
Abstract
Therapeutic hypothermia is an effective cytoprotectant and promising intervention shown to improve outcome in patients following cardiac arrest and neonatal hypoxia-ischemia. However, despite our clinical and experimental experiences, the protective molecular mechanisms of therapeutic hypothermia remain to be elucidated. Therefore, in this brief overview we discuss both the clinical evidence and molecular mechanisms of therapeutic hypothermia in order to provide further insights into this promising intervention.
Similar content being viewed by others


Introduction
Clinicians have investigated the application of therapeutic hypothermia on the human body for centuries. Increasing clinical evidence from meta-analysis of large randomized controlled trials and experimental data advocates the induction of therapeutic hypothermia as a tool to achieve neuroprotection [1]. Clinical indications for therapeutic hypothermia as a protection strategy include: myocardial infarction [2],[3], cardiopulmonary bypass in adults [4], pediatric open heart surgery [5],[6], stroke [7], neonatal hypoxia-ischemia [8]-[10], and traumatic brain injury [11],[12].
To date, the strongest evidence for its efficacy exists for various clinical situations following cardiac arrest and neonatal hypoxia-ischemia. However, the effective application of hypothermia requires a thorough understanding of the injury mechanisms as well as its protective mechanisms. Therefore, we discuss in this brief overview both the clinical factors and molecular mechanisms of therapeutic hypothermia in order to provide further insights into this promising intervention.
Review
Therapeutic hypothermia: clinical evidence
Early experiences with therapeutic hypothermia in the 1940s thru 1960s falsely assumed that the protective effects were only due to the temperature-dependent reduction in metabolism, which leads to lower oxygen and glucose demands [13]. Therefore, patients were routinely subjected to deep hypothermia (<30°C) with varying durations ranging from 2 to 10 days [14]. Animal experiments in the 1980s led to the breakthrough discovery that using mild to moderate hypothermia (31°C to 35°C) resulted in improved neurological outcome with fewer and less severe side effects [15]. More importantly, these findings led to the realization that hypothermia-induced neuroprotection is not only limited to decreased oxygen and glucose demands, but the mechanisms involved are indeed much more intricate.
Three large multicenter randomized studies of newborn infants with hypoxic ischemic encephalopathy suggest a beneficial effect in this patient population. Gluckman et al. demonstrated an improved outcome that persisted at 18 months of life in term infants suffering from moderate neonatal encephalopathy who were subjected to head cooling (CoolCap, Natus, San Carlos, CA, USA) for 72 h [10]. A second trial demonstrated whole-body cooling to 33.5°C for 72 h reduced the risk of mortality or moderate to severe disability in infants with moderate or severe encephalopathy surveyed at 18 to 22 months of age [16]. A third published Total Body Hypothermia for Neonatal Encephalopathy (TOBY) trial also showed benefits from similar whole-body cooling in newborns with perinatal asphyxia [17]. The study showed that hypothermia did not significantly reduce the rate of mortality or severe disability but resulted in improved neurologic outcomes in infants assessed at 18 months of age. However, the criteria for optimal candidates for therapeutic hypothermia have yet to be defined, and long-term follow-up (beyond 18 months of age) to assess the persistent and lifelong benefits are needed.
Current investigations also include the Infant Cooling Evaluation (ICE) trial to investigate the effect of moderate whole-body hypothermia to 33.5°C for 72 h in newborns with HIE [18] and the Therapeutic Hypothermia after Pediatric Cardiac Arrest (THAPCA) trials, a 30-site randomized clinical trial investigating the effectiveness of therapeutic hypothermia versus therapeutic normothermia after in-hospital (THAPCA-IH) or out-of-hospital (THAPCA-OH) cardiac arrest in children [19],[20].
Clinical issues
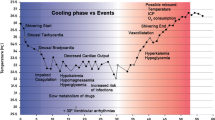
Clinical issues regarding optimal target temperature, rate of cooling, duration of cooling, rate of rewarming, as well as optimal treatment window need further investigation. With the introduction of therapeutic hypothermia, it is important to differentiate between the ‘induction phase’ , when the temperature drops; the ‘maintenance phase’ , when the target temperature is achieved and maintained at the desired level; and the ‘rewarming phase’ , when the patient is slowly rewarmed back to normothermia.
