
Abstract
Background
Genetic variants in the coding region could directly affect the structure and expression levels of genes and proteins. However, the importance of variants in the non-coding region, such as microRNAs (miRNAs), remain to be elucidated. Genetic variants in miRNA-related sequences could affect their biogenesis or functionality and ultimately affect disease risk. Yet, their implications and pleiotropic effects on many clinical conditions remain unknown.
Methods
Here, we utilised genotyping and hospital records data in the UK Biobank (N = 423,419) to investigate associations between 346 genetic variants in miRNA-related sequences and a wide range of clinical diagnoses through phenome-wide association studies. Further, we tested whether changes in blood miRNA expression levels could affect disease risk through colocalisation and Mendelian randomisation analysis.
Results
We identified 122 associations for six variants in the seed region of miRNAs, nine variants in the mature region of miRNAs, and 27 variants in the precursor miRNAs. These included associations with hypertension, dyslipidaemia, immune-related disorders, and others. Nineteen miRNAs were associated with multiple diagnoses, with six of them associated with multiple disease categories. The strongest association was reported between rs4285314 in the precursor of miR-3135b and celiac disease risk (odds ratio (OR) per effect allele increase = 0.37, P = 1.8 × 10–162). Colocalisation and Mendelian randomisation analysis highlighted potential causal role of miR-6891-3p in dyslipidaemia.
Conclusions
Our study demonstrates the pleiotropic effect of miRNAs and offers insights to their possible clinical importance.
Similar content being viewed by others


Background
MicroRNAs (miRNAs) are small non-coding ribonucleic acids (RNA) of approximately 22 nucleotides that regulate gene expression and play critical roles in determining whether genes are active and how much protein is produced [1]. Mature miRNAs can perform their functions either in the cytoplasm or be released from the cell to the circulation and body fluids [2] to serve as chemical messengers and facilitate cell-to-cell communications [3]. These miRNAs can bind to target mRNAs to suppress translation [1] through inhibiting translation or affect degradation of mRNAs [4, 5]. A single miRNA can bind to multiple target genes, leading to a complex regulatory mechanism. Conversely, each gene could be targeted by multiple miRNAs [6].
Previous studies have explored the role of miRNAs in complex disorders through various approaches, including observational and experimental studies. Experimental studies typically assess molecular changes following administration miRNA mimics or antagonists and an assessment of the expression of potential target genes [7,8,9,10]. Candidate-based studies still dominate the studies of miRNAs in complex disorders, although unbiased high-throughput approaches are increasingly reported [11, 12]. With over 2000 miRNAs currently identified in humans [13], there is an opportunity for a comprehensive investigation to identify novel candidate miRNAs in complex diseases. Among observational studies, relatively few have leveraged genetic association data for circulatory miRNAs. The use of genetic data to study miRNAs could minimise the effect of confounders and avoid reverse causation since the genetic variants are fixed during conception.
Similar to other genes, the non-coding sequences of miRNAs are also subject to genetic variation, which could exist in the seed, mature, or precursor sequences of miRNAs [14], albeit the density is lower than the other parts of human genome [15,16,17], indicating that these regions are evolutionary conserved. The genetic variants in the seed region could interfere with the interaction between miRNA and target mRNA and, thus, are expected to be more functional [15, 18]. Meanwhile, those residing outside the seed region can affect binding beyond the seed region and the strength of inhibition of miRNA target [19], influence the biogenesis of mature miRNAs [20], or the processing of primary miRNA to precursor miRNAs [21]. Since a single miRNA could have multiple target genes [22], genetic variants residing in the miRNA-related sequence could affect the expression of target genes and downstream biological processes.
Extensive research has investigated the associations between genetic variants in miRNA-related sequences, i.e., seed, mature, or precursor regions with specific type of disorders including cancers [23, 24] and cardiometabolic traits [24, 25]. We previously showed that pleiotropy is common for miRNAs [26], which highlights the value of investigating many phenotypes rather than a single trait or disease for each miRNA. However, no systematic investigation of genetic variants in miRNAs on a wide range of traits has been published thus far.
In this study, genetic variants residing in different regions of miRNAs were used to proxy miRNA expression levels and link them with an extensive range of clinical conditions in the UK Biobank through phenome-wide association studies (PheWAS). We further tested for the presence of an effect of miRNAs on clinical conditions through colocalisation and Mendelian randomisation analysis. This approach offered the first large-scale systematic analysis to investigate the effects of variants in miRNA sequences on human diseases in a hypothesis-free manner and discover novel associations with possible clinical importance.
Methods
Study population
This study used individual-level data from the UK Biobank, a large prospective cohort of ~ 500,000 participants in the UK [27]. The UK Biobank recruited individuals aged 40–69 years old living in the UK between 2006 and 2010. This cohort collects extensive phenotype and genotype data, including longitudinal follow-up provided in the hospital episode statistics (HES) data.
