) and inhibiting the development of CD4+ T cells. Loss of TOX results in a considerable reduction in CD4+ T cells in mice, and CD4+ T cells cannot fully mature, thus losing some of their function [33]. TOX is associated with different types of cells in the thymus and is highly expressed in CD4 + CD8+ double-positive thymocytes but not in CD4-CD8- double-negative cells. Therefore, based on the different types of T cells that form in the thymus, T cells can be divided as TOX-dependent T cells (NKT cells, regulatory T cells), including CD4 + CD8+ double-positive thymus cells, and TOX-independent T cells (γδ T cells), including CD4-CD8- double-negative thymocytes [34]. TOX2 can alter chromatin and affect its function, promoting T follicular helper (TFH) cell development. Binding of the transcription factor B-cell lymphoma 6 (Bcl-6) to the TOX2 locus fosters TOX2 expression, and the binding of TOX2 with DNA can alter the structure of chromatin to express relevant genes (such as Bcl-6, Cxcr5, and Pdcd1) and pathways (IL-6-STAT3, Notch, and Wnt). In TOX2 knockout mice, the formation of TFH cells decreased, impacting the development of B cells in the germinal center [8].
TOX is related to T cell senescence and exhaustion
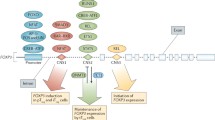
T cell exhaustion is a term used to describe T cells under chronic antigen stimulation that alter or lose their effector function, and this is related to abnormal expression of immune checkpoint proteins in T cells [35,36,37]. Recently, many studies have shown that TOX is a crucial transcription factor involved in exhaustion of CD8+ T cells [1, 2, 4, 32, 38,39,40,41]. For example, in tumor-infiltrating CD8+ T cells from human melanoma and non-small cell lung cancer (NSCLC) samples, TOX had increased expression in CD8+ T cells with high expression of programmed cell death protein 1 (PD-1) [42]. The mechanism by which TOX promotes CD8+ T cell exhaustion may be similar to when antigens continuously stimulate CD8+ T cells where they activate calcium and NFAT signaling, thereby inducing the expression of the TOX in the nucleus. Once the pathway has begun, sustained TOX expression leads to chromosome recombination and changes in RNA transcription, inhibiting its differentiation into effector T cells and entering a state of exhaustion. In the exhaustion stage, the expression of immune checkpoint proteins such as PD-1, T cell immunoglobulin mucin-domain-containing-3 (Tim-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT), and cytotoxic T lymphocyte-associated molecule-4 (CTLA-4) and transcription factors, such as Eomes and TCF1, is increased, while the secretion of cytokines is impeded (Fig. 2) [2, 4, 32, 38,39,40,41,42]. Our recent study also showed that TOX is highly co-expressed with PD-1, Tim-3, and CD244 in CD3+ T cells, particularly in CD3 + CD8+ cells in patients with B cell non-Hodgkin’s lymphoma (B-NHL). Furthermore, the proportions of both TOX+ and TOX + PD-1+ regulatory T cells (Tregs) are also significantly increased in B-NHL patients [43]. This finding may further confirm that TOX contributes to promoting T cell exhaustion in lymphocytic malignancies.

Schematic model of persistent antigen binding and continuous stimulation of CD8+ T cells. Persistent antigens activate calcium and NFAT signaling, inducing the expression of TOX in the nucleus. TOX remodels the chromatin and increases RNA transcription, which results in an increase in immune checkpoint inhibitor proteins and a decrease in cytokines. APC: antigen presenting cell, TCR: T cell receptor
Alteration of TOX in lymphocytic malignancies
Genetic alterations in TOX have been characterized in lymphocytic malignancies, including acute lymphoblastic leukemia (ALL) and lymphoma. According to results from the Cancer Cell Line Encyclopedia database, compared with other cancers, TOX is highly expressed in ALL, particularly in T cell-ALL (T-ALL) [5]. The high expression of TOX is under regulation of the transcription factors T cell acute lymphoblastic leukemia protein 1 (TAL1) and LIM domain only 1/2 (LMO1/2) and binds to the repair factors KU70/KU80 to inhibit their function, causing abnormal non-homologous end joining (NHEJ) repair, thus inhibiting DNA repair in T-ALL cells and reducing genome stability. When TOX was knocked out in T-ALL cells, NHEJ repair could be restored (Fig. 3) [5]. It is well known that instability in the genome may be responsible for the development of T-ALL. TOX is even positively expressed in most ALL cases. In contrast, TOX deletion has also been reported in ALL patients. Molecular analysis of 205 cases with ALL demonstrated that approximately 4% of patients lacked TOX [44]. In addition, TOX deletion was found mostly during relapse for pediatric and adult ALL patients [45, 46]. Moreover, it was found in primary central nervous system lymphomas (PCNSL) that approximately 32% of patients have TOX deletion [47]. The role of TOX deletion in such patients remains unclear. Hence, it is worth investigating whether deletion is related to reduced T cell proliferation and anti-tumor function. Interestingly, the expression of TOX appears to be different in different types of lymphoma. In T cell lymphomas, TOX is highly expressed in mycosis fungoides (MF), precursor T lymphoblastic lymphoma, and angioimmunoblastic T-cell lymphoma (AITL), but it is expressed at a low level in peripheral T-cell lymphoma cell lymphoma (PTCL) and anaplastic large cell lymphoma (ALCL). In B-cell lymphoma, TOX is overexpressed in precursor B lymphoblastic lymphoma (B-LBL), diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL), and a small proportion of Burkitt lymphoma (BL) patients. There was no or only rare TOX expression in chronic lymphocytic leukemia (CLL), classical Hodgkin lymphoma, and myeloma [17].

