
Abstract
With the development of next-generation sequencing technology, de novo variants (DNVs) with deleterious effects can be identified and investigated for their effects on birth defects such as congenital heart disease (CHD). However, statistical power is still limited for such studies because of the small sample size due to the high cost of recruiting and sequencing samples and the low occurrence of DNVs. DNV analysis is further complicated by genetic heterogeneity across diseased individuals. Therefore, it is critical to jointly analyze DNVs with other types of genomic/biological information to improve statistical power to identify genes associated with birth defects. In this review, we discuss the general workflow, recent developments in statistical methods, and future directions for DNV analysis.
Similar content being viewed by others

Introduction
Birth defects are structural changes present at birth that can affect one part or several parts of the body, such as the heart and brain [1]. They pose significant challenges for both individual health and public health. Learning about the causes of birth defects is crucial for improving the quality of support and resources to help individuals and families affected. It is estimated that around 240,000 infants globally do not survive past their first 28 days every year due to birth defects, with these conditions also leading to the deaths of an additional 170,000 children aged one month to five years [2]. There are several possible causes of birth defects, including genetic changes, adverse reactions to medications, exposure to substances or chemicals, or complications during pregnancy. It is estimated that about 20% of birth defects are caused by genetic factors [3]. There are in general three general categories of genetic causes: chromosomal abnormalities, single-gene defects, and multifactorial influences [4]. Efforts have been made to identify genetic causes of birth defects [5]. The role of rare variants in disease genetics has been unraveled with the development of whole exome sequencing (WES) and whole genome sequencing (WGS) technologies. In this review, we exclusively focus on the statistical methods that can be applied to de novo single nucleotide variants (SNVs) and small insertions/deletions (indels). They are referred to as de novo variants (DNVs), within the purview of birth defects research. Compared to other rare variants, DNVs represent an extreme case, given their very low occurrence and large effect size. On average, an individual may carry approximately 100 DNVs in the genome, with about 1 variant affecting the exome [6,7,8,9]. DNVs have been considered strong supporting evidence for pathogenicity based on the American College of Medical Genetics and Genomics classification guidelines and provide important insights into the genetic cause of diseases [10].
Mounting evidence has pinpointed the importance of conducting DNV analyses to identify risk genes for birth defects such as congenital heart disease (CHD), congenital diaphragmatic hernia (CDH), orofacial cleft (OFC) [9, 11,12,13,14,15], and some early onset neurodevelopmental disorders such as autism [16,17,18,19]. In the study of WES data from 2,645 proband-parent trios by the Pediatric Cardiac Genomics Consortium (PCGC), Jin et al. found that DNVs accounted for 8% of cases and inferred that DNVs in about 440 genes contributed to CHD [13]. In the largest genetic exploration of coding DNVs affected by nonsyndromic OFCs to date, Bishop et al. analyzed the contribution of coding DNVs from WGS to OFC risks in 756 proband-parent trios and identified multiple promising genes that had not been reported before, such as ZFHX3 and ZFHX4 [9]. In 2021, Qiao et al. conducted an analysis of coding DNVs sequenced from 827 CDH proband-parent trios. They confirmed an overall enrichment of damaging DNVs in constrained genes (ExAC [20] pLI score > 0.5) and identified LONP1 and ALYREF as candidate CDH-associated genes with a false discovery rate (FDR) of 0.05 [15]. These results shed new insights into the disease etiology of birth defects, call upon the application of statistical methods that can analyze the enrichment of DNV in other birth defect cohorts, and motivate the development of novel statistical methods to improve the power of identifying genes associated with birth defects.
In this review, we first summarize the general workflow of conducting DNV analysis, including data preprocessing, mutation rate calculation, and DNV enrichment analysis. Next, we introduce several integrative statistical methods that can further incorporate DNVs with other types of variants or biological information to boost the power of risk gene identification (Table 1). In addition, we discuss several potential future directions for DNV analysis in birth defects.
General workflow of DNV analysis
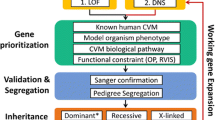
In this section, we summarize the general workflow of DNV analysis into three steps: data preprocessing, mutation rate calculation, and enrichment analysis. An illustration of the general workflow from step 1 to step 3 is shown in Fig. 1.
Step 1: data preprocessing
After samples are sequenced via WES or WGS, DNVs need to be called from unmapped sequencing reads. In 2021, Diab et al. published a detailed protocol for analyzing germline DNVs from WES [21]. Briefly, two steps are needed before trio DNV analysis. In the first step, binary alignment/map (BAM) files are generated from unmapped sequencing reads. In the second step, variants are called based on the GATK best practices [22]. Compared with other types of variant calling, DNVs are required to be jointly called in trios, where pedigree files for trios will be generated. For each trio, VCF files are generated based on GATK best practices and further processed, including steps such as splitting multi-allelic sites and left normalization by BCFtools [23]. Then, the generated variants are annotated and filtered based on minor allele frequencies and alternate allele ratios in probands and parents. After the above analyses, all candidate DNV calls are manually verified using the integrative genomics viewer (IGV) [24]. Before proceeding with enrichment analysis, variants are further classified into loss of function (LoF), damaging missense (Dmis), and other groups using annotation tools such as ANNOVAR [25, 26]. Step-by-step procedures can be referred to in the original protocol [21].