Participants were asked to give blood samples at enrolment from which the DNA was extracted. Genotyping was conducted at Affymetrix Research Services Laboratory. Quality control of the data was carried out at the Wellcome Trust Centre for Human Genetics [28]. Genome-wide genotyping was performed on all UK Biobank participants, covering ~ 805,000 markers. Marker-based quality control was conducted using 463,844 participants of European ancestries, where they were tested for batch effects, plate effects, Hardy–Weinberg Equilibrium (HWE), sex effects, array effects, and discordance across replicates. Any marker that failed in at least one of those tests was set to have a missing genotype call.
Imputation in the UK Biobank was conducted using UK10K and 1000 Genomes Phase 3 reference panels, consisting of 87,696,888 bi-allelic markers in 12,570 haplotypes for interim data release (~ 150,000 samples). Further, the HRC reference panel consisting of 39,235,157 markers in 64,976 haplotypes was incorporated in the remaining samples' imputation. The major histocompatibility complex (MHC) region in chromosome six was imputed separately using a specific algorithm [29]. The UCSC genome annotation database for the Genome Reference Consortium Human Build 37 (GRCh37) assembly of the human genome was used to assign the dbSNP Reference SNP (rs) IDs.
In our study, one of each pair or relatives was excluded based on a kinship coefficient of > 0.088, the proposed lower-limit kinship-coefficient threshold for second-degree relatives [30]. Analysis was restricted to participants who identified as “White”. Additionally, individuals who asked to withdraw from the cohort until the time of analysis were excluded (Additional file 1: Fig. S1).
Selection of candidate variants
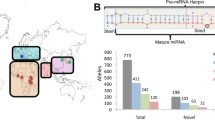
An overview of our study is presented in Fig. 1. We used a database from the most recent version of miRNASNP-v3 (http://bioinfo.life.hust.edu.cn/miRNASNP/#!/) [31] to obtain the list of candidate variants residing in miRNA genes. The dataset was available from containing 46,826 unique variants residing in 1897 precursor miRNAs and corresponding to 2625 mature miRNAs.

Study overview. We conducted PheWAS on variants in miRNAs using the UK Biobank data (N = 423,419). Putative target genes for miRNAs with FDR-significant findings in their seed regions were identified by leveraging databases for target genes of miRNAs and the genome-wide association studies (GWAS) hits for relevant traits. Further, we tested whether changes in miRNA expression could affect disease risk through colocalisation and Mendelian randomisation
Our analysis focused on well-imputed common variants. From the list of variants, 370 of them were available in the imputed genetic data in the UK Biobank. There were 350 variants remained after filtering for info score > 0.7 and minor allele frequency > 0.01. Multi-allelic variants were further excluded, resulting in 346 variants included in our analysis (Additional file 1: Table S1). In total, we tested for 43 variants in the seed region of 44 miRNAs, 45 variants in the mature region of 66 miRNAs, and 238 variants in 208 precursor miRNAs (Fig. 1).
Phenome-wide association studies (PheWAS)
PheWAS was conducted separately for each candidate variant using the PheWAS package in R [32]. We used the hospital episode statistics data in the UK Biobank including a total of 8,404,826 episodes. There were 372,256 participants with at least one episode of diagnosis in all hospital inpatient records. ICD (ninth and tenth editions) codes from hospital episode statistics data were aligned into phecodes to identify clinically related phenotypes and assign a case–control status for each participant [33]. The analysis was conducted for phecodes with at least 200 cases [34]. Logistic regression was performed for each genetic variant with adjustment for age, sex, genotyping array, and the first five genetic principal components to account for population stratification. In this study, we accounted for multiple testing by controlling for false discovery rate (FDR), which estimates the proportion of falsely rejected hypothesis among all tests [35]. This method was chosen because hospital diagnoses are not entirely independent of each other.
Target gene and enrichment analysis
For findings in the seed region of miRNAs, putative target genes for miRNAs that might be involved in the disease process were identified by leveraging the GWAS summary statistics for the traits of interest and target genes from TargetScan v7.2 [36] and miRTarBase [37]. Enrichment analysis for target genes were conducted for miRNAs and their counterparts following the method applied in our previous work [26].
Colocalisation and Mendelian randomisation (MR)
Previous expression quantitative trait loci (eQTL) analysis on blood or plasma levels of miRNAs were used to check whether the genetic variants affect the level of corresponding miRNAs or acting as miRNA expression quantitative trait loci (miR-eQTLs) [38, 39]. For associations with evidence of miR-eQTLs, we performed colocalisation using summary statistics of miR-eQTLs in the Rotterdam Study [38] and summary statistics from large GWAS of the trait of interest. For each miRNA-disease pair, we used genomic region extending 200 kb on either side of mature miRNA position according to miRBase [13]. We implemented a Bayesian framework to test for the presence of shared causal variant and calculated posterior probability to declare as evidence of colocalisation implemented in coloc package in R [40].