Mechanism of TOX function in T cell malignancies. a TOX binds to the repair factors KU70/KU80 and inhibits their function, causing abnormal NHEJ repair, leading to DNA repair inhibition. b TOX and GATA3 facilitate the downstream gene ThPOK to inhibit the expression of RUNX3, which leads to inhibition of its tumor-suppressive function
TOX inhibition for immunotherapy
TOX and TOX2 induced T cell exhaustion in different tumor mouse models and cancer patients as well as in patient hematological malignancies [4, 39,40,41,42,43]. TOX is associated with larger tumor size, lower differentiation, later TNM stage, and facilitating endocytic recycling of PD-1 [5]. Moreover, TOX knockdown in a mouse hepatocellular carcinoma (HCC) model or in tumor-infiltrating CD8+ T cells resulted in significant reduction in immune checkpoint proteins, including PD-1, CTLA-4, TIM-3, and TIGIT, ameliorating T cell exhaustion and promoting the antitumor effects of antigen-specific CD8+ T cells [32,33,34]. Moreover, in a chimeric antigen receptor-T cell (CAR-T) therapy tumor model, it was found that CAR-T cells doubly deficient for both TOX and TOX2 (TOX DKO) are more effective than either TOX-deficient or TOX2-deficient CAR-TILs (tumor-infiltrating lymphocytes) in suppressing tumor growth and prolonging the survival of tumor-bearing mice. TOX DKO CAR TILs demonstrated decreased expression of immune checkpoint proteins, increased cytokine secretion, and enhanced antitumor effects [3]. In contrast, knockdown of TOX in T-ALL cells by shRNAs exhibited disruption of the cell cycle and higher levels of apoptosis [5]. Up-regulating TOX may inhibit the expression of tumor suppressor genes, which may be a reason for tumor maintenance. It is known that reduction or deletion of the tumor suppressor gene RUNX3 occurs in many tumors [6, 48, 49]. The TOX-RUNX3 pathway inhibits the expression of RUNX3 when TOX is highly expressed, thereby affecting the tumor suppression function [50]. In cutaneous T-cell lymphoma, TOX and GATA3 can facilitate the expression of the downstream gene ThPOK, inhibiting the expression of RUNX3 (Fig. 3) [51]. Similarly, in Sézary syndrome (SS) cells, the expression of RUNX3 is reversed, and cell viability is reduced after TOX is suppressed by siRNA. Overall, targeted inhibition of TOX and TOX2 may include two therapeutic approaches for lymphocytic malignancies. First, directly inhibiting the proliferation of tumor cells as a targeted approach. Second, reversing T cell exhaustion to restore the anti-tumor function. Moreover, based on the finding that TOX expression was negatively correlated with the effects of PD-1 immunotherapy [40], it has been speculated that combined anti-TOX or anti-TOX2 and immune checkpoint inhibitor therapy may be a new approach for tumor immunotherapy.