Hypothermia induces stress responses, such as shivering, that result in increased oxygen consumption and metabolic rate in non-sedated patients. Animal experiments suggest that the neuroprotective effects of hypothermia are negated if cooling is used on non-sedated animals [21],[22]. Other side effects include increased risk of infection, cold diuresis and hypovolemia, electrolyte disorders, insulin resistance, impaired drug clearance, and mild coagulopathy [15].
Therapeutic hypothermia: molecular mechanisms
Therapeutic hypothermia-induced cellular protection against anoxic brain injury is a global process affecting multiple molecular and cellular mechanisms. Cooling results in a 6% to 10% decrease in cerebral metabolism for every 1°C reduction in body temperature [23]. Therefore, a thorough understanding of the underlying mechanisms of protection induced by therapeutic hypothermia is vital for designing appropriate and effective treatments. Insufficient knowledge of the physiological changes and side effects that occur during mild (34°C to 35.9°C), moderate (32°C to 33.9°C), moderate-deep (30°C to 31.9°C), and deep (<30°C) hypothermia is likely to lead to lower therapeutic efficacy or even failure of treatment [15].
Ischemic brain injury, reperfusion injury, and secondary brain damage are the three main categories of temperature-dependent injury processes that can be effectively mitigated by mild to moderate hypothermia. Due to the broad effects of hypothermia, it is more clinically effective than treatments that focus on blocking just one of these processes.
Variable factors such as type of injury (traumatic versus pure ischemia) and patient physiology (genetic factors, age, gender, etc.) all contribute to the complicated injury processes. Although the window of opportunity to initiate hypothermic treatment as well as the duration of cooling to achieve full efficacy may vary, the success or failure of therapeutic hypothermia treatment is dependent on the following four key factors [24],[15]:
-
1.
Rate of cooling - rapid initiation of cooling immediately after injury resulted in better outcomes in animal studies. A concept summarized by the phrase ‘time is brain’.
-
2.
Duration of cooling - dependent of severity of injury and the time allotted to achieve target temperature.
-
3.
Rate of rewarming - slow rewarming is critical to not reactivate initial injury processes.
-
4.
Management and prevention of side effects.
Apoptosis and mitochondrial dysfunction
Ischemia/reperfusion injuries can lead to cellular necrosis or apoptosis, also referred to as programmed cell death. The development of apoptosis is dependent on several cellular processes, including mitochondria dysfunction, activation of caspase enzymes, and other cellular energy metabolism disorders. Hypothermia has been observed to affect almost all of these injury processes, thereby preventing the initiation or interrupting the early stages of the apoptotic pathway [25]. The window of opportunity to initiate therapeutic hypothermia to mitigate apoptosis after anoxia is broad, as apoptosis begins relatively late and continues for a long duration. Therefore, mitigating the effects of these injury processes of the apoptotic pathway is a viable treatment for neuroprotection in patients.
Interrupted blood supply to the brain causes an immediate reduction in ATP and phosphocreatine, initiating a switch in intracellular metabolism to anaerobic glycolysis [26]. As a result, intracellular levels of inorganic phosphate, lactate, and H+ are dramatically increased, leading to both intracellular and extracellular acidosis and calcium (Ca2+) influx into the cells [27]. To further exacerbate the injury process, acidosis and the lack of ATP inhibit cellular mechanisms normally responsible for controlling excessive intracellular Ca2+, such as ATP-dependent Na+/K+ pumps and Na+, K+, and Ca+ channels [28]. The excess influx of Ca2+ eventually leads to mitochondria dysfunction and activation of the caspase-9 intrinsic apoptotic pathway. Data from experimental studies have proven that hypothermia has a beneficial impact on the ion pump dysfunction and reduces the influx of calcium into the cells, thereby decreasing neurotoxicity [28],[29].