Colocalisation method estimates the posterior probabilities (PP) of four scenarios/hypotheses: H0: neither trait has a genetic association in the region, H1: only trait 1 (miRNA) has a genetic association in the region, H2: only trait 2 (disease) has a genetic association in the region, H3: both traits are associated with different causal variants, H4: both traits are associated and share a single causal variant. Default priors were used in all analyses. Results with PP H4 > 0.5 were reported and those with PP H4 > 0.7 were considered significant. We also extracted SNP with the highest SNP PP H4 (posterior probability that the SNP being causal conditional on H4 being true) as the likely shared causal variant between miRNAs and the clinical conditions.
For findings with evidence of colocalisation, we further conducted MR analysis using the same genetic variants used for PheWAS and estimating the MR effect size using the Wald ratio method. The genetic association estimates between the instruments and miRNAs were taken from the Rotterdam Study [38], whereas between the instruments and clinical outcomes were taken from the UK Biobank.
Results
Study participants
This analysis used individual-level data from the UK Biobank, a large prospective cohort of ~ 500,000 participants in the UK [27]. After excluding related participants, those who self-identified as non-White, and withdrawn participants, 423,419 participants were included (Additional file 1: Fig. S1). Of those, 54% were female. The mean (SD) of age of participants was 56.8 (7.9) years (Table 1). The hospital-based diagnoses coded in both ICD9/10 were mapped into 1805 phecodes to enable conducting PheWAS, of which 905 phecodes had at least 200 cases across 16 disease groups (Table 2).
Phenome-wide association studies
We included 346 common genetic variants in our analysis, corresponding to 43 variants in the seed region of 44 miRNAs, 45 variants in the mature region of 66 miRNAs, and 238 variants in 208 precursor miRNAs (Additional file 1: Table S1). The minor allele frequencies of tested variants ranged from 0.03 to 0.33 (in seed region), 0.01–0.42 (in mature region), 0.01–0.46 (in precursor miRNAs). PheWAS was conducted for each of those 346 variants by testing against 905 phecodes with at least 200 cases. At FDR < 0.05 (P < 2.15 × 10–5), there were 122 significant associations for 35 miRNAs, consisting of six variants in the seed region, nine variants in the mature region, and 27 variants in the precursor sequences (Fig. 2, Additional file 1: Table S2). Of 35 miRNAs with significant findings, nineteen were associated with multiple diagnoses, with six of them associated with multiple disease categories. The strongest associations were reported between rs4285314 in the precursor gene of miR-3135b and celiac disease risk (odds ratio (OR) per effect allele increase = 0.37, P = 1.8 × 10–162). The strong association found in current study highlighted the value of genetic studies using large population-based cohort.

Circular plot shows the links between genetic variants in miRNAs (coloured outer circle in bottom half) and clinical diagnoses (grey outer circle in upper half). Each line represents FDR-significant association between genetic variants in miRNAs and clinical diagnoses belonging to a particular disease group
Fifty-seven associations (46.7%) identified in our analysis were for seven variants in the major histocompatibility complex (MHC) region (Fig. 2, Additional file 1: Table S2). Each variant in this region was associated with multiple diagnoses in at least two disease groups (Additional file 1: Fig. S2), the majority were immune-related disorders (Fig. 3, Additional file 1: Table S2). Examples are between rs2276448 and celiac disease (OR 0.49, P = 1.7 × 10–32), ankylosing spondylitis (OR 0.57, P = 1.2 × 10–6), and allergy to antibiotics (OR 1.08, P = 1.2 × 10–7).

Forest-plots for FDR-significant associations of genetic variants located in the MHC region, including for miR-6891 and miR-3135b precursors. Dots represent odds ratio (OR) and 95%CI of the association between the variant (shown by different colour) and clinical conditions. The chromosomal position and corresponding effect alleles are presented in Additional file 1: Table S2
Outside the MHC region, 12 associations were with variants in seed, 15 in mature, and 38 in the precursor regions of miRNAs (Fig. 4). Several variants in the seed region were associated with hypertension, namely rs2168518 in miR-4513 (OR 0.96, P = 8.5 × 10–10), rs2070960 in miR-3620-5p (OR 0.95, P = 3.7 × 10–7), rs2925980 in miR-7854-3p (OR 0.97, P = 7.0 × 10–6), and rs11382316 in miR-316 (OR 1.03, P = 1.7 × 10–6). The rs2168518 polymorphism in miR-4513 was additionally associated with musculoskeletal disorders, including osteoarthritis (OR 1.04, P = 1.1 × 10–5). To note, the associations between rs2168518 and hypertension were significant after more stringent corrections for multiple testing using Bonferroni method (P < 0.05/(346 × 905) (Fig. 4a, Additional file 1: Table S2).