Conclusion and further directions
From the initial importance of TOX in the immune system for the development of various malignancies, the function of TOX and the possibility of its clinical application have been increasingly explored. In recent years, reversing T cell exhaustion has been the focus of immunotherapy. How to convert exhausted T cells into active T cells is a problem that many scientists are trying to solve. In hematological malignancies, CAR-T cell therapy has impressive therapeutic efficacy [52]; however, it also faces the drawback that CAR-T cells lose their effects due to rapid exhaustion after injection into patients [37, 52]. On this issue, TOX might be used as a specific target to reverse T cell exhaustion by reducing its expression activity, which can maintain the effects of CAR-T cells in patients for a longer period of time. Moreover, TOX has different characteristics in different lymphocytic malignancies, and the role of TOX in T cell exhaustion in patients with myeloid leukemia is worth further investigation [53]. Overall, TOX and TOX2 might be potential immune biomarkers and targets for hematological malignancy immunotherapy. Moreover, for patients with deleted TOX, it remains an open question how to design TOX-based targeted therapies.
Availability of data and materials
The material supporting the conclusions of this review are included within the article.
Abbreviations
- AILT:
-
Angioimmunoblastic T-cell lymphoma
- ALCL:
-
Anaplastic large cell lymphoma
- ALL:
-
Acute lymphoblastic leukemia
- Bcl-6:
-
B-cell lymphoma 6
- BL:
-
Burkitt lymphoma
- B-LBL:
-
B lymphoblastic lymphoma
- B-NHL:
-
B cell non-Hodgkin’s lymphoma
- CAR-T:
-
Chimeric antigen receptor-T cell
- CLL:
-
Chronic lymphocytic leukemia
- CTLA-4:
-
Cytotoxic T lymphocyte-associated molecule-4
- DLBCL:
-
Diffuse large B cell lymphoma
- FL:
-
Follicular lymphoma
- GWAS:
-
Genome-wide association studies
- HCC:
-
Hepatocellular carcinoma
- HMG-box:
-
High mobility group box
- iPSC:
-
Induced pluripotent stem cells
- LMO1/2:
-
LIM domain only 1/2
- MF:
-
Mycosis fungoides
- NHEJ:
-
Non-homologous end joining
- NK cells:
-
Natural killer cells
- NSCLC:
-
Non-small cell lung cancer
- PCNSL:
-
Primary central nervous system lymphomas
- PD-1:
-
Programmed cell death protein 1
- PP1:
-
Protein phosphatase 1
- PTCL:
-
Peripheral T-cell lymphoma cell lymphoma
- RUNX3:
-
Runt-related transcription factor 3
- SS:
-
Sézary syndrome
- TAL1:
-
T cell acute lymphoblastic leukemia protein 1
- T-ALL:
-
T cell-ALL
- TFH :
-
T follicular helper cell
- TILs:
-
Tumor-infiltrating lymphocytes
- Tim-3:
-
T cell immunoglobulin mucin-domain-containing-3
- TOX:
-
Thymocyte selection-associated HMG BOX
- Tregs:
-
Regulatory T cells
References
Sekine T, Perez-Potti A, Nguyen S, Gorin JB, Wu VH, Gostick E, et al. TOX is expressed by exhausted and polyfunctional human effector memory CD8(+) T cells. Sci Immunol. 2020;5(49):eaba7918.
Cheng Y, Shao Z, Chen L, Zheng Q, Zhang Q, Ding W, Zhang M, Yu Q, Gao D, et al. Role, function and regulation of the thymocyte selection-associated high mobility group box protein in CD8(+) T cell exhaustion. Immunol Lett. 2021;229:1–7. https://doi.org/10.1016/j.imlet.2020.11.004.
Aliahmad P, Seksenyan A, Kaye J. The many roles of TOX in the immune system. Curr Opin Immunol. 2012;24(2):173–7. https://doi.org/10.1016/j.coi.2011.12.001.
Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A, Wu CJ, Lu LF, Hogan PG, Bhandoola A, Rao A, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A. 2019;116(25):12410–5. https://doi.org/10.1073/pnas.1905675116.
Lobbardi R, Pinder J, Martinez-Pastor B, Theodorou M, Blackburn JS, Abraham BJ, Namiki Y, Mansour M, Abdelfattah NS, Molodtsov A, Alexe G, Toiber D, de Waard M, Jain E, Boukhali M, Lion M, Bhere D, Shah K, Gutierrez A, Stegmaier K, Silverman LB, Sadreyev RI, Asara JM, Oettinger MA, Haas W, Look AT, Young RA, Mostoslavsky R, Dellaire G, Langenau DM, et al. TOX regulates growth, DNA repair, and genomic instability in T-cell acute lymphoblastic leukemia. Cancer Discov. 2017;7(11):1336–53. https://doi.org/10.1158/2159-8290.CD-17-0267.