Inflammation
Ischemia/reperfusion injury stimulates innate immune responses, which can lead to secondary brain injury. It triggers the release of pro-inflammatory cytokines (i.e. IL-1β, TNFα, and IL-6) [30], the chemokine MCP1 [31] and pro-inflammatory mediators (ROS and NOS) in microglia, and circulating leukocytes [32]. Hypothermia attenuates many aspects of this pro-inflammatory immune response, but it also reduces the expression of some anti-inflammatory cytokines (IL-10 and TGFβ) [33]. Animal studies showed controversial effects of hypothermia on NF-κB, a transcription factor that plays a central role in regulating the inflammatory responses [34]-[37]. Yenari et al. showed that mild hypothermia decreased NF-κB translocation and activation in rodents [37]. In contrast to Fairchild et al. who showed in cell culture studies after moderate hypothermia a prolonged accumulation of NF-κB in the nucleus. These findings were explained with a delayed phosphorylation of IκBα, the cytosolic inhibitory protein of NF-κB. The Janus kinase (JAK) and signal transducer and activator of transcription pathway (STAT) is a common signaling pathway used by many cytokines. Ischemia leads to a JAK2 and STAT3 activation mediated by IL-6. Inhibiting this pathway resulted in a decreased number of apoptotic cells [34]. Mild hypothermia attenuates STAT3 expression which might be a possible mechanism of hypothermia-induced neuroprotection [38].
Blood-brain barrier disruption
Mild to moderate hypothermia has been shown to significantly reduce blood-brain barrier disruptions following ischemia-reperfusion injury by decreasing vascular permeability, resulting in decreased edema formation [39]-[41]. Specifically, hypothermia suppresses the activation of matrix metalloproteinases (MMPs) responsible for degradation of the extracellular matrix while increasing the expression of endogenous MMP inhibitors, such as metalloproteinase inhibitor 2 (TIMP2) [42]. As a result, the structural proteins and cells that constitute the BBB are preserved and the opening of water channels is prevented.
Free radical production
The inflammatory response induced by ischemia/reperfusion injury usually coincides with the production of free oxygen radicals such as superoxide (O2−), peroxynitrite (NO2−), hydrogen peroxide (H2O2), and hydroxyl radicals (OH−) [43]. The extent of free radical production following ischemia/reperfusion is so large that protective cellular antioxidant mechanisms are overwhelmed, resulting in peroxidation of lipids, proteins, and nucleic acids [27]. Although hypothermia cannot completely diminish free radical production in injured cells, it can significantly reduce the amount of free radicals enough to allow for the endogenous antioxidant mechanisms to mitigate the oxidative damage. In addition, the suppression of free radical production has been observed to be linearly proportional to the decrease in temperature [28].
Rescue gene RBM3
Although hypothermia reduces cellular metabolism and down-regulates global protein synthesis in mammalian cells, a small subset of cold-shock proteins are induced by low temperature. RNA-binding motif protein 3 (RBM3), a member of the glycine-rich RNA-binding protein (GRP) family, is one of the first proteins synthesized in response to hypothermia [44] and hypoxia [45]. Additionally, RBM3 expression is elevated in response to NMDA-type glutamate receptor activation [46]. Mild hypothermia is sufficient to induce the expression of RBM3 [44], while elevated temperature has been shown to suppress RBM3 expression [47]. We observed that RBM3 expression is induced by moderate hypothermia (33.5°C) in murine organotypic hippocampal slice cultures (OHSCs) and hippocampal neurons (HT-22), but not by deep hypothermia (17°C) nor in microglia cells (BV-2) at both cooling temperatures [48].
Although many studies on RBM3 have been focused on their regulation in non-neuronal cells in response to hypothermia and other stress factors [44], there is growing interest in their role as effectors in therapeutic hypothermia-induced neuroprotection. RBM3 is widely expressed during the early stages of brain development, especially in the first to second postnatal weeks, where it is dynamically regulated [49]. Recent studies have concluded that RBM3 has anti-apoptotic functions and can enhance cell proliferation [50]. RBM3 is induced by hypoxia independent of HIF-1 [45], which itself is suppressed by mild hypothermia [51]. Furthermore, RBM3 has been shown to play a major role in promoting translation in neuronal cells, and recently, RBM3 up-regulation in neuronal cells in response to hypothermia has been implicated in hypothermia-induced neuroprotection [52].