Enhanced volcano plots for PheWAS results of SNPs in seed (a), mature (b), or precursor genes (c) of miRNAs. Plots were only created for variants with at least one FDR-significant finding and shown for those with P > 1 × 10–50. Full results with allele information are provided in Additional file 1: Table S2. The X-axis denotes effect estimates (log odds ratio) for corresponding effect alleles. Y axis indicates − log10 of the association p values between each variant and clinical condition. Different colours of the dots represent different variants. Different shapes show different disease groups. Thresholds of significance are indicated by dashed blue (nominal), red (FDR < 0.05), and purple (Bonferroni or P < 0.05/(346 × 905)) lines
In the mature region, two variants in miR-3939 were associated with hypothyroidism, namely rs368791729 (OR 1.08, P = 8.1 × 10–11) and rs370955537 (OR 1.08, P = 8.1 × 10–11). Multiple variants were associated with hypertension, namely rs3817551 in miR-7107-3p (OR 0.97, P = 1.1 × 10–6) and rs4687672 in miR-8064 (OR 1.03, P = 2.9 × 10–6). Additionally, rs4687672 was also associated with type 2 diabetes (OR 1.05, P = 4.8 × 10–6). Rs11614913 in the mature region of miR-196a was previously associated with survival in non-small cell lung cancers [41], decreased risk of breast cancer [42], and type 2 diabetes [43]. Here, we additionally showed rs11614913 being associated with reflux esophagitis (OR 1.06, P = 3.3 × 10–6) (Fig. 4b, Additional file 1: Table S2). Of the 38 associations for 22 variants in precursor genes of 22 miRNAs, the strongest association was between rs547611708 in the precursor of miR-1225 and atrial fibrillation and flutter (OR 1.32, P = 3.6 × 10–9) (Fig. 4c, Additional file 1: Table S2).
Target gene and enrichment analysis
As genetic variants in the seed region of miRNAs are likely to affect the miRNA-target mRNA binding, putative target genes for miRNAs with findings in their seed region were investigated by leveraging the genome-wide association studies (GWAS) hits for relevant traits. To this end, we used the genetic variants located in the target genes of miRNAs and tested their association with corresponding traits.
Among associations in the seed region of six miRNAs (miR-3161, miR-3620-5p, miR-4467, miR-4513, miR-6891-3p, miR-7854-3p) (Additional file 1: Table S2), genome-wide significant hits from previous GWAS with publicly available summary statistics on blood pressure [44], cholesterol levels [45], and osteoarthritis [46] were used in combination with predicted and validated target genes from TargetScan v7.2 [36] and miRTarBase [37]. Target genes that might mediate the association between miR-3620-5p, miR-3161, and miR-4513 with hypertension and between miR-6891-3p and cholesterol levels were identified (Additional file 1: Table S3). We also found the proportion of target genes associated with blood pressure is higher than expected for miR-3620-3p (P = 0.01).
miRNA expression quantitative trait loci look-up
We used summary statistics of miRNA expression quantitative trait loci (miR-eQTLs) [38, 39] to check whether the studied variants are associated with the blood or plasma levels of their corresponding miRNAs. The genetic variants in the precursor region of miRNAs were mapped into both -3p and -5p forms of mature miRNAs. Out of 346 variants tested in current analysis, 33 variants were nominally significant of which 13 variants were found to be miR-eQTLs of corresponding miRNAs after Bonferroni correction (P < 0.05/346) (Additional file 1: Table S4).
Colocalisation and Mendelian randomisation analysis
We conducted colocalisation and Mendelian randomisation (MR) analysis for findings with significant miR-eQTLs and the clinical conditions where large GWAS summary statistics are available. We used summary statistics for type 2 diabetes from DIAGRAM [47], type 1 diabetes from Forgetta et al. [48], and lipid traits from Global Lipid Genetics Consortium (GLGC) (total cholesterol (TC), triglyceride (TG), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C)) [45] to conduct colocalisation for miR-6891-3p and miR-6821-5p (Additional file 1: Table S5). We found evidence for colocalisation between miR-6891-3p and total cholesterol (PP H4 > 0.9). Suggestive evidence was also found between miR-6891-3p and triglycerides (PP H4 > 0.6) and HDL-C (PP H4 > 0.6). The same shared causal variant was identified for association between miR-6891-3p and TC and HDL-C (rs2596501), but not for TG (rs3130614) (Additional file 1: Table S5, Fig. S3). Further, our single-instrument MR analysis suggested protective effect of miR-6891-3p on the risk of hyperlipidaemia (OR 0.69, P = 1.0 × 10–5) using the UK Biobank as the outcome dataset. This finding was replicated using the largest GWAS from GLGC as the outcome dataset [49], supporting the lowering effect of miR-6891-3p on TC, TG, LDL-C, and non-HDL-C (Table 3).
Discussion
This study presented an agnostic investigation of 346 common genetic variants residing in miRNA-related sequences against a wide range of clinical diagnoses. We highlighted the top findings between variants in the MHC region with a range of diseases, the majority belonging to immune-related disorders. We demonstrated the value of phenome-wide analysis in unravelling the pleiotropy of miRNAs by studying a wide range of conditions beyond what has been previously investigated. For example, rs2168518 in the seed region of miR-4513 was known mainly for its association with the risk of cardiometabolic phenotypes, including fasting glucose, LDL-C, total cholesterol, and risk of coronary artery disease [50]. In the current analysis, rs2168518 was also associated with osteoarthritis and intervertebral disc disorders. As another example, rs11614913, located in the mature region of miR-196a-3p, which was also known to be related to cardiometabolic traits [43, 51] and the risk of several cancers [41, 42], was associated with reflux esophagitis in this analysis. Several findings belong to miRNAs that were more recently discovered in humans, such as miR-5094, miR-6821, miR-7107-3p, and miR-8064 and, thus, have not been extensively studied in the literature.
