
Abstract
Background
Cancer predisposition is most often studied in the context of single cancers. However, inherited cancer predispositions can also give rise to multiple primary cancers. Yet, there is a paucity of studies on genetic predisposition in multiple primary cancers, especially those outside of well-defined cancer predisposition syndromes. This study aimed to identify germline variants associated with dual primary cancers of the breast and lung.
Methods
Exome sequencing was performed on germline DNA from 55 Singapore patients (52 [95%] never-smokers) with dual primaries in the breast and lung, confirmed by histopathology. Using two large control cohorts: the local SG10K_Health (n = 9770) and gnomAD non-cancer East Asians (n = 9626); and two additional local case cohorts of early-onset or familial breast cancer (n = 290), and lung cancer (n = 209), variants were assessed for pathogenicity in accordance with ACMG/AMP guidelines. In particular, comparisons were made with known pathogenic or likely pathogenic variants in the ClinVar database, pathogenicity predictions were obtained from in silico prediction software, and case–control association analyses were performed.
Results
Altogether, we identified 19 pathogenic or likely pathogenic variants from 16 genes, detected in 17 of 55 (31%) patients. Six of the 19 variants were identified using ClinVar, while 13 variants were classified pathogenic or likely pathogenic using ACMG/AMP guidelines. The 16 genes include well-known cancer predisposition genes such as BRCA2, TP53, and RAD51D; but also lesser known cancer genes EXT2, WWOX, GATA2, and GPC3. Most of these genes are involved in DNA damage repair, reaffirming the role of impaired DNA repair mechanisms in the development of multiple malignancies. These variants warrant further investigations in additional populations.
Conclusions
We have identified both known and novel variants significantly enriched in patients with primary breast and lung malignancies, expanding the body of known cancer predisposition variants for both breast and lung cancer. These variants are mostly from genes involved in DNA repair, affirming the role of impaired DNA repair in the predisposition and development of multiple cancers.
Similar content being viewed by others


Introduction
Multiple primary cancers (MPCs) refer to two or more malignancies arising independently in different organs of an individual that are not due to recurrence or metastasis. MPCs occur at a frequency of between 2 to 17% within 20 years of follow-up [1]. An estimated 1% (6,269/620,429) of female primary breast cancer patients develop primary lung cancer, based on data obtained from the Surveillance Epidemiology, and End Results (SEER) Program [2]. Genetic susceptibility, malignancy resulting from previous anticancer treatments, lifestyle factors such as smoking and alcohol consumption and environmental influences are some factors that increase the risk for MPCs [1]. For example, breast cancer patients who have undergone radiotherapy are at an increased risk of having a second malignancy of lung cancer [3, 4].
MPCs occur in various combinations, such as breast and ovarian cancer in hereditary breast and ovarian cancer [5] or colon and endometrial cancer in Lynch syndrome [6]. However, for other MPCs, such as for breast and lung cancer, there is a paucity of information on candidate cancer predisposition genes. To date, there has been a study that has identified GSN as a candidate gene significantly associated with the development of lung cancer in patients with a history of breast cancer, based on germline whole-exome sequencing (WES) of 28 cases [7].
In a landmark study on MPCs, whole genome sequencing (WGS) was performed on 460 individuals from 440 families to identify variants in 83 known cancer-predisposition genes. Tumor combinations included breast and colorectal cancer (51/883; 5.8%); breast and ovarian (34/883; 3.9%); breast and thyroid (23/883; 2.6%); and breast and lung cancer (12/883; 1.4%) [8]. Pathogenic variants were identified in moderate- and high-risk cancer predisposition genes in 15.2% of probands (67/440), but none of these patients had been diagnosed with primary breast and lung cancer [8]. More recently, a study has assessed genetic susceptibility to MPCs (n = 6429) across 36 organ sites, through WES of two multi-ancestry study populations [9]. A total of 22 variant-phenotype associations were identified, which included rare and common variants. Gene-based burden test showed that 9.52% and 6.78% of individuals with breast and lung cancer had predicted loss of function variants in BRCA1 and BRCA2, respectively. Hence, there are few in-depth WGS or WES studies focusing on the combination of primary malignancies of the breast and lung. The rationale for this study was to elucidate the underlying genetic predisposition to account for the occurrence of these dual malignancies.
Given that lung cancer is one of the most common second primary in breast cancer patients [10], we have performed WES on germline DNA from a cohort of 55 patients with both primary breast and primary lung cancer to identify candidate predisposition variants. Variants were classified as pathogenic or likely pathogenic for cancer predisposition in accordance with ACMG/AMP guidelines, using annotations from the ClinVar database or by evaluating the guidelines. The overall study design is illustrated in Fig. 1. We have identified 19 pathogenic or likely pathogenic variants in 16 genes, detected in 17 of our 55 (31%) patients. Most of these 16 genes are well-known and involved in DNA damage repair, such as BRCA2, TP53, and RAD51D.

Overview of the study design
Results
Clinicopathological characteristics of our study cohort
Information on the demographics, age of onset, ethnicity, family history, and clinicopathological characteristics of the 55 patients with primary breast and lung cancers is provided in Table 1. A large proportion of patients in our study (94.5%) never smoked. Slightly more than half (54.5%) of the patients’ breast and lung cancers occurred at the same right or left side. Thirty-eight (69.1%) subjects developed breast cancer before lung cancer; radiotherapy to the breast or chest wall had been administered to 11 of them, with 6 of these patients having ipsilateral breast and lung cancers. Six (10.9%) patients had bilateral breast cancers and four (7.3%) patients had synchronous dual lung primaries, including one with bilateral breast cancer. Of the 45 lung adenocarcinoma never-smoker patients with known EGFR status, 33 (73.3%) had EGFR-mutated tumors (Table 1). We did not observe an association between hormone receptor status with BRCA1, BRCA2 or EGFR mutation status in lung adenocarcinoma patients (Additional file 1: Table S1).