Drug-induced hypothermia
Drug-induced hypothermia may serve as an alternative to physical hypothermia, which can be technically tedious to apply and may cause serious side effects. Animal studies have shown that hypothermia is induced by synthetic cannabinoid CB1 agonists WIN55212-2 and HU-210. Intramuscular injection of WIN55212-2 induced rapid and prolonged hypothermia in a dose-dependent manner in rats [53], and HU-210 was observed to be protective against ischemic damage by reducing infarct volumes and motor dysfunction [54]. In the same study, HU-210 injected after ischemic onset resulted in deeper and lengthier hypothermia.
Conclusions
Temperature management continues to play an important role in the treatment of patients suffering from neurological injuries. Therapeutic hypothermia and it's proven ability to attenuate post-ischemic injury represents a quantum leap in the clinical setting [15], but more work is needed, particularly in infants and children suffering from asphyxial cardiac arrest. Additionally, the induced hypothermic effects of cannabinoid CB1 receptor agonists, WIN55212-2 and HU-210, as alternatives to or in combination with therapeutic hypothermia also warrant further investigation. The full beneficial effect of cooling remains to be discovered as we optimize the process by expanding our knowledge of the basic mechanisms, verify the treatment windows, and improve the cooling and monitoring devices in the coming years.
References
Drury PP, Gunn ER, Bennet L, Gunn AJ: Mechanisms of hypothermic neuroprotection. Clin Perinatol 2014, 41(1):161–175. 10.1016/j.clp.2013.10.005
Holzer M, Behringer W: Therapeutic hypothermia after cardiac arrest and myocardial infarction. Best Pract Res Clin Anaesthesiol 2008, 22(4):711–728. 10.1016/j.bpa.2008.02.001
Oommen SS, Menon V: Hypothermia after cardiac arrest: beneficial, but slow to be adopted. Cleve Clin J Med 2011, 78(7):441–448. 10.3949/ccjm.78a.10157
Svensson LG, Crawford ES, Hess KR, Coselli JS, Raskin S, Shenaq SA, Safi HJ: Deep hypothermia with circulatory arrest. Determinants of stroke and early mortality in 656 patients. J Thorac Cardiovasc Surg 1993, 106(1):19–28. discussion 28–31
Bellinger DC, Wypij D, Kuban KC, Rappaport LA, Hickey PR, Wernovsky G, Jonas RA, Newburger JW: Developmental and neurological status of children at 4 years of age after heart surgery with hypothermic circulatory arrest or low-flow cardiopulmonary bypass. Circulation 1999, 100(5):526–532. 10.1161/01.CIR.100.5.526
2005 international consensus on cardiopulmonary resuscitation and emergency cardiovascular care science with treatment recommendations. Part 6: paediatric basic and advanced life support Resuscitation 2005, 67(2–3):271–291.
Maier CM, Ahern K, Cheng ML, Lee JE, Yenari MA, Steinberg GK: Optimal depth and duration of mild hypothermia in a focal model of transient cerebral ischemia: effects on neurologic outcome, infarct size, apoptosis, and inflammation. Stroke 1998, 29(10):2171–2180. 10.1161/01.STR.29.10.2171
Eicher DJ, Wagner CL, Katikaneni LP, Hulsey TC, Bass WT, Kaufman DA, Horgan MJ, Languani S, Bhatia JJ, Givelichian LM, Sankaran K, Yager JY: Moderate hypothermia in neonatal encephalopathy: efficacy outcomes. Pediatr Neurol 2005, 32(1):11–17. 10.1016/j.pediatrneurol.2004.06.014
Edwards AD, Azzopardi DV: Therapeutic hypothermia following perinatal asphyxia. Arch Dis Child Fetal Neonatal Ed 2006, 91(2):F127-F131. 10.1136/adc.2005.071787
Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, Whitelaw A, Gunn AJ: Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 2005, 365(9460):663–670. 10.1016/S0140-6736(05)17946-X