Nearly half of the associations identified in our analysis were for variants in the MHC region. We highlighted potential causal associations between miR-6891-3p and hyperlipidaemia through colocalisation and MR analysis. miR-6891-3p is located in the MHC region, known as the most-dense region in the human genome that contains a rich density of genes, is highly polymorphic, and has complex linkage disequilibrium (LD) structure that differs across populations [52]. The proteins encoded by human leukocyte antigen (HLA) genes in this region are essential in immune response, contributing to the pleiotropic role of this region in infectious, inflammatory, autoimmune diseases [53], and neurological disorders [54]. The genetic variants in the MHC region were previously shown to contribute substantially to genome-wide association analysis across an extended range of phenotypes in the UK Biobank [55]. Current findings validate our approach by replicating the known pleiotropic nature of this region [56, 57]. Three variants in miR-6891-3p with significant PheWAS findings also affect the expression of HLA-B [58], the host gene where miR-6891-3p resides in its intron [59], in addition to many other nearby genes. The extensive LD structure in the MHC region makes it challenging to assign causal variants since multiple variants in high LD could have nearly equivalent statistical associations [52, 60]. For a particular disease risk, there could be interactive effects of different alleles within the same locus or across different loci (epistasis) [61, 62].
Our colocalisation and MR analysis indicated the presence of shared causal variant and potential aetiological effect of miR-6891-3p on the risk of hyperlipidaemia. While one of the assumptions for colocalisation is that the region is densely genotyped and that the true causal variant is among these genotyped variants [40], the complex LD structure in the MHC region and the absence of substantial proportion of well-imputed variants in the tested region could have led to undetected shared causal variants between miR-6891-3p and other conditions reported here. A previous study showed miR-6891-3p targeting TLR4, thereby inhibiting inflammatory response in osteoarthritis [63]. For its counterpart, the target site of miR-6891-5p in IGHA1 and IGHA2 was identified, supporting the role of miR-6891-5p in IgA deficiency [64]. In vitro experiments showed that rs4351242 reduced the level of miR-3135b and was associated with age-related macular degeneration [8], while rs4285314 was associated with the risk of rheumatoid arthritis in the Chinese population [65]. We highlighted putative target genes relevant to the diseases for the associations identified in seed region, in line with the concept that target genes for miRNAs tend to be clustered according to their function [26]. These candidate target genes might be of interest for further studies to dissect underlying mechanisms. Our findings also indicate the value of using population-based genetic and clinical data to conduct a hypothesis-generating approach and complement those from experimental studies.
This analysis has several strengths by taking advantage of PheWAS as a powerful method to discover novel associations beyond what is known in the literature and reveal the pleiotropy of a genetic variant and miRNAs [66]. First, the UK Biobank has been deeply phenotyped and is genotyped using similar genotyping arrays, ensuring uniformity in the data and making it less likely to find spurious associations. Nevertheless, for some traits such as cancers, the UK Biobank might not have sufficient number of cases. To maximise power in our analysis, we utilised the full set of UK Biobank participants with available genotype and clinical diagnoses data, excluding related individuals and those who have withdrawn their consent. Although there is a higher proportion of females in our study (54%), we adjusted for sex, among other covariates in our analysis, which should have minimised the effect of confounding due to sex. Second, the use of ICD classification could minimise the misclassification of cases and controls compared to a standard self-reported based assignment. Third, clinical data could best represent the clinical importance of genotype to phenotype associations. A genetic variant could be associated with phenotype, but that does not always imply its clinical relevance.
This study has several limitations. First, the current investigation is limited to clinical conditions that belong to binary traits. PheWAS could also be implemented on different outcomes, such as laboratory measurement or imaging data [67]. Second, the current analysis is limited to miRNAs with common variants. Among 346 variants tested, only a small proportion was found to affect corresponding mature miRNA levels. The remaining variants might affect the functionality rather than the level of miRNAs. As we are focusing on common variants, this observation may also imply that miRNAs have a tight constraint on their expression such that common variants do not show many effects. Most candidate variants are rare and, thus, are not available in our current dataset. This highlights the need to analyse the effect of rare variants on miRNA expression and their consequences on human diseases, such as by using whole-genome sequencing data.
Third, it is important to ascertain whether the same genetic variant affects miRNA level in tissues that are relevant to the diseases. When tissue-specific miR-eQTLs become available, we could check whether the same candidate variant affects the expression of miR-6891-3p in adipose tissue and conduct colocalisation and MR analysis using this data. An experimental study could also test whether the presence of certain allele of the candidate variant influences the corresponding miRNA abundance through transfection experiments [8, 68]. Our study could help in prioritising candidate miRNAs for in-depth candidate-based experiments in relevant cell or tissue type.