Step 2: mutation rate calculation
After variant calling is completed, the next step is to estimate the per gene mutation rate. In 2012, Neale et al. [27] developed a statistical model for estimating the expected mutation rate in the exome. They assess the mutation rates of all possible trinucleotide contexts within the intergenic region of the human genome. They considered variations in two ways: comparing the fixed genomic difference to chimpanzees and baboons [28], and variations identified through the 1000 Genomes Project [29]. The mutation rate for the exome was estimated by summing up the mutation rates for all bases captured by the exome, and that of each functional annotation class was determined by summing the mutation rates of variants belonging to that class.
In 2014, Samocha et al. extended Neal’s framework to calculate each gene’s expected rate for different types of mutation [30]. There are two steps of the framework. First, the sequence context is used to estimate the probability of each base mutating to another base. Second, based on the change of trinucleotide, the outcome of each type of base change is identified, including synonymous, missense, nonsense, essential splice site and frameshift mutations. These probabilities are added up to obtain a mutation rate per gene for different types of mutations. The input includes bed files where each row represents a specific genomic region from exome capture, trio information, and sequencing coverage of WES samples that can be calculated using Mosdepth [31]. More specifically, bed files should be first converted into sequence data that contain four base information (ATCG) and subsequently transformed into a probability table. Then, annotations are added to the table using ANNOVAR. Next, the per-base mutation rate is adjusted by sequencing depth. For each base, the number of trios with 10x or greater coverage is counted. The numbers are adjusted with a coefficient ranging from 0.9 to 1, which assigns greater weights to bases with higher sequence depths.
Step 3: enrichment analysis of DNVs
Enrichment analysis of DNVs aims to find elevated or excessive gene burden due to DNVs. The goal is to assess whether there is a significant accumulation or enrichment of DNVs within specific genes. Statistical methods for enrichment analysis test whether the observed DNVs occur more frequently than expected by chance in a gene.
Current methods for DNV enrichment analysis are mostly developed based on a Poisson framework, which assumes the number of observed DNV counts follows a Poisson distribution. Some methods focus only on DNVs, and others can incorporate information from other types of variants or other biological information. Among methods that focus on DNVs, Ware et al. proposed one of the first models based on the statistical framework of Samocha et al. to analyze coding DNVs and named the R package of the model DenovolyzeR [8, 30]. The framework assumes the number of observed DNV counts in a single gene (\(m\)) follows a Poisson distribution, and a certain type of variants (e.g. LoF) with the expected counts calculated as two times the product of sample size and mutation rate of the corresponding type. It uses a Poisson exact test to compare the observed counts of the type of variants with the expected counts:
where \(\lambda\) is the mean of the distribution.
DenovolyzeR provides a pre-calculated mutability table from Samocha et al. that can be used to conduct four types of enrichment analyses [8, 30]. These include (1) assessment of genome-wide burden of different types of DNVs, (2) assessment of burden of genes with multiple DNVs, (3) assessment of whether a single gene carries an excess number of mutations, and (4) assessment of whether a gene set is enriched with DNVs. Details of the analysis step can be found in the original protocol [8].
DeNovoWEST (De Novo Weighted Enrichment Simulation Test) is another method proposed for DNV enrichment analysis that uses a simulation-based statistical test to detect gene-specific enrichment of DNVs [32]. It includes two components: an overall enrichment test that includes all nonsynonymous DNVs and a clustering test that assesses the enrichment of missense variants. The overall enrichment test is a simulation-based test that calculates the probability of observing the severity of a gene higher than expected, considering all possible numbers of DNVs per gene:
,
where \(S\) denotes the gene severity score, \(s\) denotes the observed severity score, and \(K\) is the number of DNVs in the gene. The severity score of each DNV is an empirically estimated positive predictive value of being pathogenic based on its predicted protein consequence, CADD score, and selective constraint against heterozygous protein truncating variants in the gene, and it is calculated by summing the severity scores of mutations within this gene. The upper limit of k is chosen as 250 as this number is far larger than the observed DNV counts in real data. When \(k=\text{0,1}\), \(P(S\ge s|k\left)P\right(K=k)\) can be calculated analytically. When \(k\ge 2\), \(P(K=k)\) can be calculated analytically under the null assumption that DNVs follow a Poisson distribution, and \(P(S\ge s|k)\) can be estimated using a simulation-based approach. The estimated value of the probability \(P\left(S\ge s\right)\)is defined as pEnrich.