Han YX, Liang DY. The role of the tumor suppressor RUNX3 in giant cell tumor of the bone. Int J Oncol. 2012;40(3):673–8. https://doi.org/10.3892/ijo.2011.1249.
Vong QP, Leung WH, Houston J, Li Y, Rooney B, Holladay M, Oostendorp RAJ, Leung W, et al. TOX2 regulates human natural killer cell development by controlling T-BET expression. Blood. 2014;124(26):3905–13. https://doi.org/10.1182/blood-2014-06-582965.
Xu W, Zhao X, Wang X, Feng H, Gou M, Jin W, et al. The Transcription Factor Tox2 Drives T Follicular Helper Cell Development via Regulating Chromatin Accessibility. Immunity. 2019;51(5):826–39 e825.
Yuan SH, Qiu Z, Ghosh A. TOX3 regulates calcium-dependent transcription in neurons. Proc Natl Acad Sci U S A. 2009;106(8):2909–14. https://doi.org/10.1073/pnas.0805555106.
Bastien D, Bellver Landete V, Lessard M, Vallières N, Champagne M, Takashima A, et al. IL-1α gene deletion protects Oligodendrocytes after spinal cord injury through Upregulation of the survival factor Tox3. J Neurosci. 2015;35(30):10715–30. https://doi.org/10.1523/JNEUROSCI.0498-15.2015.
Han YJ, Zhang J, Zheng Y, Huo D, Olopade OI. Genetic and epigenetic regulation of TOX3 expression in breast Cancer. PLoS One. 2016;11(11):e0165559. https://doi.org/10.1371/journal.pone.0165559.
Zeng D, Lin H, Cui J, Liang W. TOX3 is a favorable prognostic indicator and potential immunomodulatory factor in lung adenocarcinoma. Oncol Lett. 2019;18(4):4144–52. https://doi.org/10.3892/ol.2019.10748.
He Y, Liu H, Chen Q, Shao Y, Luo S. Relationships between SNPs and prognosis of breast cancer and pathogenic mechanism. Mol Genet Genomic Med. 2019;7(9):e871. https://doi.org/10.1002/mgg3.871.
Vanheer L, Song J, De Geest N, Janiszewski A, Talon I, Provenzano C, et al. Tox4 modulates cell fate reprogramming. J Cell Sci. 2019;132:20.
Ding L, Paszkowski-Rogacz M, Winzi M, Chakraborty D, Theis M, Singh S, Ciotta G, Poser I, Roguev A, Chu WK, Choudhary C, Mann M, Stewart AF, Krogan N, Buchholz F, et al. Systems analyses reveal shared and diverse attributes of Oct4 regulation in pluripotent cells. Cell systems. 2015;1(2):141–51. https://doi.org/10.1016/j.cels.2015.08.002.
O'Flaherty E, Kaye J. TOX defines a conserved subfamily of HMG-box proteins. BMC Genomics. 2003;4(1):13. https://doi.org/10.1186/1471-2164-4-13.
Maestre L, Garcia-Garcia JF, Jimenez S, Reyes-Garcia AI, Garcia-Gonzalez A, Montes-Moreno S, et al. High-mobility group box (TOX) antibody a useful tool for the identification of B and T cell subpopulations. PLoS One. 2020;15(2):e0229743. https://doi.org/10.1371/journal.pone.0229743.
Arora M, Kumari S, Singh J, Chopra A, Chauhan SS. Expression pattern, regulation, and clinical significance of TOX in breast cancer. Cancer Immunol Immunother. 2021;70(2):349–63. https://doi.org/10.1007/s00262-020-02689-3.
Wilkinson B, Chen JY, Han P, Rufner KM, Goularte OD, Kaye J. TOX: an HMG box protein implicated in the regulation of thymocyte selection. Nat Immunol. 2002;3(3):272–80. https://doi.org/10.1038/ni767.
Yun S, Lee SH, Yoon SR, Kim MS, Piao ZH, Myung PK, Kim TD, Jung H, Choi I, et al. TOX regulates the differentiation of human natural killer cells from hematopoietic stem cells in vitro. Immunol Lett. 2011;136(1):29–36. https://doi.org/10.1016/j.imlet.2010.11.008.
Aliahmad P, de la Torre B, Kaye J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat Immunol. 2010;11(10):945–52. https://doi.org/10.1038/ni.1930.