Marion DW, Penrod LE, Kelsey SF, Obrist WD, Kochanek PM, Palmer AM, Wisniewski SR, DeKosky ST: Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med 1997, 336(8):540–546. 10.1056/NEJM199702203360803
Li P, Yang C: Moderate hypothermia treatment in adult patients with severe traumatic brain injury: a meta-analysis. Brain Inj 2014, 28(8):1036–1041. 10.3109/02699052.2014.910609
Fay T: Early experiences with local and generalized refrigeration of the human brain. J Neurosurg 1959, 16(3):239–259. discussion 259–260 10.3171/jns.1959.16.3.0239
Shulman K, Rosomoff HL: Effect of hypothermia on mortality in experimental injury to the brain. Am J Surg 1959, 98: 704–705. 10.1016/0002-9610(59)90495-7
Polderman KH: Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med 2009, 37(7 Suppl):S186-S202. 10.1097/CCM.0b013e3181aa5241
Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, Fanaroff AA, Poole WK, Wright LL, Higgins RD, Finer NN, Carlo WA, Duara S, Oh W, Cotten CM, Stevenson DK, Stoll BJ, Lemons JA, Guillet R, Jobe AH: Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med 2005, 353(15):1574–1584. 10.1056/NEJMcps050929
Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, Kapellou O, Levene M, Marlow N, Porter E, Thoresen M, Whitelaw A, Brocklehurst P: Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med 2009, 361(14):1349–1358. 10.1056/NEJMoa0900854
Jacobs SE, Morley CJ, Inder TE, Stewart MJ, Smith KR, McNamara PJ, Wright IM, Kirpalani HM, Darlow BA, Doyle LW: Whole-body hypothermia for term and near-term newborns with hypoxic-ischemic encephalopathy: a randomized controlled trial. Arch Pediatr Adolesc Med 2011, 165(8):692–700. 10.1001/archpediatrics.2011.43
Pemberton VL, Browning B, Webster A, Dean JM, Moler FW: Therapeutic hypothermia after pediatric cardiac arrest trials: the vanguard phase experience and implications for other trials. Pediatr Crit Care Med 2013, 14(1):19–26. 10.1097/PCC.0b013e31825b860b
Moler FW, Silverstein FS, Meert KL, Clark AE, Holubkov R, Browning B, Slomine BS, Christensen JR, Dean JM: Rationale, timeline, study design, and protocol overview of the therapeutic hypothermia after pediatric cardiac arrest trials. Pediatr Crit Care Med 2013, 14(7):e304-e315. 10.1097/PCC.0b013e31828a863a
Thoresen M, Satas S, Loberg EM, Whitelaw A, Acolet D, Lindgren C, Penrice J, Robertson N, Haug E, Steen PA: Twenty-four hours of mild hypothermia in unsedated newborn pigs starting after a severe global hypoxic-ischemic insult is not neuroprotective. Pediatr Res 2001, 50(3):405–411. 10.1203/00006450-200109000-00017
Tooley JR, Satas S, Porter H, Silver IA, Thoresen M: Head cooling with mild systemic hypothermia in anesthetized piglets is neuroprotective. Ann Neurol 2003, 53(1):65–72. 10.1002/ana.10402
Hagerdal M, Harp J, Nilsson L, Siesjo BK: The effect of induced hypothermia upon oxygen consumption in the rat brain. J Neurochem 1975, 24(2):311–316. 10.1111/j.1471-4159.1975.tb11881.x
Lampe JW, Becker LB: State of the art in therapeutic hypothermia. Annu Rev Med 2011, 62: 79–93. 10.1146/annurev-med-052009-150512
Xu L, Yenari MA, Steinberg GK, Giffard RG: Mild hypothermia reduces apoptosis of mouse neurons in vitro early in the cascade. J Cereb Blood Flow Metab 2002, 22(1):21–28. 10.1097/00004647-200201000-00003
Small DL, Morley P, Buchan AM: Biology of ischemic cerebral cell death. Prog Cardiovasc Dis 1999, 42(3):185–207. 10.1016/S0033-0620(99)70002-2
Belch JJ: Free radicals and their scavenging in stroke. Scott Med J 1992, 37(3):67–68.