ClinVar-recorded pathogenic or likely pathogenic variants
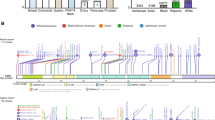
We identified six variants in five patients which were recorded as pathogenic or likely pathogenic variants by multiple submitters on ClinVar in the context of cancer predisposition syndromes (Fig. 2). Expectedly, these six variants were all in known cancer genes related to DNA damage repair (gene set "DNA repair" in the Gene Ontology database): FANCA, PALB2, BRCA2, BRIP1, RAD51D, and TP53.

ClinVar-recorded pathogenic or likely pathogenic variants found in 55 patients with dual breast and lung cancer
Newly classified pathogenic or likely pathogenic variants
Applying the ACMG/AMP guidelines [11] on all variants except for the six ClinVar-recorded pathogenic or likely variants, we newly classified seven pathogenic variants and six likely pathogenic variants in 13 genes, in 12 patients (Table 2). These guidelines include evidence of pathogenicity from computational and predictive data (Additional file 1: Tables S2–S5), as well as from population data (Additional file 1: Table S6). The 12 patients with newly classified pathogenic or likely pathogenic variants do not overlap with the five patients with ClinVar-recorded pathogenic or likely pathogenic variants.
As with the ClinVar-recorded variants, most of these genes are well-known cancer genes or DNA damage repair genes: TP53, MSH2, FANCA, ERCC6, BRIP1, WRN, FANCI, KMT2A, and PTEN (Additional file 1: Table S7); though some are lesser known: EXT2, WWOX, GATA2, and GPC3. The EXT2, WWOX, and GATA2 variants were backed by very strong (“PVS1”) computational and predictive data, as there are pathogenic stop-gain variants for each gene in ClinVar associated with hereditary cancer-predisposing syndrome, malignant tumor of the esophagus, and susceptibility to acute myeloid leukemia, respectively (Additional file 1: Table S2). The GPC3 variant was backed by moderate (“PM5”) computational and predictive data as it modifies the same amino acid residue as another pathogenic missense variant in ClinVar associated with Wilms tumor (Additional file 1: Table S4).
Despite each variant being present in only one heterozygous patient in the dual primary breast and lung cohort of 55 patients, they were all enriched in that case cohort when compared to gnomAD non-cancer East Asians (n = 9626) or SG10K_Health, a local cohort of healthy volunteers (n = 9770) as per the ACMG/AMP guidelines. As expected of rare variants, only five of these 13 variants were also detected in a local cohort of lung cancer patients (n = 209) of which only three were enriched in that lung cancer cohort versus the two control cohorts. However, none of these 13 variants could be found in a local cohort of early-onset or familial breast cancer patients (n = 290).
Some variants satisfied at least one pathogenicity criteria, but not enough to be classified as pathogenic or likely pathogenic. In total, there are 33 such variants of uncertain significance (VUS) (Additional file 1: Table S8).
Clinical features of patients with pathogenic or likely pathogenic variants
Altogether, 17 of 55 (31%) patients had pathogenic or likely pathogenic variants (Table 3). Of these, 6 variants, which are clinically annotated as pathogenic/likely pathogenic in ClinVar, were detected in 9% (5/55) of patients, and 13 newly classified variants were found in 22% (12/55) of patients (Additional file 1: Table S9). Notably, each of these variants is unique to a single patient, and 2 patients were carriers for two variants each: Patient H3168 had stop gain variants in FANCA and PALB2, and had a sister with lung cancer, and another sister with breast cancer. Patient H4326 had a nonsynonymous variant in MSH2 and a non-frameshift deletion in PTEN, and had a family history of two sisters having lung cancer. There was no observed association between the presence of pathogenic or likely pathogenic variants and EGFR mutation status (Additional file 1: Table S10).
Stop gain and nonsynonymous variants were the most prevalent among our patient cohort, followed by splice site variants. Among patients with pathogenic/likely pathogenic variants, 59% (10/17) of patients had a family history of cancer. Specifically, only 4 patients had a family history of breast and/or lung cancer. Sixteen of our 17 patients identified as ethnic Chinese, with one patient, H2552, who is of Indian ethnicity. Detailed clinical features of the 17 patients with pathogenic/likely pathogenic variants are presented in Table 3.
Discussion
There are limited WGS or WES studies on patients with multiple primary cancers. To the best of our knowledge, we report here the largest WES on patients with dual primary cancers of the breast and lung. Overall, our study uncovered that 31% (17/55) of our patients with dual primary breast and lung cancer harbor pathogenic or likely pathogenic variants in cancer associated genes.
The majority of the patients in our cohort had developed breast cancer before lung cancer. This is in concordance with the findings of other studies that have observed that primary lung cancer occurred after a previous primary breast cancer [10], which may be related to the higher survival probability after diagnosis of breast cancer compared to lung cancer. The interval between the development of a second primary lung cancer after breast cancer was less than 10 years in 25.5% of our cohort, while 34.5% of our patients developed lung cancer 10 to 20 years later. This was in contrast to a WES study comprising 60 breast cancer survivors from Italy, where a second primary lung cancer developed after approximately 7 years [7]. That cohort had 46.7% smokers whereas 94.5% of our cohort were never-smokers.