The missense enrichment test is performed the same as the overall enrichment test, except that only missense variants are included in the score calculation. The proportion of the simulated scores for missense variants that are larger than the observed scores is defined as pMisEnrich. For the clustering test, the clustering distance is determined as the geometric mean coding distance between all potential pairs of DNVs [33]. The observed distance of missense DNVs is then compared with the expected distance from simulated missense DNVs, and the probability of missense variants being as or more clustered than the null model is defined as pClustering [34]. pMisEnrich and pClustering are then combined using Fisher’s method to obtain pMEC.
pMEC = combined (pMisEnrich, pClustering).
The final testing p-values for DeNovoWest are obtained by taking the minimum of pEnrich and pMEC. Bonferroni correction is used to account for multiple testing comparisons.
pDeNovoWEST = min(pEnrich, pMEC).

General workflow of DNV analysis. In step 1, sequencing data from trios are preprocessed and annotated. In step 2, mutation rates are calculated for each gene. In step 3, enrichment analysis is conducted to identify risk genes
Integrative analysis of DNVs and other types of variants
Studies on DNVs often lack statistical power due to their relatively low frequency. Efforts have been made to increase statistical power by incorporating other biological information.
To integrate other types of variants with DNVs, He et al. proposed a hierarchical Bayesian framework named the transmission and de novo association (TADA) test to incorporate the information from both inherited and DNVs [17]. Assuming subjects can be classified as carrying two alleles, TADA denotes alleles with deleterious mutation as a, and alleles without as A, and \(\gamma\) denotes the relative risk of Aa compared with AA. TADA tests the null hypothesis of \(\gamma =1\) against the alternative hypothesis of \(\gamma \ne 1\) for all genes. TADA has gained success in its application in multiple studies [35,36,37,38,39]. However, it requires external information or prior knowledge to estimate hyperparameters. In 2017, Nguyen et al. adopted a fully Bayesian framework to extend TADA and named their method extTADA [40]. However, both TADA and extTADA do not consider the recessive mode of inheritance in their models.
In 2021, Li et al. proposed a TADA-R model built upon TADA to include the recessive disease model, including both the cases of homozygotes and compound heterozygotes [41]. By applying TADA-R to CHD, they identified 15 significant genes, many of which were not implicated in previously published studies.
Multiple methods have been proposed to incorporate functional annotation to boost the statistical power of risk gene identification. fitDNM [42] is a method that tests the excess de novo load of genes by deriving score statistics from a retrospective likelihood that incorporates functional information quantitatively rather than classifies variants into different functional categories like TADA. They integrated the probabilities of a mutation functionally impacting the gene when characterizing the distribution of DNVs in an affected individual and estimated these probabilities using scores that are predicted externally. Specifically, they defined the probability that the mutation is damaging to protein function when characterizing the distribution of DNVs at a locus, and set the probability of LoF variants predicted by SnpEff [43] as 1, the probability of missense variants as their PolyPhen-2 [28] scores, and that of synonymous variants as 0. It was observed that fitDNM had increased power compared to Poisson tests and TADA tests with DNVs only in the simulation studies and real data analyses.
Similar to fitDNM, Hu et al. developed a likelihood ratio test named VARPRISM that incorporates variant prioritization to test associations of DNVs [44]. Although VARPRISM shares a few features with fitDNM, it utilizes different strategies to incorporate functional information. VARPRISM employs the likelihood ratio of a variant being damaging versus neural by incorporating a conservation-controlled amino acid substitution matrix (CASM) from VAAST 2.0 [45], while fitDNM requires probabilities of given mutations being damaging from an external resource instead of being estimated directly from data. Hu et al. showed that VARPRISM had better power than fitDNM with two simulated datasets.
Efforts have also been made to integrate information from noncoding regions. TADA-A is a statistical framework that models mutation counts for each position in the genome with Poisson distribution [46]. The model can combine genomic annotation information from both coding and non-coding regions. Furthermore, TADA-A supports meta-analysis of multiple DNV studies by fitting a background mutation model for each study to adjust for potential technical factors. However, TADA-A only focuses on regulatory sequences close to genes without considering those distal to transcription start sites. Also, it uses a linear model to combine information from different annotations, which may not be as powerful as using a non-linear model such as deep neural networks [46, 47].
HeartENN (Heart Effect Neural Network) is developed to identify noncoding DNV burden in CHD. It is extended from a deep learning-based framework for predicting the effects of non-coding variants named DeepSEA [48, 49]. HeartENN uses two neural network-based epigenomic models for human and mouse to predict genome-wide features based on heart-specific chromatin profiles. It was found that noncoding variants prioritized by HeartENN score (score ≥ 0.1) had significant enrichment of known human CHD genes in CHD cases.