Fourth, since our study focused on participants of European ancestries, it is essential to replicate our findings in different ancestral groups. Incorporating diverse ancestries could generate more transferrable findings for a wider population. Finally, despite using the largest GWAS on miRNA to date, we were not able to study all candidates and larger sample size might be needed to identify strong instruments for miRNAs. Therefore, we cannot rule out a possibility of undetected causal relationship between miRNAs and the diseases reported.
Conclusions
Our study offers an opportunity to investigate the effect of variants in miRNAs and discover their clinical importance using population-level data. We identified a potential causal effect of miR-6891-3p on the risk of hyperlipidaemia, as consistently supported by PheWAS, colocalisation, and MR analysis, highlighting the value of using large population genetic and clinical data to study the causal role of miRNAs in human diseases.
Availability of data and materials
Access to the individual-level data in UK Biobank is dependent on an application directly to the UK Biobank through its standard application process (https://www.ukbiobank.ac.uk/).
Abbreviations
- DIAGRAM:
-
Diabetes genetics replication and meta-analysis
- eQTL:
-
Expression quantitative trait loci
- GLGC:
-
Global lipids genetics consortium
- GWAS:
-
Genome-wide association study
- HDL-C:
-
High-density lipoprotein cholesterol
- HES:
-
Hospital episode statistics
- ICD:
-
International classification of disease
- LD:
-
Linkage disequilibrium
- LDL-C:
-
Low-density lipoprotein cholesterol
- MHC:
-
Major histocompatibility region
- miRNA:
-
MicroRNA
- OR:
-
Odds ratio
- PheWAS:
-
Phenome-wide association study
- RNA:
-
Ribonucleic acid
- SE:
-
Standard error
- TC:
-
Total cholesterol
- TG:
-
Triglycerides
References
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Cortez MA, Bueso-Ramos C, Ferdin J, Lopez-Berestein G, Sood AK, Calin GA. MicroRNAs in body fluids—the mix of hormones and biomarkers. Nat Rev Clin Oncol. 2011;8(8):467–77.
Peng C, Wang Y. MicroRNAs as new players in endocrinology. Front Endocrinol. 2018;9:459.
Mendell JT. MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle. 2005;4(9):1179–84.
He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–31.
Wu S, Huang S, Ding J, Zhao Y, Liang L, Liu T, et al. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene. 2010;29(15):2302–8.
Ghanbari M, Ikram MA, de Looper HW, Hofman A, Erkeland SJ, Franco OH, et al. Genome-wide identification of microRNA-related variants associated with risk of Alzheimer’s disease. Sci Rep. 2016;6(1):1–9.
Ghanbari M, Darweesh SK, de Looper HW, van Luijn MM, Hofman A, Ikram MA, et al. Genetic variants in microRNAs and their binding sites are associated with the risk of Parkinson disease. Hum Mutat. 2016;37(3):292–300.
Ghanbari M, Franco OH, de Looper HW, Hofman A, Erkeland SJ, Dehghan A. Genetic variations in microRNA-binding sites affect microRNA-mediated regulation of several genes associated with cardio-metabolic phenotypes. Circ Cardiovasc Genet. 2015;8(3):473–86.
Ghanbari M, Iglesias AI, Springelkamp H, van Duijn CM, Ikram MA, Dehghan A, et al. A genome-wide scan for MicroRNA-related genetic variants associated with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2017;58(12):5368–77.
Ciccacci C, Di Fusco D, Cacciotti L, Morganti R, D’Amato C, Greco C, et al. MicroRNA genetic variations: association with type 2 diabetes. Acta Diabetol. 2013;50:867–72.
Kaur A, Mackin ST, Schlosser K, Wong FL, Elharram M, Delles C, et al. Systematic review of microRNA biomarkers in acute coronary syndrome and stable coronary artery disease. Cardiovasc Res. 2020;116(6):1113–24.
Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–62.
Gong J, Tong Y, Zhang H, Wang K, Hu T, Shan G, et al. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum Mutat. 2012;33(1):254–63.
Cammaerts S, Strazisar M, De Rijk P, Del Favero J. Genetic variants in microRNA genes: impact on microRNA expression, function, and disease. Front Genet. 2015;6:186.
Han M, Zheng Y. Comprehensive analysis of single nucleotide polymorphisms in human microRNAs. PLoS ONE. 2013;8(11):e78028.
Saunders MA, Liang H, Li W. Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci. 2007;104(9):3300–5.
Bhattacharya A, Cui Y. Systematic prediction of the impacts of mutations in microRNA seed sequences. J Integr Bioinform. 2017;14(1):20170001.
Sun G, Yan J, Noltner K, Feng J, Li H, Sarkis DA, et al. SNPs in human miRNA genes affect biogenesis and function. RNA. 2009;15(9):1640–51.