Hall ED: Brain attack. Acute therapeutic interventions. Free radical scavengers and antioxidants. Neurosurg Clin N Am 1997, 8(2):195–206.
Siesjo BK, Bengtsson F, Grampp W, Theander S: Calcium, excitotoxins, and neuronal death in the brain. Ann N Y Acad Sci 1989, 568: 234–251. 10.1111/j.1749-6632.1989.tb12513.x
Wang Q, Tang XN, Yenari MA: The inflammatory response in stroke. J Neuroimmunol 2007, 184(1–2):53–68. 10.1016/j.jneuroim.2006.11.014
Meybohm P, Gruenewald M, Zacharowski KD, Albrecht M, Lucius R, Fosel N, Hensler J, Zitta K, Bein B: Mild hypothermia alone or in combination with anesthetic post-conditioning reduces expression of inflammatory cytokines in the cerebral cortex of pigs after cardiopulmonary resuscitation. Crit Care 2010, 14(1):R21. 10.1186/cc8879
Perrone S, Szabo M, Bellieni CV, Longini M, Bango M, Kelen D, Treszl A, Negro S, Tataranno ML, Buonocore G: Whole body hypothermia and oxidative stress in babies with hypoxic-ischemic brain injury. Pediatr Neurol 2010, 43(4):236–240. 10.1016/j.pediatrneurol.2010.05.009
Matsui T, Kakeda T: IL-10 production is reduced by hypothermia but augmented by hyperthermia in rat microglia. J Neurotrauma 2008, 25(6):709–715. 10.1089/neu.2007.0482
Fairchild KD, Singh IS, Carter HC, Hester L, Hasday JD: Hypothermia enhances phosphorylation of I{kappa}B kinase and prolongs nuclear localization of NF-{kappa}B in lipopolysaccharide-activated macrophages. Am J Physiol Cell Physiol 2005, 289(5):22. 10.1152/ajpcell.00152.2005
Gibbons H, Dragunow M: Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Res 2006, 1084: 1–15. 10.1016/j.brainres.2006.02.032
Han HS, Karabiyikoglu M, Kelly S, Sobel RA, Yenari MA: Mild hypothermia inhibits nuclear factor-[kgr]B translocation in experimental stroke. J Cereb Blood Flow Metab 2003, 23(5):589–598. 10.1097/01.WCB.0000059566.39780.8D
Webster CM, Kelly S, Koike MA, Chock VY, Giffard RG, Yenari MA: Inflammation and NF[kappa]B activation is decreased by hypothermia following global cerebral ischemia. Neurobiol Dis 2009, 33(2):301–312. 10.1016/j.nbd.2008.11.001
Choi JS, Park J, Suk K, Moon C, Park YK, Han HS: Mild hypothermia attenuates intercellular adhesion molecule-1 induction via activation of extracellular signal-regulated kinase-1/2 in a focal cerebral ischemia model. Stroke Res Treat 2011, 2011: 846716.
Huang ZG, Xue D, Preston E, Karbalai H, Buchan AM: Biphasic opening of the blood-brain barrier following transient focal ischemia: effects of hypothermia. Can J Neurol Sci 1999, 26(4):298–304.