Radiotherapy may result in increased risk of subsequent lung cancer on the same side as the breast cancer where the radiation was administered [3, 4], though the risk may be lower with modern radiotherapy techniques. Of 194,981 women with nonmetastatic invasive breast cancer who had been treated with mastectomy, women who had also been treated with radiotherapy had a moderate increased risk for developing ipsilateral lung carcinoma after 10 years, but there was no increased risk for the contralateral lung [12]. Of the 11 patients in our study who had received radiotherapy for the treatment of breast cancer, only 6 had ipsilateral breast and lung cancer. Thus, the lack of predominance of patients having received radiotherapy later developing lung cancer on the same side supports the fact that these cancers arose independently without this "iatrogenic cause". Hence other factors such as intrinsic genetic predisposition may play a role in the development of these lung cancers.
There is an ongoing unmet need in identifying risk alleles for lung and breast cancers to optimize genetic screening strategies. This is especially important in lung cancer screening in Asia where ongoing efforts to determine the optimal strategy especially in “at risk” never smokers. A recent study has identified germline pathogenic variants in 4.3% (222/5,118) of patients who had undergone genomic profiling. Of these 222 patients, pathogenic variants in DNA damage repair pathway genes were detected in 193 patients, including in BRCA2 (n = 54) [13]. Germline susceptibility genes warrant further investigation as to their role in risk prediction models.
Among the 55 patients in our cohort who had dual cancers in the breast and lung, we identified variants in 10 DNA damage repair genes in 12 patients, which included FANCA, PALB2, BRCA2, BRIP1, RAD51D, TP53, ERCC6, MSH2, WRN, and FANCI, with PALB2, BRCA2, RAD51D, TP53 and PTEN being known breast cancer susceptibility genes [14,15,16,17]. Germline variants in many of these genes are frequently observed in cancer patients referred for genetic testing, including those with multiple primary malignancies. For example, Lynch syndrome is often associated with pathogenic variants in MSH2, while pathogenic variants in BRCA1 and BRCA2 are commonly associated with hereditary breast and ovarian cancer [15, 18, 19].
Our study extends the findings of previous research proposing that DNA damage repair pathways contribute to cancer development, including in breast and lung cancers [20]. Specifically, our results suggest that DNA damage repair pathways may play a critical role in the development of dual primary breast and lung cancer, as a significant proportion of the variants we found are implicated in DNA repair. For example, the FANCA and FANCI genes which have been implicated in breast cancer, had pathogenic variants detected in both breast cancer patients and individuals with hereditary breast and ovarian cancer (HBOC) [21,22,23,24]. We also identified pathogenic variants in other DNA repair genes such as WRN and ERCC6. WRN is linked to Werner syndrome, a rare disease characterized by premature aging and increased predisposition to cancer. Multiple studies have reported an association between WRN variants and hereditary breast cancer as well as HBOC [23, 25, 26]. Additionally, germline variants and genetic polymorphisms in the ERCC6 gene have been associated with early onset breast cancer and an elevated risk of lung cancer, respectively [27, 28].
The exact mechanism by how germline variants in DNA damage repair genes lead to MPCs remains unclear. However, exposure to carcinogens and treatments like chemotherapy and radiation used to treat the first cancer can exacerbate the effects of these variants, increasing the risk of developing a second primary cancer [29]. A recent study has also established associations between air pollution and lung cancer in non-smokers or light smokers [30].
In addition, our study identified variants in several lesser-known cancer genes, including EXT2, WWOX, GATA2, and GPC3. To the best of our knowledge, the specific germline variants found in our study have not been previously reported in any cancer-related studies. However, other germline alterations have been detected in these genes. For example, in EXT2, germline variants (NP_000392.3:p.Trp79* and NP_997005.1:p.Trp46*) are associated with hereditary multiple osteochondromas, a non-cancerous condition that can develop into chondrosarcoma [31]. Another study reported that germline variants (p.W606X, IVS1762-1G > A, p.T507fs, and p.T642fs) in EXT2 were found in 4 out of 1026 patients with non-small cell lung cancer [32]. Interestingly, a female patient with early-onset multiple primary tumors was reported to have a germline homozygous WWOX variant (NC_000016.9:g.79245877_79245881dup) [33]. Multiple germline variants in GATA2 predispose individuals to familial myelodysplastic/acute leukemia syndrome [34, 35], while germline GPC3 variants are observed in Simpson-Golabi-Behmel syndrome, an overgrowth syndrome that increases the risk of developing Wilms tumor, hepatoblastoma, and neuroblastoma [36, 37].
None of the 19 pathogenic or likely pathogenic variants identified in the dual primary breast and lung cohort were found in the local cohort of 290 early-onset or familial breast cancer patients (Additional file 1: Table S6). This latter cohort comprised of BRCA-negative cases. The absence of these dual primary breast and lung variants in the breast cancer cohort might be explained by the high genetic heterogeneity of these variants, with each of these 19 variants detected in only one dual breast and lung cancer patient. Such high genetic heterogeneity has been observed in BRCA-negative familial high-grade serous ovarian carcinoma patients, where WES had detected rare loss-of-function variants that occurred in less than 0.5% of individuals [38]. Our findings suggest that patients with dual breast and lung cancer could have additional genes involved in cancer predisposition, other than genes known to be associated with breast cancer predisposition.