Gong J, Liu C, Liu W, Wu Y, Ma Z, Chen H, et al. An update of miRNASNP database for better SNP selection by GWAS data, miRNA expression and online tools. Database. 2015;2015:bav029.
Duan R, Pak C, Jin P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum Mol Genet. 2007;16(9):1124–31.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33.
Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10(6):389–402.
Chhichholiya Y, Suryan AK, Suman P, Munshi A, Singh S. SNPs in miRNAs and target sequences: role in cancer and diabetes. Front Genet. 2021;12:793523.
Zhang Y, Bai R, Liu C, Ma C, Chen X, Yang J, et al. MicroRNA single-nucleotide polymorphisms and diabetes mellitus: a comprehensive review. Clin Genet. 2019;95(4):451–61.
Mustafa R, Ghanbari M, Evangelou M, Dehghan A. An enrichment analysis for cardiometabolic traits suggests non-random assignment of genes to microRNAs. Int J Mol Sci. 2018;19(11):3666.
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779.
Biobank U. Genotyping of 500,000 UK Biobank participants. Description of sample processing workflow and preparation of DNA for genotyping. 2015; 11.
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–9.
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26(22):2867–73.
Liu C, Fu X, Xia M, Zhang Q, Gu Z, Guo A. miRNASNP-v3: a comprehensive database for SNPs and disease-related variations in miRNAs and miRNA targets. Nucleic Acids Res. 2021;49(D1):D1276–81.
Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics. 2014;30(16):2375–6.
Wei W, Bastarache LA, Carroll RJ, Marlo JE, Osterman TJ, Gamazon ER, et al. Evaluating phecodes, clinical classification software, and ICD-9-CM codes for phenome-wide association studies in the electronic health record. PLoS ONE. 2017;12(7):e0175508.
Verma A, Bradford Y, Dudek S, Lucas AM, Verma SS, Pendergrass SA, et al. A simulation study investigating power estimates in phenome-wide association studies. BMC Bioinform. 2018;19(1):120.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol). 1995;57(1):289–300.
Agarwal V, Bell GW, Nam J, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005.
Huang H, Lin Y, Li J, Huang K, Shrestha S, Hong H, et al. miRTarBase 2020: updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res. 2020;48(D1):D148–54.
Mustafa R, Mens M, van Hilten A, Huang J, Roshchupkin GV, Huan T, et al. An atlas of genetic regulation and disease associations of microRNAs. medRxiv. 2022.
Nikpay M, Beehler K, Valsesia A, Hager J, Harper M, Dent R, et al. Genome-wide identification of circulating-miRNA expression quantitative trait loci reveals the role of several miRNAs in the regulation of cardiometabolic phenotypes. Cardiovasc Res. 2019;115(11):1629–45.
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383.
Hu Z, Chen J, Tian T, Zhou X, Gu H, Xu L, et al. Genetic variants of miRNA sequences and non–small cell lung cancer survival. J Clin Investig. 2008;118(7):2600–8.
Hoffman AE, Zheng T, Yi C, Leaderer D, Weidhaas J, Slack F, et al. microRNA miR-196a-2 and breast cancer: a genetic and epigenetic association study and functional analysis. Can Res. 2009;69(14):5970–7.
Buraczynska M, Zukowski P, Wacinski P, Ksiazek K, Zaluska W. Polymorphism in microRNA-196a2 contributes to the risk of cardiovascular disease in type 2 diabetes patients. J Diabetes Complicat. 2014;28(5):617–20.
Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50(10):1412–25.
Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274.
Tachmazidou I, Hatzikotoulas K, Southam L, Esparza-Gordillo J, Haberland V, Zheng J, et al. Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat Genet. 2019;51(2):230–6.
Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50(11):1505–13.
Forgetta V, Manousaki D, Istomine R, Ross S, Tessier M, Marchand L, et al. Rare genetic variants of large effect influence risk of type 1 diabetes. Diabetes. 2020;69(4):784–95.
Graham SE, Clarke SL, Wu KH, Kanoni S, Zajac GJ, Ramdas S, et al. The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600(7890):675–9.
Ghanbari M, de Vries PS, de Looper H, Peters MJ, Schurmann C, Yaghootkar H, et al. A genetic variant in the seed region of mi R-4513 shows pleiotropic effects on lipid and glucose homeostasis, blood pressure, and coronary artery disease. Hum Mutat. 2014;35(12):1524–31.
Ghanbari M, Sedaghat S, De Looper HW, Hofman A, Erkeland SJ, Franco OH, et al. The association of common polymorphisms in mi R-196a2 with waist to hip ratio and mi R-1908 with serum lipid and glucose. Obesity. 2015;23(2):495–503.
De Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006;38(10):1166–72.
Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, et al. Gene map of the extended human MHC. Nat Rev Genet. 2004;5(12):889–99.
Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42(9):781–5.
Canela-Xandri O, Rawlik K, Tenesa A. An atlas of genetic associations in UK Biobank. Nat Genet. 2018;50(11):1593–9.