Integrative analysis of DNVs and other sources of biological information
Muti-trait methods
Extended from extTADA, Nguyen et al. developed a multi-trait Bayesian framework that can jointly analyze two traits named mTADA [50]. There are four hypotheses of the model: the gene is associated with neither trait (\({H}_{0}\)), the gene is only associated with the first trait (\({H}_{1}\)), the gene is only associated with the second trait (\({H}_{2}\)), and the gene is associated with both traits (\({H}_{3}\)). The input data includes DNVs from the two cohorts and mutation rates of genes that can be calculated from an external framework. The mTADA framework assumes the DNV counts for both traits follow Poisson distributions. When gene i is associated with trait \(k\) (k = 1 or 2), the rate parameter in the Poisson distribution of its DNV count is multiplied by a relative risk parameter \({\gamma }_{ik}\). \({\gamma }_{ik}\) is assumed to follow a Gamma distribution with two parameters \({\stackrel{-}{\gamma }}_{k}\) (mean relative risk) and \({\beta }_{k}\) (to control the variance of the relative risk). Based on the four hypotheses, the corresponding posterior probabilities for genes can be calculated from Markov chain Monte Carlo. However, the hyperpriors of mTADA cannot be estimated from the data but rely on running extTADA first.
M-DATA is another multi-trait method that shares the same hypotheses as mTADA, but uses an Expectation-Maximization algorithm to estimate parameters and infer risk gene status [51]. Compared to mTADA, this method uses an alternative way to characterize the effects of variants in different functional groups by linking variant-level and gene-level functional annotations to the relative risk of de novo genotype in the model. However, M-DATA requires users to preselect functional annotations before inputting the data and the algorithm cannot prioritize the functional annotations automatically. In addition, if the underlying functional annotation effect size is small, the power improvement of M-DATA compared with models without integrating functional annotations will be minor.
Quantifying the genetic association of DNVs between different genetic disorders is crucial because it can lead to a better understanding of the common molecular foundations these disorders may share. While recent research has shown that certain genes and biological pathways are commonly affected by DNVs in various disorders, current methods tend to only consider genes that are statistically significant across multiple disorders and cannot fully capture the complexity of genetic associations due to the polygenic nature of diseases and incomplete penetrance. EncoreDNM is a novel statistical method that quantifies the overall genetic sharing of DNVs between two disorders for different variant types [52]. Instead of using the Bayesian framework, it constructs mixed-effects Poisson regression models to evaluate the correlation between two traits by providing the estimated correlation and p-values from statistical inference. This method is designed for testing global genetic architecture from DNV information across traits but does not provide a way to prioritize specific risk genes.
Network-assisted models
Risk genes identified from DNV studies have been shown to enrich in a protein-protein interaction (PPI) network in the post-association analysis [50]. Two methods have been proposed to integrate network information with DNV data based on the assumption that neighboring genes are more likely to have similar disease association statuses.
DAWN is a post-association method that takes association results from TADA p-values and gene-gene interaction network estimated from expression data as input [53, 54]. In real data application, Liu et al. identified 333 genes that plausibly affect autism risk by integrating association results from WES data and brain gene expression data [54].
Compared to DAWN, N-DATA is a model that does not require summary statistics results from other methods such as TADA [55]. It directly incorporates PPI information into the prior risk gene status based on the Poisson mixture distribution. After applying N-DATA to real DNV data from the CHD study, Xie et al. identified 46 candidate genes with at least one DNV in the study cohort. Among these genes, they discovered that some genes can only be identified after integrating the network information compared with existing genes that can be identified using the baseline model without integrating the network. Visualizing the 46 genes in the PPI network, they found three main gene clusters formed that are biologically interpretable within the network, including one cluster related to transcriptional regulation and early cell growth or differentiation processes, one cluster related to RNA splicing, and the third cluster related to protein synthesis. This further demonstrates the improvement of power after incorporating network information into the framework. In simulation studies, Xie et al. showed that the performance of N-DATA and DAWN was comparable when the signals in the network became stronger. However, with more and more network databases available, these methods did not provide a way to incorporate multiple types of interactions or to prioritize network information.
Integrative analysis of DNVs and expression data
VBASS is an integration model that incorporates bulk or single-cell expression data into the analysis of DNVs based on a Bayesian framework to discover risk genes [56]. It constructs a model of disease risk based on expression profiles, which are estimated using deep neural networks. It simultaneously trains the neural network weights and determines the parameters for the Gamma-Poisson likelihood model of DNV counts based on both gene expression data and genetic data. Different from previous methods, it has the key assumption that the prior probability of a gene being a risk candidate should be specific at the gene level and could be inferred from gene expression information in relevant tissues. Therefore, it takes the gene expression profiles as a vector into its probabilistic model and estimates parameters in the model using deep neural networks. In addition, it can also incorporate RNA sequencing data of relevant organs or cell types other than single-cell expression profiles. The performance of VBASS is highly dependent on the quality of the gene expression data. One practical issue that may hamper incorporating gene expression information with DNVs in birth defect studies is that gene expression data of early developmental human organs are hard to acquire.