We have compared our variants against those detected by other studies on MPCs. One study had performed WES on 28 of 60 patients from Italy with breast and lung cancer, and had identified one gene, GSN, to be significantly enriched [7]. However, GSN variants were not observed in our study. This discrepancy could be attributed to differences in the ethnic distribution between our study and the previous study, or to the small sample sizes of both studies highlighting the impact of genetic heterogeneity on variant detection. A second study on patients with MPCs had focused only on 83 known cancer predisposition genes. However, pathogenic variants in these 83 genes were not observed in our patients with primary breast and lung cancer [8]. In a third study, gene-based burden testing showed that there was an increase in carriers with BRCA1 and BRCA2 loss of function variants for breast cancer patients with an additional lung cancer [9]. A pathogenic BRCA2 variant was also found in one of our 55 patients.
Our study has also identified a high rate of VUS, with 63% (33/52) of variants detected in 31 out of 55 patients (56%) patients (Additional file 1: Table S9). This is comparable to the overall VUS rate of 49.9% reported in another cohort of Singaporean patients who underwent genetic testing [39]. These findings align with our previous studies on Asian populations, which have consistently shown higher VUS rates compared to populations of European descent, possibly due to the underrepresentation of Asians in reference databases [40,41,42]. However, as more data becomes available, VUS can be reclassified [43, 44]. Previous studies have reported VUS reclassification rates of 6–15%, with a Singaporean study reporting a reclassification rate of 6.7% over a 6-year period [39]. Of these reclassifications, 94.1% of VUS were downgraded to benign or likely benign variants, while 5.9% were upgraded to pathogenic or likely pathogenic variants. Such reclassifications have a significant impact on the risk assessment and management of patients with suspected or confirmed hereditary cancer, particularly in understudied populations.
A limitation of this study is the small sample size (n = 55) utilized in this cohort, which limits the statistical power of the study, and this may have led to no associations observed between the occurrence of our pathogenic variants and clinicopathological features. However, this is inevitable since the occurrence of dual primary breast and lung cancer is relatively uncommon. Another limitation was that we were unable to perform segregation analysis and validation analysis on additional patients with primary cancers of both the breast and lung, due to the unavailability of DNA samples. Therefore, confirmation of the variants identified in this study in larger cohorts of patients with dual cancers of the breast and lung is warranted.
Conclusions
In summary, through employing WES, we have identified variants that are significantly enriched in patients with primary malignancies in both the breast and lung. This study represents the largest cohort of patients with multiple primaries in the breast and lung, that have been sequenced. Altogether, we have identified 19 pathogenic or likely pathogenic variants in 16 genes, detected in 17 patients. Ten of these 16 genes are DNA damage repair genes, underscoring the importance of these genes in patients with dual malignancies of the breast and lung.
Materials and methods
Patient samples
An overview of our study design is shown in Fig. 1. Peripheral blood or saliva samples were obtained from 55 patients with primary breast and primary lung cancer from the National Cancer Centre Singapore. The later-occurring cancer was verified as a primary cancer by histopathology. Written informed consent was obtained from all patients and the study was approved by the SingHealth Centralised Institutional Review Board (CIRB Ref: 2018/2147 and 2018/2963). The samples were retrospectively selected.
Whole-exome sequencing (WES), quality control, and variant annotation
Genomic DNA was extracted from blood or saliva samples. Saliva samples were used when blood samples were not available. Altogether, 15 of our 55 cases used DNA extracted from saliva samples. The Agilent SureSelect Human All Exon V6 kit (Agilent Technologies, CA, USA) was used to prepare sequencing libraries from DNA samples and the Illumina Novaseq 6000 platform was used for 150 bp paired-end sequencing of up to 100X by a service provider, Novogene AIT Singapore.
For germline variant calling, Genome Analysis Toolkit (GATK) v4.2.6.0 with the hg38 reference genome was used [45]. Each sample was individually aligned, recalibrated for improved quality scores using GATK’s Base Quality Score Recalibration (BQSR), individually re-assembled to form haplotypes (GVCF files), then jointly-called to obtain genotypes (VCF file). Thereafter, technical artifacts were removed using GATK’s Variant Quality Score Recalibration (VQSR). In addition, all case variants reported in this study were visually checked with Integrative Genomics Viewer (IGV), and variants with low read depths or imbalanced variant allele frequencies were omitted. Case variants were annotated for functional consequence using ANNOVAR [46], REVEL v1.3 using pre-computed scores, [47] CADD v1.6 scores using the CADD offline scoring program [48], SIFT 4G using the pre-computed SIFT 4G annotator [49], PolyPhen-2 using the pre-computed variant call format (VCF) database file [50], and MutationTaster using the MutationTaster2021 webserver [51]. Case variants were lifted-over from hg38 to hg19 using GATK LiftoverVcf (Picard) with UCSC chain files for PolyPhen-2 and MutationTaster annotation as these services only supported hg19.