Kim C, Kim YJ, Choi W, Jang H, Hwang MY, Jung S, et al. Phenome-wide association study of the major histocompatibility complex region in the Korean population identifies novel association signals. Hum Mol Genet. 2022;31:2655–67.
Liu J, Ye Z, Mayer JG, Hoch BA, Green C, Rolak L, et al. Phenome-wide association study maps new diseases to the human major histocompatibility complex region. J Med Genet. 2016;53(10):681–9.
Võsa U, Claringbould A, Westra H, Bonder MJ, Deelen P, Zeng B, et al. Large-scale cis-and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53:1300–10.
Hinske LC, Franca GS, Torres HA, Ohara DT, Lopes-Ramos CM, Heyn J, et al. miRIAD—integrating microRNA inter-and intragenic data. Database. 2014;2014:99.
Ioannidis J, Thomas G, Daly MJ. Validating, augmenting and refining genome-wide association signals. Nat Rev Genet. 2009;10(5):318–29.
Lincoln MR, Ramagopalan SV, Chao MJ, Herrera BM, DeLuca GC, Orton S, et al. Epistasis among HLA-DRB1, HLA-DQA1, and HLA-DQB1 loci determines multiple sclerosis susceptibility. Proc Natl Acad Sci. 2009;106(18):7542–7.
Hughes T, Adler A, Kelly JA, Kaufman KM, Williams AH, Langefeld CD, et al. Evidence for gene–gene epistatic interactions among susceptibility loci for systemic lupus erythematosus. Arthritis Rheum. 2012;64(2):485–92.
Zhang P, Sun J, Liang C, Gu B, Xu Y, Lu H, et al. lncRNA IGHCγ1 acts as a ceRNA to regulate macrophage inflammation via the miR-6891–3p/TLR4 Axis in osteoarthritis. Mediat Inflamm. 2020. https://doi.org/10.1155/2020/9743037.
Chitnis N, Clark PM, Kamoun M, Stolle C, Brad Johnson F, Monos DS. An expanded role for HLA genes: HLA-B encodes a microRNA that regulates IgA and other immune response transcripts. Front Immunol. 2017;8:583.
Guo S, Jin Y, Zhou J, Zhu Q, Jiang T, Bian Y, et al. MicroRNA variants and HLA-miRNA interactions are novel rheumatoid arthritis susceptibility factors. Front Genet. 2021;12:747274.
Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31(12):1102–11.
Verma A, Leader JB, Verma SS, Frase A, Wallace J, Dudek S, et al. Integrating clinical laboratory measures and ICD-9 code diagnoses in phenome-wide association studies. In: Biocomputing 2016: proceedings of the Pacific symposium. World Scientific; 2016. p. 168–79.
Beehler K, Nikpay M, Lau P, Dang A, Lagace TA, Soubeyrand S, et al. A common polymorphism in the FADS1 Locus Links miR1908 to low-density lipoprotein cholesterol through BMP1. Arterioscler Thromb Vasc Biol. 2021;41(8):2252–62.
Acknowledgements
We would like to thank all participants of the UK Biobank. Explicit informed consent was obtained from all participants when they enrolled in the UK Biobank. Access to the UK Biobank was provided through application 52569. We thank Loukas Zagkos for helping with the visualisation of the results.
Funding
RM is supported by the President’s Ph.D. Scholarship from Imperial College London. AD is funded by a Wellcome Trust seed award (206046/Z/17/Z). This project is partly supported by the Erasmus MC Fellowship Grant (EMCF20213) of MG. VK is funded by Academy of Finland Project 326291 and the European Union’s Horizon 2020 Grant Agreement No. 848158 (EarlyCause). The funders have no involvement in the study design, analysis, and interpretation of data.
Author information
Authors and Affiliations
Contributions
RM and AD designed the study. RM conducted statistical analysis. VK helped in the methodology and statistical analysis. MG, ME, and AD oversaw the research. RM wrote the first draft of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The UK Biobank has approval from the North-West Multi-centre Research Ethics Committee (MREC) as a Research Tissue Bank (RTB) approval.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1:
The list of 346 genetic variants tested in PheWAS. Table S2: 122 FDR-significant hits in PheWAS. Table S3: Candidate target genes whose interaction might be implicated by the presence of SNPs in seed region of miRNAs. Table S4: Genetic variants in miRNAs that affect plasma levels of corresponding mature miRNAs (P<0.05). Table S5: Results of colocalisation analysis. Fig. S1: Selection of study participants. Fig. S2: Each tested variant in MHC is associated with multiple disease groups. Fig. S3: Regional plot for colocalisation analysis between miR-6891-3p and total cholesterol (a), triglycerides (b), and high-density lipoprotein cholesterol (HDL-C). Variants were filtered based on imputation quality (Rsq>0.7). The most likely shared causal variant is labelled.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mustafa, R., Ghanbari, M., Karhunen, V. et al. Phenome-wide association study on miRNA-related sequence variants: the UK Biobank. Hum Genomics 17, 104 (2023). https://doi.org/10.1186/s40246-023-00553-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-023-00553-w