Future directions
Sex-aware models
There is accumulating evidence of sex bias in neurodevelopmental disorders. For instance, the diagnosed male-to-female ratio of autism spectrum disorder (ASD) is three to four times. The female protective effect can be attributed to genetic, hormonal, and environmental factors. Limited studies have focused on studying the sex-based mechanisms related to DNVs.
In 2017, Turner et al. conducted sex-based enrichment DNV analysis for neurodevelopmental disorders using ∼ 8,825 sequenced parent-offspring trios in denovo-db as the discovery cohort [57, 58]. The discovery cohort identified 17 female-only significant genes, 18 male-only significant genes, and 19 shared significant genes. Among the 54 genes identified, 25 genes were replicated in the 18,778 trios from the GeneDx cohort [58]. They not only observed significant enrichment on the X chromosome for females but identified potential sex-biased genes on autosomal chromosomes.
Similar to ASD, the prevalence and disease mechanism of birth defects can vary based on sex. For instance, a review study surveyed 21 articles and confirmed a significant gender variation in specific CHD subgroups [59]. These findings call for more DNV studies to test sex-based enrichment in birth defects and suggest the potential to develop methods that can incorporate sex-specific mechanisms.
Integration of common variants with DNVs
Common variants have been shown to play an important role in complex human traits and diseases. Despite the success of identifying risk genes using rare variants such as DNVs, it is worth exploring the joint and different contributions of both rare variants and common variants. Different hypotheses have been made about the contribution of common and rare variants in complex human diseases. In a study on UK Biobank participants, Lu et al. found that rare pathogenic variants were more prevalent among patients with a low polygenic risk score (PRS) affected by diseases including breast cancer, colorectal cancer, type 2 diabetes, osteoporosis, and short stature [60]. Studies in ASD have also suggested that DNVs and common variants have addictive effects [61, 62]. Another study has suggested that genes harboring schizophrenia-associated common variants and genes harboring DNVs both contribute to a core set of biologically important pathways and networks and the interactions of these genes may play a part in the risk of schizophrenia [63]. To better prioritize suggestive loci from OFC GWAS, Bishop et al. hypothesized DNVs near a GWAS peak could provide evidence in support of suggestive loci without reaching formal significance [9]. After evaluating genes within 1 Mb (± 500 kb) of both suggestive and significant loci from two recent OFC GWAS studies, they found 37 protein-altering DNVs were within these genes and several of them were located in genes implicating OFC development. These results suggest a potential future direction in integrating common variants and DNVs in a general framework for birth defects.
Integration of epigenetic information with DNVs
Epigenetic changes modify the activation of certain genes without changing the DNA sequence, and they play an essential role in human development and disease etiology [64]. There are different classes of epigenetic information including DNA methylation, histone modification, and noncoding RNA action [65]. DNVs and epigenetic interactions may interact to influence gene expression and contribute to disease development and progression [66]. For instance, DNV in the RING1 gene was identified in a 13-year-old girl with neurodevelopmental disabilities. RING1 encodes an E3-ubiquitin ligase that is involved in the epigenetic control of transcription during development. The mutant RING1 retained catalytic activity but was unable to ubiquitylate histone H2A. The animal model suggested that animals with the same mutation or complete knockout of RING1 ortholog had defects in histone H2A ubiquitylation. RING1 mutations are likely a cause of human neurodevelopmental disorders where epigenetic effects play an important role [67].
Epigenetic regulation mechanisms have also been identified for CHD. For instance, researchers conducted a case-control study using exome sequencing to compare the occurrence of DNVs in genes related to the modification of histone proteins in individuals with severe CHD and those without the condition. They discovered an excess of DNVs in the genes responsible for writing, erasing, or reading H3K4 methylation or H2BK120 ubiquitination required for H3K4 methylation. This indicates a potential pathogenic role of abnormal histone methylation in CHD [11, 68].
Studies have also shown that integrating genetic data with epigenetic information could better elucidate functional insights of complex diseases [69,70,71]. For example, Andrews et al. found ASD-associated SNPs in GWAS are enriched for tissue-specific meQTLs in fetal brain and peripheral blood [70].
These studies collectively demonstrate the potential of integrating epigenetic information with DNVs to understand the complex mechanisms underlying birth defects.
Integration of protein structural information with DNVs
Protein Data Bank (PDB) is a worldwide repository that stores 3D structural information about biological macromolecules. Despite the efforts in experiments, only 35% of human proteins are mapped to a PDB entry, and frequently, these entries represent only fragments or segments of protein sequences rather than the whole proteins [72, 73]. The release of AlphaFold2 expands the coverage of human protein structures to 98.5%, with 58% of them with high confidence [73]. The enlarged knowledge of protein structures can help us elucidate the molecular mechanisms of more variants. Leveraging protein structures from protein structure databases, three statistical methods - POINT [74], PSCAN [75], and POKEMON [76] - have been developed to characterize the association between rare missense variants and phenotypes by integrating 3D spatial distance of variants within protein structures.