Identification of ClinVar pathogenic or likely pathogenic variants
The weekly release of the ClinVar database dated 18 February 2023 was used. Relevant cancer conditions or cancer predisposition syndromes were manually selected from the list of all conditions in ClinVar (Additional file 1: Table S11). Variants were defined as pathogenic and likely pathogenic variants when the variants were interpreted as pathogenic or likely pathogenic by an expert panel, practice guidelines, or multiple submitters with named criteria and without conflicting interpretations. Hereafter, only pathogenic and likely pathogenic variants satisfying these levels of evidence in this list of relevant conditions are considered in our analysis as “ClinVar variants”; while other variants such as those evidenced only by single submitters or with conflicting interpretations are ignored.
ACMG/AMP computational and predictive data criteria
Case variants were evaluated for five criteria within the ACMP/AMP group of Computational and Predictive data criteria as described previously [11].
For PVS1, defined as a predicted null variant in a gene where loss of function is a known mechanism of disease, we first selected genes with pathogenic ClinVar variants functionally annotated by ANNOVAR as splice site changes, frameshift indels, start-losses, or stop-gains. Then, we selected case variants with the same functional annotations, also in those selected genes.
For PS1, defined as the same amino acid change as an established pathogenic variant, we selected case variants sharing at least one amino acid change with a pathogenic ClinVar variant: the case variant and ClinVar variants must change the same residue to the same amino acid.
For PM5, defined as a novel missense change at an amino acid residue where a different pathogenic missense change has been seen before, we selected case variants sharing at least one modified residue with a pathogenic ClinVar variant: the case variant and ClinVar variant must change the same residue, but to different amino acids.
For PM4, defined as a protein length changing variant, we selected case variants functionally annotated as non-frameshift indels in non-repeat regions (identified by the UCSC Genome Browser track “simpleRepeats,” last updated 18 October 2022) or as stop-losses, in genes with at least one pathogenic or likely pathogenic ClinVar variant.
For PP3, defined as a variant having multiple lines of computational evidence supporting a deleterious effect on the gene/gene product, we selected case variants with no benign predictions and at least two pathogenic/deleterious predictions from REVEL, CADD, SIFT, PolyPhen-2 (HumDiv-trained Polyphen-2 model or HumVar-trained Polyphen-2 model), and MutationTaster. For REVEL, a genomic variant was considered pathogenic if all its protein changes had REVEL scores greater than 0.68, corresponding to a specificity of 95%); or benign if any of its protein changes had REVEL scores less than or equal to 0.32, for symmetry with the 0.68 pathogenicity threshold. For CADD, a genomic variant was considered pathogenic if it had PHRED-scaled score greater than 20, corresponding to the top 0.1% of variants by pathogenicity.
ACMG/AMP population data criteria
Case variants satisfying any of the five pathogenicity criteria above were further evaluated for three criteria within the ACMP/AMP group of Population Data criteria.
For BS1, defined as a variant having a minor allele frequency (MAF) that is too high for a disorder, we excluded variants with a minor allele frequency greater than 1% in either gnomAD (EAS) or SG10K_Health control cohorts. The gnomAD (EAS) control cohort refers to the 9,626 non-cancer exomes and whole-genomes of the gnomAD v2.1.1 East Asian (EAS) subpopulation [52]. The SG10K_Health control cohort comprises the whole genome sequences of 9,770 healthy Chinese, Indian, and Malay Singaporeans [53].
For PM2, defined as a variant that is absent in population databases, we selected variants not recorded in both gnomAD (EAS) and SG10K_Health control cohorts. This is different and more conservative than selecting variants with zero allele count, as a variant can be recorded with zero allele count in the EAS subpopulation if it is present in any other gnomAD subpopulation. Instead, an unrecorded variant in gnomAD (EAS) represents a variant not found in any gnomAD subpopulation.
For PS4, defined as a variant having a prevalence in affected individuals that is statistically increased over controls, we select variants statistically enriched in cases at p < 0.05, in at least one of six pairwise case–control association analyses between three case cohorts and the two control cohorts gnomAD (EAS) and SG10K_Health; but exclude any variant statistically enriched in controls in any of the six comparisons.
The three case cohorts used are: (1) the 55 patients with dual primary breast and lung cancers, subjected to WES in this study; (2) a cohort of 290 patients with early-onset (≤ 40 years old) or familial breast cancer from Singapore [40]; and (3) a lung cancer case cohort of 209 lung adenocarcinoma patients from Singapore [54].
Case–control association analyses were performed using a two-tailed Fisher’s Exact Test. To enable the statistical test of unrecorded variants in the two control cohorts, allele numbers (the number of alleles that were successfully genotyped at that position) were linearly interpolated from the two closest variants within 150 bp upstream and downstream of the unrecorded variant.
Identification of newly classified pathogenic or likely pathogenic variants
Following the ACMG/AMP guideines [11] case variants were classified as pathogenic if they satisfied pathogenicity criteria PVS1 and PS4, or PS1 and PS4. Case variants were classified as likely pathogenic if they satistified pathogenicity criteria PVS1 and PM2, PS1 and PM2, PS4 and PM4, or PS4 and PM5. Variants which satisfied some pathogenicty criteria but could not be classified as pathogenic or likely pathogenic were classified as variants of uncertain significance. Any variant satisfying benign criterion BS1 was excluded from consideration.
Statistical analysis, software, and additional databases
The Fisher’s Exact Test was performed using the R stats package [55]. The protein domains shown for illustration were obtained from the NCBI Conserved Domain Database, accessed 11 April 2023 [56]. Pathways were obtained using the online “Investigate Human Gene Sets” tool of the Gene Set Enrichment Analysis website, computing overlaps with the Gene Ontology Biological Process database without correction for the universe set of genes and pathways, accessed 12 April 2023 [57].