Jurkovich GJ, Pitt RM, Curreri PW, Granger DN: Hypothermia prevents increased capillary permeability following ischemia-reperfusion injury. J Surg Res 1988, 44(5):514–521. 10.1016/0022-4804(88)90156-4
Lee JE, Yoon YJ, Moseley ME, Yenari MA: Reduction in levels of matrix metalloproteinases and increased expression of tissue inhibitor of metalloproteinase-2 in response to mild hypothermia therapy in experimental stroke. J Neurosurg 2005, 103(2):289–297. 10.3171/jns.2005.103.2.0289
Baumann E, Preston E, Slinn J, Stanimirovic D: Post-ischemic hypothermia attenuates loss of the vascular basement membrane proteins, agrin and SPARC, and the blood-brain barrier disruption after global cerebral ischemia. Brain Res 2009, 1269: 185–197. 10.1016/j.brainres.2009.02.062
Globus MY, Alonso O, Dietrich WD, Busto R, Ginsberg MD: Glutamate release and free radical production following brain injury: effects of posttraumatic hypothermia. J Neurochem 1995, 65(4):1704–1711. 10.1046/j.1471-4159.1995.65041704.x
Danno S, Nishiyama H, Higashitsuji H, Yokoi H, Xue JH, Itoh K, Matsuda T, Fujita J: Increased transcript level of RBM3, a member of the glycine-rich RNA-binding protein family, in human cells in response to cold stress. Biochem Biophys Res Commun 1997, 236(3):804–807. 10.1006/bbrc.1997.7059
Wellmann S, Buhrer C, Moderegger E, Zelmer A, Kirschner R, Koehne P, Fujita J, Seeger K: Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J Cell Sci 2004, 117(Pt 9):1785–1794. 10.1242/jcs.01026
Hsu HK, Shao PL, Tsai KL, Shih HC, Lee TY, Hsu C: Gene regulation by NMDA receptor activation in the SDN-POA neurons of male rats during sexual development. J Mol Endocrinol 2005, 34(2):433–445. 10.1677/jme.1.01601
Zeng Y, Kulkarni P, Inoue T, Getzenberg RH: Down-regulating cold shock protein genes impairs cancer cell survival and enhances chemosensitivity. J Cell Biochem 2009, 107(1):179–188. 10.1002/jcb.22114
Tong G, Endersfelder S, Rosenthal LM, Wollersheim S, Sauer IM, Buhrer C, Berger F, Schmitt KR: Effects of moderate and deep hypothermia on RNA-binding proteins RBM3 and CIRP expressions in murine hippocampal brain slices. Brain Res 2013, 1504: 74–84. 10.1016/j.brainres.2013.01.041
Pilotte J, Cunningham BA, Edelman GM, Vanderklish PW: Developmentally regulated expression of the cold-inducible RNA-binding motif protein 3 in euthermic rat brain. Brain Res 2009, 1258: 12–24. 10.1016/j.brainres.2008.12.050
Wellmann S, Zelmer A, Nitsch C, Bucher HU, Buhrer C: The Rna-binding protein RBM3 is involved in hypothermia induced neuroprotection. Pediatr Res 2010, 68: 101–102. 10.1203/00006450-201011001-00193
Tanaka T, Wakamatsu T, Daijo H, Oda S, Kai S, Adachi T, Kizaka-Kondoh S, Fukuda K, Hirota K: Persisting mild hypothermia suppresses hypoxia-inducible factor-1 alpha protein synthesis and hypoxia-inducible factor-1-mediated gene expression. Am J Physiol Regul Integr Comp Physiol 2010, 298(3):R661-R671. 10.1152/ajpregu.00732.2009
Chip S, Zelmer A, Ogunshola OO, Felderhoff-Mueser U, Nitsch C, Buhrer C, Wellmann S: The RNA-binding protein RBM3 is involved in hypothermia induced neuroprotection. Neurobiol Dis 2011, 43(2):388–396. 10.1016/j.nbd.2011.04.010
Rawls SM, Cabassa J, Geller EB, Adler MW: CB1 receptors in the preoptic anterior hypothalamus regulate WIN 55212–2 [(4,5-dihydro-2-methyl-4(4-morpholinylmethyl)-1-(1-naphthalenyl-carbonyl)-6H-pyrrolo[3,2,1ij]quinolin-6-one]-induced hypothermia. J Pharmacol Exp Ther 2002, 301(3):963–968. 10.1124/jpet.301.3.963
Leker RR, Gai N, Mechoulam R, Ovadia H: Drug-induced hypothermia reduces ischemic damage: effects of the cannabinoid HU-210. Stroke 2003, 34(8):2000–2006. 10.1161/01.STR.0000079817.68944.1E
Acknowledgments
We thank Sylvia Wowro for proofreading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
KRLS and GT contributed equally to the drafting of this manuscript. FB edited the manuscript and gave the final approval of the version to be published. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schmitt, K.R., Tong, G. & Berger, F. Mechanisms of hypothermia-induced cell protection in the brain. Mol Cell Pediatr 1, 7 (2014). https://doi.org/10.1186/s40348-014-0007-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-014-0007-x