In addition to spatial distance, studies have also shown the importance of functional features, physiochemical features, interaction features, and others. Multiple studies showed differences of these features between pathogenic missense variants and variants that are commonly found in the general population [77,78,79]. For instance, Iqbal et al. surveyed 40 structural features and found significant enrichment of multiple features under different categories in pathogenic missense variants from Clinvar [80] and HGMD [81] compared with general population variants from gnomAD [78, 82] in 1,330 disease-associated genes. Chen et al. demonstrated that ASD cases exhibited an enrichment of de novo missense variants with disruptive impacts on protein interactions, and these variants often affect hub proteins and disturb their interactions [83]. These findings suggest the potential benefit of new methods that can integrate different categories of protein structural features such as secondary structures, residue exposure levels, and PPIs into the modeling of de novo missense variants.
Conclusions
In this review, we have summarized different statistical methods that can be applied to identify risk genes for birth defects by analyzing DNVs. Most of the methods characterize DNV counts within a gene using a Poisson distribution and estimate parameters using likelihood-based approaches or Bayesian methods. The output of these methods includes p-values, posterior probabilities, Bayesian q-values, and others in correspondence to different modeling approaches. The identified risk genes by these methods can help guide future biological experiments and clinical studies to further understand disease mechanisms. In the meantime, the complexity of genetic architectures, the interplay between genetic and environmental factors, and the rare nature of DNVs pose challenges to the current field. Moreover, the methods that can integrate multi-layers of multi-omics data, such as integrating transcription and methylation data with DNVs remains in the early stage of development. There is also a critical need for improved computational models that can effectively incorporate the heterogeneity of birth defects, and for databases that can capture the phenotypic spectrum associated with DNVs in a standardized way. Addressing these challenges requires interdisciplinary collaboration and the development of innovative analytical tools capable of dissecting the intricate biological networks underlying birth defects. To better elucidate the etiology of birth defects, we discussed several potential future directions, including incorporating information on sex, common variants, epigenetic information, and protein structures with DNVs. These future directions offer abundant possibilities, inviting researchers to unlock the mysteries of genetic etiology and developmental biology for birth defects and paving the way for personalized therapeutic strategies.
Data availability
Not applicable.
Abbreviations
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
- SNV:
-
Single nucleotide variant
- DNV:
-
De novo variant
- CHD:
-
Congenital heart disease
- CDH:
-
Congenital diaphragmatic hernia
- OFC:
-
Orofacial cleft
- FDR:
-
False discovery rate
- LoF:
-
Loss of function
- Dmis:
-
Deleterious missense
- IGV:
-
Integrative genomics viewer
- PPI:
-
Protein-protein interaction
- ASD:
-
Autism spectrum disorder
- PRS:
-
Polygenic risk score
- PDB:
-
Protein Data Bank
References
Centers for Disease Control and Prevention. What are Birth Defects? Available from: cdc.gov/ncbddd/birthdefects/facts.html#references.
Congenital disorders. World Health Organization. 2023 [cited 2023 Jan 3]. Available from: https://www.who.int/news-room/fact-sheets/detail/birth-defects.
Birth defects. Cleveland Clinic. [cited 2023 Sep 30]. Available from: https://my.clevelandclinic.org/health/diseases/12230-birth-defects.
Queremel Milani DA, Tadi P, Genetics. Chromosome Abnormalities. StatPearls Publishing; 2023.
Khokha MK, Mitchell LE, Wallingford JB. White paper on the study of birth defects. Birth Defects Res. 2017;109:180–5.
Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 2010;107:961–8.
Conrad DF, Keebler JEM, DePristo MA, Lindsay SJ, Zhang Y, Casals F, et al. Variation in genome-wide mutation rates within and between human families. Nat Genet. 2011;43:712–4.
Ware JS, Samocha KE, Homsy J, Daly MJ. Interpreting de novo variation in Human Disease using denovolyzeR. Curr Protoc Hum Genet. 2015;87:7251–15.
Bishop MR, Diaz Perez KK, Sun M, Ho S, Chopra P, Mukhopadhyay N, et al. Genome-wide Enrichment of De Novo Coding Mutations in Orofacial Cleft trios. Am J Hum Genet. 2020;107:124–36.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–3.
Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–6.
Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–601.
Qi H, Yu L, Zhou X, Wynn J, Zhao H, Guo Y, et al. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. 2018;14:e1007822.
Qiao L, Xu L, Yu L, Wynn J, Hernan R, Zhou X, et al. Rare and de novo variants in 827 congenital diaphragmatic hernia probands implicate LONP1 as candidate risk gene. Am J Hum Genet. 2021;108:1964–80.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99.
He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013;9:e1003671.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
Zhou X, Feliciano P, Shu C, Wang T, Astrovskaya I, Hall JB, et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet. 2022;54:1305–19.