Availability of data and materials
The gnomAD dataset was analyzed in this study and is accessible at https://gnomad.broadinstitute.org/. Access to the SG10K_Health dataset can be requested from the Singapore National Precision Medicine program. The analysis pipeline used to generate these results is accessible at https://github.com/ning-y/nccs-lcc4.
Change history
14 August 2023
A Correction to this paper has been published: https://doi.org/10.1186/s40246-023-00518-z
Abbreviations
- ACMG/AMP:
-
American College of Medical Genetics and Association for Molecular Pathology
- EAS:
-
East-Asian
- GATK:
-
Genome analysis toolkit
- gnomAD:
-
Genome aggregation database
- IGV:
-
Integrative genomics viewer
- MAF:
-
Minor allele frequency
- MPCs:
-
Multiple primary cancers
- VCF:
-
Variant call format
- VUS:
-
Variants of uncertain significance
- WES:
-
Whole-exome sequencing
- WGS:
-
Whole-genome sequencing
References
Vogt A, Schmid S, Heinimann K, Frick H, Herrmann C, Cerny T, et al. Multiple primary tumours: challenges and approaches, a review. ESMO Open. 2017;2(2): e000172.
Wang R, Yin Z, Liu L, Gao W, Li W, Shu Y, et al. Second primary lung cancer after breast cancer: a population-based study of 6269 women. Front Oncol. 2018;8:427.
Prochazka M, Granath F, Ekbom A, Shields PG, Hall P. Lung cancer risks in women with previous breast cancer. Eur J Cancer. 2002;38(11):1520–5.
Kirova YM, Gambotti L, De Rycke Y, Vilcoq JR, Asselain B, Fourquet A. Risk of second malignancies after adjuvant radiotherapy for breast cancer: a large-scale, single-institution review. Int J Radiat Oncol Biol Phys. 2007;68(2):359–63.
Go RC, King MC, Bailey-Wilson J, Elston RC, Lynch HT. Genetic epidemiology of breast cancer and associated cancers in high-risk families. I. Segregation analysis. J Natl Cancer Inst. 1983;71(3):455–61.
Watson P, Vasen HF, Mecklin JP, Järvinen H, Lynch HT. The risk of endometrial cancer in hereditary nonpolyposis colorectal cancer. Am J Med. 1994;96(6):516–20.
Coco S, Bonfiglio S, Cittaro D, Vanni I, Mora M, Genova C, et al. Integrated somatic and germline whole-exome sequencing analysis in women with lung cancer after a previous breast cancer. Cancers (Basel). 2019;11(4):441.
Whitworth J, Smith PS, Martin J-E, West H, Luchetti A, Rodger F, et al. Comprehensive cancer-predisposition gene testing in an adult multiple primary tumor series shows a broad range of deleterious variants and atypical tumor phenotypes. Am J Hum Genet. 2018;103(1):3–18.
Cavazos TB, Kachuri L, Graff RE, Nierenberg JL, Thai KK, Alexeeff S, et al. Assessment of genetic susceptibility to multiple primary cancers through whole-exome sequencing in two large multi-ancestry studies. BMC Med. 2022;20(1):332.
Hayat MJ, Howlader N, Reichman ME, Edwards BK. Cancer statistics, trends, and multiple primary cancer analyses from the surveillance, epidemiology, and end results (SEER) program. Oncologist. 2007;12(1):20–37.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–23.
Zablotska LB, Neugut AI. Lung carcinoma after radiation therapy in women treated with lumpectomy or mastectomy for primary breast carcinoma. Cancer. 2003;97(6):1404–11.
Mukherjee S, Bandlamudi C, Hellmann MD, Kemel Y, Drill E, Rizvi H, et al. Germline pathogenic variants impact clinicopathology of advanced lung cancer. Cancer Epidemiol Biomark Prev. 2022;31(7):1450–9.
Tung N, Domchek SM, Stadler Z, Nathanson KL, Couch F, Garber JE, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. 2016;13(9):581–8.
Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3(9):1190–6.
Kurian AW, Hughes E, Handorf EA, Gutin A, Allen B, Hartman AR, et al. Breast and ovarian cancer penetrance estimates derived from germline multiple-gene sequencing results in women. JCO Precis Oncol. 2017;1:1–12.
Daly MB, Pilarski R, Yurgelun MB, Berry MP, Buys SS, Dickson P, et al. NCCN guidelines insights: genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 1.2020: featured updates to the NCCN guidelines. J Natl Comp Cancer Netw. 2020;18(4):380–91.
Bonadona V, Bonaïti B, Olschwang S, Grandjouan S, Huiart L, Longy M, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305(22):2304–10.
Balmaña J, Stockwell DH, Steyerberg EW, Stoffel EM, Deffenbaugh AM, Reid JE, et al. Prediction of MLH1 and MSH2 mutations in Lynch syndrome. JAMA. 2006;296(12):1469–78.
Hopkins JL, Lan L, Zou L. DNA repair defects in cancer and therapeutic opportunities. Genes Dev. 2022;36(5–6):278–93.
Girard E, Eon-Marchais S, Olaso R, Renault AL, Damiola F, Dondon MG, et al. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int J Cancer. 2019;144(8):1962–74.
Del Valle J, Rofes P, Moreno-Cabrera JM, López-Dóriga A, Belhadj S, Vargas-Parra G, et al. Exploring the role of mutations in Fanconi anemia genes in hereditary cancer patients. Cancers (Basel). 2020;12(4):829.