Nagase T, Ishikawa K, Suyama M, Kikuno R, Miyajima N, Tanaka A, Kotani H, Nomura N, Ohara O, et al. Prediction of the coding sequences of unidentified human genes. XI. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 1998;5(5):277–86. https://doi.org/10.1093/dnares/5.5.277.
Lee JH, You J, Dobrota E, Skalnik DG. Identification and characterization of a novel human PP1 phosphatase complex. J Biol Chem. 2010;285(32):24466–76. https://doi.org/10.1074/jbc.M110.109801.
Hosking BM, Wyeth JR, Pennisi DJ, Wang SC, Koopman P, Muscat GE. Cloning and functional analysis of the Sry-related HMG box gene, Sox18. Gene. 2001;262(1–2):239–47. https://doi.org/10.1016/S0378-1119(00)00525-4.
Bowles J, Schepers G, Koopman P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol. 2000;227(2):239–55. https://doi.org/10.1006/dbio.2000.9883.
Yuan S, Liu Z, Xu Z, Liu J, Zhang J. High mobility group box 1 (HMGB1): a pivotal regulator of hematopoietic malignancies. J Hematol Oncol. 2020;13(1):91. https://doi.org/10.1186/s13045-020-00920-3.
Wang S, Zhang Y. HMGB1 in inflammation and cancer. J Hematol Oncol. 2020;13(1):116. https://doi.org/10.1186/s13045-020-00950-x.
Tan G, Huang C, Chen J, Zhi F. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J Hematol Oncol. 2020;13(1):149. https://doi.org/10.1186/s13045-020-00985-0.
Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799(1–2):101–13. https://doi.org/10.1016/j.bbagrm.2009.09.008.
Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5(5):504–14. https://doi.org/10.1016/j.stem.2009.08.018.
Jin S, Yang Z, Hao X, Tang W, Ma W, Zong H. Roles of HMGB1 in regulating myeloid-derived suppressor cells in the tumor microenvironment. Biomarker Res. 2020;8(1):21. https://doi.org/10.1186/s40364-020-00201-8.
Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, Wherry EJ, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571(7764):211–8. https://doi.org/10.1038/s41586-019-1325-x.
Aliahmad P, O'Flaherty E, Han P, Goularte OD, Wilkinson B, Satake M, Molkentin JD, Kaye J, et al. TOX provides a link between calcineurin activation and CD8 lineage commitment. J Exp Med. 2004;199(8):1089–99. https://doi.org/10.1084/jem.20040051.
Aliahmad P, Kaye J. Development of all CD4 T lineages requires nuclear factor TOX. J Exp Med. 2008;205(1):245–56. https://doi.org/10.1084/jem.20071944.
Kasakovski D, Xu L, Li Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J Hematol Oncol. 2018;11(1):91. https://doi.org/10.1186/s13045-018-0629-x.
Kong Y, Jia B, Zhao C, Claxton DF, Sharma A, Annageldiyev C, Fotos JS, Zeng H, Paulson RF, Prabhu KS, Zheng H, et al. Downregulation of CD73 associates with T cell exhaustion in AML patients. J Hematol Oncol. 2019;12(1):40. https://doi.org/10.1186/s13045-019-0728-3.
Zhang H, Zhao P, Huang H. Engineering better chimeric antigen receptor T cells. Exp Hematol Oncol. 2020;9(1):34. https://doi.org/10.1186/s40164-020-00190-2.
Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, Heuston EF, Kirby M, Anderson S, Cheng J, Khan O, Handon R, Reilley J, Fioravanti J, Hu J, Gossa S, Wherry EJ, Gattinoni L, McGavern DB, O’Shea JJ, Schwartzberg PL, Wu T, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol. 2019;20(7):890–901. https://doi.org/10.1038/s41590-019-0403-4.
Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, Zamarin D, Walther T, Snyder A, Femia MR, Comen EA, Wen HY, Hellmann MD, Anandasabapathy N, Liu Y, Altorki NK, Lauer P, Levy O, Glickman MS, Kaye J, Betel D, Philip M, Schietinger A, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571(7764):270–4. https://doi.org/10.1038/s41586-019-1324-y.
Wang X, He Q, Shen H, Xia A, Tian W, Yu W, Sun B, et al. TOX promotes the exhaustion of antitumor CD8(+) T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol. 2019;71(4):731–41. https://doi.org/10.1016/j.jhep.2019.05.015.
Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y, Eisenberg V, Wohlleber D, Steiger K, Merkler D, Delorenzi M, Knolle PA, Cohen CJ, Thimme R, Youngblood B, Zehn D, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571(7764):265–9. https://doi.org/10.1038/s41586-019-1326-9.
Kim K, Park S, Park SY, Kim G, Park SM, Cho JW, Kim DH, Park YM, Koh YW, Kim HR, Ha SJ, Lee I, et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020;12(1):22. https://doi.org/10.1186/s13073-020-00722-9.
Huang S, Liang C, Zhao Y, Deng T, Tan J, Lu Y, et al. Increased TOX expression concurrent with PD-1, Tim-3, and CD244 in T cells from patients with non-Hodgkin lymphoma. Asia-Pac J Clin Oncol. 2021; doi.org/10.1111/ajco.13545.
Bardet V, Couque N, Cattolico L, Hetet G, Devaux I, Duprat S, Gressin L, Vilmer E, Cavé H, Grandchamp B, et al. Molecular analysis of nonrandom 8q12 deletions in acute lymphoblastic leukemia: identification of two candidate genes. Genes Chrom Cancer. 2002;33(2):178–87. https://doi.org/10.1002/gcc.10014.
Safavi S, Hansson M, Karlsson K, Biloglav A, Johansson B, Paulsson K. Novel gene targets detected by genomic profiling in a consecutive series of 126 adults with acute lymphoblastic leukemia. Haematologica. 2015;100(1):55–61. https://doi.org/10.3324/haematol.2014.112912.
Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322(5906):1377–80. https://doi.org/10.1126/science.1164266.
Schwindt H, Vater I, Kreuz M, Montesinos-Rongen M, Brunn A, Richter J, Gesk S, Ammerpohl O, Wiestler OD, Hasenclever D, Deckert M, Siebert R, et al. Chromosomal imbalances and partial uniparental disomies in primary central nervous system lymphoma. Leukemia. 2009;23(10):1875–84. https://doi.org/10.1038/leu.2009.120.
Lee CW, Ito K, Ito Y. Role of RUNX3 in bone morphogenetic protein signaling in colorectal cancer. Cancer Res. 2010;70(10):4243–52. https://doi.org/10.1158/0008-5472.CAN-09-3805.
Ito K, Lim AC, Salto-Tellez M, Motoda L, Osato M, Chuang LS, et al. RUNX3 attenuates beta-catenin/T cell factors in intestinal tumorigenesis. Cancer Cell. 2008;14(3):226–37. https://doi.org/10.1016/j.ccr.2008.08.004.
Dulmage BO, Akilov O, Vu JR, Falo LD, Geskin LJ. Dysregulation of the TOX-RUNX3 pathway in cutaneous T-cell lymphoma. Oncotarget. 2019;10(33):3104–13. https://doi.org/10.18632/oncotarget.5742.
Xiong Y, Castro E, Yagi R, Zhu J, Lesourne R, Love PE, Feigenbaum L, Bosselut R, et al. Thpok-independent repression of Runx3 by Gata3 during CD4+ T-cell differentiation in the thymus. Eur J Immunol. 2013;43(4):918–28. https://doi.org/10.1002/eji.201242944.
Yu J, Wu X, Yan J, Yu H, Xu L, Chi Z, Sheng X, Si L, Cui C, Dai J, Ma M, Xu T, Kong Y, Guo J, et al. Anti-GD2/4-1BB chimeric antigen receptor T cell therapy for the treatment of Chinese melanoma patients. J Hematol Oncol. 2018;11(1):1. https://doi.org/10.1186/s13045-017-0548-2.
Chen C, Liang C, Wang S, Chio C, Zhang Y, Zeng C, et al. Expression patterns of immune checkpoints in acute myeloid leukemia. J Hematol Oncol. 2020;13(1):28. https://doi.org/10.1186/s13045-020-00853-x.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the National Natural Science Foundation of China (Nos. 82070152, 81770152, and 81570143), and the Guangzhou Science and Technology Project (Nos. 201807010004 and 201803040017).
Author information
Authors and Affiliations
Contributions
YQL and SHC: contributed to the concept development, reviewed the design, and revised the manuscript. CFL: reviewed the references, wrote the manuscript, and prepared the figures. SXH and YJZ helped to review the references and prepared figures. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, C., Huang, S., Zhao, Y. et al. TOX as a potential target for immunotherapy in lymphocytic malignancies. Biomark Res 9, 20 (2021). https://doi.org/10.1186/s40364-021-00275-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-021-00275-y