Lek M, Exome Aggregation Consortium, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Diab NS, King S, Dong W, Allington G, Sheth A, Peters ST, et al. Analysis workflow to assess de novo genetic variants from human whole-exome sequencing. STAR Protoc. 2021;2:100383.
Van der Auwera GA, O’Connor BD. Genomics in the Cloud: using Docker, GATK, and WDL in Terra. O’Reilly Media, Inc.; 2020.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10:giab008.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–66.
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–5.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–50.
Pedersen BS, Quinlan AR. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2018;34:867–8.
Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–62.
Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–8.
Short PJ, McRae JF, Gallone G, Sifrim A, Won H, Geschwind DH, et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature. 2018;555:611–6.
Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007.
Du Y, Li Z, Liu Z, Zhang N, Wang R, Li F, et al. Nonrandom occurrence of multiple de novo coding variants in a proband indicates the existence of an oligogenic model in autism. Genet Med. 2020;22:170–80.
Li K, Fang Z, Zhao G, Li B, Chen C, Xia L, et al. Cross-disorder analysis of DE Novo mutations in neuropsychiatric disorders. J Autism Dev Disord. 2022;52:1299–313.
Li Q, Zhao L, Zeng Y, Kuang Y, Guan Y, Chen B, et al. Large-scale analysis of de novo mutations identifies risk genes for female infertility characterized by oocyte and early embryo defects. Genome Biol. 2023;24:68.
Cirnigliaro M, Chang TS, Arteaga SA, Pérez-Cano L, Ruzzo EK, Gordon A, et al. The contributions of rare inherited and polygenic risk to ASD in multiplex families. Proc Natl Acad Sci U S A. 2023;120:e2215632120.
Nguyen HT, Bryois J, Kim A, Dobbyn A, Huckins LM, Munoz-Manchado AB, et al. Integrated bayesian analysis of rare exonic variants to identify risk genes for schizophrenia and neurodevelopmental disorders. Genome Med. 2017;9:114.
Li M, Zeng X, Jin C, Jin SC, Dong W, Brueckner M, et al. Integrative modeling of transmitted and de novo variants identifies novel risk genes for congenital heart disease. Quant Biol. 2021;9:216–27.
Jiang Y, Han Y, Petrovski S, Owzar K, Goldstein DB, Allen AS. Incorporating functional information in tests of excess De Novo Mutational load. Am J Hum Genet. 2015;97:272–83.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92.
Hu H, Coon H, Li M, Yandell M, Huff CD. VARPRISM: incorporating variant prioritization in tests of de novo mutation association. Genome Med. 2016;8:91.
Hu H, Huff CD, Moore B, Flygare S, Reese MG, Yandell M. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet Epidemiol. 2013;37:622–34.
Liu Y, Liang Y, Cicek AE, Li Z, Li J, Muhle RA, et al. A statistical Framework for Mapping Risk genes from De Novo mutations in whole-genome-sequencing studies. Am J Hum Genet. 2018;102:1031–47.
Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31:761–3.
Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods. 2015;12:931–4.
Richter F, Morton SU, Kim SW, Kitaygorodsky A, Wasson LK, Chen KM, et al. Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat Genet. 2020;52:769–77.
Nguyen T-H, Dobbyn A, Brown RC, Riley BP, Buxbaum JD, Pinto D, et al. mTADA is a framework for identifying risk genes from de novo mutations in multiple traits. Nat Commun. 2020;11:2929.
Xie Y, Li M, Dong W, Jiang W, Zhao H. M-DATA: a statistical approach to jointly analyzing de novo mutations for multiple traits. PLoS Genet. 2021;17:e1009849.
Guo H, Hou L, Shi Y, Jin SC, Zeng X, Li B et al. Quantifying concordant genetic effects of de novo mutations on multiple disorders. eLife. 2022. https://doi.org/10.7554/elife.75551.
Liu L, Lei J, Sanders SJ, Willsey AJ, Kou Y, Cicek AE et al. DAWN: a framework to identify autism genes and subnetworks using gene expression and genetics. Molecular Autism. 2014. https://doi.org/10.1186/2040-2392-5-22.
Liu L, Lei J, Roeder K, Network assisted analysis, to reveal the genetic basis of autism. Ann Appl Stat. 2015;9:1571–600.
Xie Y, Jiang W, Dong W, Li H, Jin SC, Brueckner M, et al. Network assisted analysis of de novo variants using protein-protein interaction information identified 46 candidate genes for congenital heart disease. PLoS Genet. 2022;18:e1010252.
Zhong G, Choi YA, Shen Y. VBASS enables integration of single cell gene expression data in bayesian association analysis of rare variants. Commun Biol. 2023;6:774.
Turner TN, Yi Q, Krumm N, Huddleston J, Hoekzema K, Stessman F. Denovo-db: a compendium of humande novovariants. Nucleic Acids Res. 2017;45:D804–11.