Ellingson MS, Hart SN, Kalari KR, Suman V, Schahl KA, Dockter TJ, et al. Exome sequencing reveals frequent deleterious germline variants in cancer susceptibility genes in women with invasive breast cancer undergoing neoadjuvant chemotherapy. Breast Cancer Res Treat. 2015;153(2):435–43.
Bonache S, Esteban I, Moles-Fernández A, Tenés A, Duran-Lozano L, Montalban G, et al. Multigene panel testing beyond BRCA1/2 in breast/ovarian cancer Spanish families and clinical actionability of findings. J Cancer Res Clin Oncol. 2018;144(12):2495–513.
Zins K, Frech B, Taubenschuss E, Schneeberger C, Abraham D, Schreiber M. Association of the rs1346044 polymorphism of the werner syndrome gene RECQL2 with increased risk and premature onset of breast cancer. Int J Mol Sci. 2015;16(12):29643–53.
Oliver J, Quezada Urban R, Franco Cortés CA, Díaz Velásquez CE, Montealegre Paez AL, Pacheco-Orozco RA, et al. Latin American Study of Hereditary Breast and Ovarian Cancer LACAM: a genomic epidemiology approach. Front Oncol. 2019;9:1429.
Gómez-Flores-Ramos L, Barraza-Arellano AL, Mohar A, Trujillo-Martínez M, Grimaldo L, Ortiz-Lopez R, et al. Germline variants in cancer genes from young breast cancer Mexican patients. Cancers (Basel). 2022;14(7):1647.
Ma H, Hu Z, Wang H, Jin G, Wang Y, Sun W, et al. ERCC6/CSB gene polymorphisms and lung cancer risk. Cancer Lett. 2009;273(1):172–6.
Qin N, Wang Z, Liu Q, Song N, Wilson CL, Ehrhardt MJ, et al. Pathogenic germline mutations in DNA repair genes in combination with cancer treatment exposures and risk of subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol. 2020;38(24):2728–40.
Hill W, Lim EL, Weeden CE, Lee C, Augustine M, Chen K, et al. Lung adenocarcinoma promotion by air pollutants. Nature. 2023;616(7955):159–67.
Heddar A, Fermey P, Coutant S, Angot E, Sabourin JC, Michelin P, et al. Familial solitary chondrosarcoma resulting from germline EXT2 mutation. Genes Chromosomes Cancer. 2017;56(2):128–34.
Liu M, Liu X, Suo P, Gong Y, Qu B, Peng X, et al. The contribution of hereditary cancer-related germline mutations to lung cancer susceptibility. Transl Lung Cancer Res. 2020;9(3):646–58.
Xu A, Wang W, Nie J, Lui VW, Hong B, Lin W. Germline mutation and aberrant transcripts of WWOX in a syndrome with multiple primary tumors. J Pathol. 2019;249(1):19–25.
Sokolenko AP, Suspitsin EN, Kuligina E, Bizin IV, Frishman D, Imyanitov EN. Identification of novel hereditary cancer genes by whole exome sequencing. Cancer Lett. 2015;369(2):274–88.
Feurstein S, Drazer MW, Godley LA. Genetic predisposition to leukemia and other hematologic malignancies. Semin Oncol. 2016;43(5):598–608.
Kosaki R, Takenouchi T, Takeda N, Kagami M, Nakabayashi K, Hata K, et al. Somatic CTNNB1 mutation in hepatoblastoma from a patient with Simpson–Golabi–Behmel syndrome and germline GPC3 mutation. Am J Med Genet A. 2014;164(4):993–7.
Grant CN, Rhee D, Tracy ET, Aldrink JH, Baertschiger RM, Lautz TB, et al. Pediatric solid tumors and associated cancer predisposition syndromes: workup, management, and surveillance. A summary from the APSA Cancer Committee. J Pediatr Surg. 2022;57(3):430–42.
Subramanian DN, Zethoven M, McInerny S, Morgan JA, Rowley SM, Lee JEA, et al. Exome sequencing of familial high-grade serous ovarian carcinoma reveals heterogeneity for rare candidate susceptibility genes. Nat Commun. 2020;11(1):1640.
Chiang J, Chia TH, Yuen J, Shaw T, Li ST, Binte Ishak ND, et al. Impact of variant reclassification in cancer predisposition genes on clinical care. JCO Precis Oncol. 2021;5:577–84.
Lee NY, Hum M, Amali AA, Lim WK, Wong M, Myint MK, et al. Whole-exome sequencing of BRCA-negative breast cancer patients and case-control analyses identify variants associated with breast cancer susceptibility. Hum Genom. 2022;16(1):61.
Wong ESY, Shekar S, Met-Domestici M, Chan C, Sze M, Yap YS, et al. Inherited breast cancer predisposition in Asians: multigene panel testing outcomes from Singapore. NPJ Genom Med. 2016;1:15003.
Kurian AW, Ward KC, Abrahamse P, Bondarenko I, Hamilton AS, Deapen D, et al. Time trends in receipt of germline genetic testing and results for women diagnosed with breast cancer or ovarian cancer, 2012–2019. J Clin Oncol. 2021;39(15):1631–40.
Mersch J, Brown N, Pirzadeh-Miller S, Mundt E, Cox HC, Brown K, et al. Prevalence of variant reclassification following hereditary cancer genetic testing. JAMA. 2018;320(12):1266–74.