Turner TN, Wilfert AB, Bakken TE, Bernier RA, Pepper MR, Zhang Z, et al. Sex-based analysis of De Novo variants in Neurodevelopmental disorders. Am J Hum Genet. 2019;105:1274–85.
Pugnaloni F, Felici A, Corno A-F, Marino B, Versacci P, Putotto C. Gender differences in congenital heart defects: a narrative review. Transl Pediatr. 2023;12:1753–64.
Lu T, Zhou S, Wu H, Forgetta V, Greenwood CMT, Richards JB. Individuals with common diseases but with a low polygenic risk score could be prioritized for rare variant screening. Genet Med. 2021;23:508–15.
Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet. 2017;49:978–85.
Antaki D, Guevara J, Maihofer AX, Klein M, Gujral M, Grove J, et al. A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Nat Genet. 2022;54:1284–92.
Jia P, Chen X, Fanous AH, Zhao Z. Convergent roles of de novo mutations and common variants in schizophrenia in tissue-specific and spatiotemporal co-expression network. Transl Psychiatry. 2018;8:105.
Li H, Wang J, Cruz DA, Modliszewski JL, Corcoran DL, Martínez-Magaña JJ et al. Functional annotation of the human PTSD methylome identifies tissue-specific epigenetic variation across subcortical brain regions. medRxiv. 2023 [cited 2023 May 1]. p. 2023.04.18.23288704. Available from: https://www.medrxiv.org/content/https://doi.org/10.1101/2023.04.18.23288704v1.
Loscalzo J, Handy DE. Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series). Pulm Circ. 2014;4:169–74.
Barbosa M, Joshi RS, Garg P, Martin-Trujillo A, Patel N, Jadhav B, et al. Identification of rare de novo epigenetic variations in congenital disorders. Nat Commun. 2018;9:1–11.
Pierce SB, Stewart MD, Gulsuner S, Walsh T, Dhall A, McClellan JM, et al. De novo mutation in RING1 with epigenetic effects on neurodevelopment. Proc Natl Acad Sci U S A. 2018;115:1558–63.
Wang G, Wang B, Yang P. Epigenetics in congenital heart disease. J Am Heart Assoc. 2022;11:e025163.
Bell CG. Integration of genomic and epigenomic DNA methylation data in common complex diseases by haplotype-specific methylation analysis. Per Med. 2011;8:243–51.
Andrews SV, Ellis SE, Bakulski KM, Sheppard B, Croen LA, Hertz-Picciotto I, et al. Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder. Nat Commun. 2017;8:1011.
Ma Q, Shams H, Didonna A, Baranzini SE, Cree BAC, Hauser SL, et al. Integration of epigenetic and genetic profiles identifies multiple sclerosis disease-critical cell types and genes. Commun Biol. 2023;6:342.
UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–9.
Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021;596:590–6.
Marceau West R, Lu W, Rotroff DM, Kuenemann MA, Chang S-M, Wu MC, et al. Identifying individual risk rare variants using protein structure guided local tests (POINT). PLoS Comput Biol. 2019;15:e1006722.
Tang Z-Z, Sliwoski GR, Chen G, Jin B, Bush WS, Li B, et al. PSCAN: spatial scan tests guided by protein structures improve complex disease gene discovery and signal variant detection. Genome Biol. 2020;21:217.
Jin B, Capra JA, Benchek P, Wheeler N, Naj AC, Hamilton-Nelson KL, et al. An association test of the spatial distribution of rare missense variants within protein structures identifies Alzheimer’s disease-related patterns. Genome Res. 2022;32:778–90.
Sunyaev S, Ramensky V, Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet. 2000;16:198–200.
Iqbal S, Pérez-Palma E, Jespersen JB, May P, Hoksza D, Heyne HO, et al. Comprehensive characterization of amino acid positions in protein structures reveals molecular effect of missense variants. Proc Natl Acad Sci U S A. 2020;117:28201–11.
Laddach A, Ng JCF, Fraternali F. Pathogenic missense protein variants affect different functional pathways and proteomic features than healthy population variants. PLoS Biol. 2021;19:e3001207.
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–7.
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Chen S, Wang J, Cicek E, Roeder K, Yu H, Devlin B. De novo missense variants disrupting protein-protein interactions affect risk for autism through gene co-expression and protein networks in neuronal cell types. Mol Autism. 2020;11:76.
Funding
This work was supported in part by NIH grant R03HD100883-01A1 (Y.X. and H.Z.) and R01GM134005-01A1 (H.L., and H.Z.).
Author information
Authors and Affiliations
Contributions
Y.X, R.W., and H.Z. conceptualized the ideas and wrote the main manuscript text. H.L., W.D., and G.Z. contributed to the main text. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xie, Y., Wu, R., Li, H. et al. Statistical methods for assessing the effects of de novo variants on birth defects. Hum Genomics 18, 25 (2024). https://doi.org/10.1186/s40246-024-00590-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-024-00590-z