Turner SA, Rao SK, Morgan RH, Vnencak-Jones CL, Wiesner GL. The impact of variant classification on the clinical management of hereditary cancer syndromes. Genet Med. 2019;21(2):426–30.
Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2018:201178.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16): e164.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877–85.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;76(1):7201–741.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–6.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
PRECISE. SG10K, about SG10K_Health. Available from: https://web.archive.org/web/20210624043126/https://www.npm.sg/collaborate/partners/sg10k/.
Chen J, Yang H, Teo ASM, Amer LB, Sherbaf FG, Tan CQ, et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat Genet. 2020;52(2):177–86.
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available from: https://r-project.org/.
Wang J, Chitsaz F, Derbyshire MK, Gonzales NR, Gwadz M, Lu S, et al. The conserved domain database in 2023. Nucleic Acids Res. 2023;51(D1):D384–8.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Acknowledgements
Patient recruitment, sample collection and clinical data compilation were supported by the Lung Cancer Consortium Singapore (LCCS). We thank authors from the Genome Institute of Singapore and National Cancer Centre Singapore for generating the genomic data of the local lung case cohort; the work was partly supported by National Medical Research Council (NMRC, Singapore) grants (NMRC/OFLCG/002-2018 and NMRC/TCR/007-NCC/2013). We thank the SG10K_Health Investigators for providing the SG10K_Health data generated as part of the Singapore National Precision Medicine program funded by the Industry Alignment Fund (Pre-Positioning) (IAF-PP: H17/01/a0/007). The views expressed by the author(s) are not necessarily those of the National Precision Medicine investigators or institutional partners. We thank all investigators, staff members and study participants who made the National Precision Medicine Project possible. We thank the Broad Institute for generating high-quality sequence data supported by NHGRI funds (grant # U54 HG003067) with Eric Lander as PI. The datasets used in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/gap through dbGaP accession number phs000822. We also thank Memorial Sloan Kettering Cancer Center and Massachusetts General Hospital for the samples collection. The cartoon of the sequencer in Figure 1 was obtained from The Togo Picture Gallery (© 2016 DBCLS TogoTV / CC-BY-4.0).
Funding
This research was funded by a grant from the Industry Alignment Fund – Industry Collaboration Projects funding (grant number IAF-ICP: I1801E0021), and partially supported by the NCCS Cancer Fund, both awarded to Ann Lee. Yoon-Sim Yap is supported by the Singapore National Medical Research Council (NMRC) Clinician Scientist Award (MOH-CSAINV18nov-0009). The funders had no role in study design; in the collection, analysis, and interpretation of data; in writing of the report; and in the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
Conceptualization, ASGL and YSY; methodology, NYL and MKM; validation, NYL, MH and SZ; formal analysis, NYL, MH and MKM; resources, DSWT, GGYL, EHT, DWTL, CKT, SCL, PA, MHT, JS and AS; data curation, LW; writing—original draft preparation, NYL, ASGL, MH and SZ; writing—review and editing, LW, YSY, GGYL, DSWT, ASGL, NYL and MH; visualization, NYL; supervision, ASGL; funding acquisition, ASGL. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all patients and the study was approved by the SingHealth Centralised Institutional Review Board (CIRB Ref: 2018/2147 and 2018/2963).
Consent for publication
Not applicable.
Competing interests
P.A. reports receiving travel support and/or honoraria from AstraZeneca, DKSH, Eisai, Bristol Myers Squibb, Lilly, Novartis, Pfizer, Roche, and MSD, all of which are outside the submitted work. M.-H.T. reports being a director, shareholder, and Chief Executive Officer of Lucence, outside of the submitted work. S.-C.L. reports grant support/research collaborations with Pfizer, Eisai, Taiho, ACT Genomics, Bayer, and MSD; advisory board/speaker invitations from Pfizer, Novartis, Astra Zeneca, ACT Genomics, Eli Lilly, MSD, Roche, Eisai and Daiichi-Sankyo; conference support from Amgen, Pfizer and Roche, all of which are outside the submitted work. No other disclosures were reported.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Table 1 has been updated.
Supplementary Information
Additional file 1. Table S1
: Comparison of breast cancer statuses in EGFR-mutated versus non-EGFR-mutated lung adenocarcinoma patients. Table S2. List of well-evidenced ClinVar null variants in genes with PVS1 variants. Table S3. List of ClinVar variants with the same amino acid change as PS1 variants. Table S4. List of ClinVar variants with the same modified residue as PM5 variants. Table S5. Pathogenicity predictions for various in silico prediction software for PP3 variants. Table S6. Frequency and Fisher's Exact Test results for three case cohorts and two control cohorts, for all pathogenic or likely pathogenic variants from this study. Table S7. Pathways of genes from all pathogenic and likely pathogenic variants in this study. Table S8. List of variants of uncertain significance and their corresponding ACMG/AMP criteria. Table S9. The number of variants of each type detected in each patient with dual primary breast and lung cancer. Table S10. Lack of association between the presence of pathogenic or likely pathogenic variants, and EGFR mutation statuses. Table S11: Cancers and cancer predisposition syndromes in ClinVar and their associated genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lee, NY., Hum, M., Zihara, S. et al. Landscape of germline pathogenic variants in patients with dual primary breast and lung cancer. Hum Genomics 17, 66 (2023). https://doi.org/10.1186/s40246-023-00510-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-023-00510-7