
Abstract
Background
Cuproptosis, as a copper-induced mitochondrial cell death, has attracted extensive attention recently, especially in cancer. Although some key regulatory genes have been identified in cuproptosis, the related lncRNAs have not been further studied. Exploring the prognostic and diagnostic value of cuproptosis-related lncRNAs (CRLs) in colon adenocarcinoma and providing guidance for individualized immunotherapy for patients are of great significance.
Results
A total of 2003 lncRNAs were correlated with cuproptosis genes and considered as CRLs. We screened 33 survival-associated CRLs and established a prognostic signature base on 7 CRLs in the training group. The patients in the low-risk group had better outcomes in both training group (P < 0.001) and test group (P = 0.016). More exciting, our model showed good prognosis prediction in both stage I–II (P = 0.020) and stage III–IV (P = 0.001). The nomogram model could further improve the accuracy of prognosis prediction. Interestingly, glucose-related metabolic pathways, which were closely related to cuproptosis, were enriched in the low-risk group. Meanwhile, the immune infiltration scores were lower in the high-risk group. The high-risk group was more sensitive to OSI.906 and ABT.888, while low-risk group was more sensitive to Sorafenib. Three lncRNAs, FALEC, AC083967.1 and AC010997.4, were highly expressed in serum of COAD patients, and the AUC was 0.772, 0.726 and 0.714, respectively, indicating their valuable diagnostic value.
Conclusions
Our research constructed a prognostic signature based on 7 CRLs and found three promising diagnostic markers for COAD patients. Our results provided a reference to the personalized immunotherapy strategies.
Similar content being viewed by others


Introduction
As one of the most common malignancies, colorectal cancer (CRC) is the second leading cause of cancer-related death [1]. According to the analysis of global cancer statistics in 2020, there were 1.9 million new cases and 935,000 deaths of CRC, accounting for about one-tenth of all malignant tumors [2]. CRC includes colon cancer (CC) and rectal cancer (RC), among which colorectal adenocarcinoma (COAD) is the main type. In addition to surgery, targeted therapy, chemotherapy and immunotherapy are common treatments for COAD, and approximately two-thirds of patients with stage III that receive adjuvant chemotherapy can reduce the risk of recurrence [3]. In recent years, targeted therapy and immunotherapy for COAD have also made great progress [4,5,6,7,8,9]. Therefore, it is particularly important to provide viable biomarkers for prognosis prediction and personalized treatment for COAD patients.
Cuproptosis, a novel cell death type, which is caused by the accumulation of intracellular copper triggering the aggregation of mitochondrial lipoylated proteins and the destabilization of Fe–S cluster proteins [10]. Because mitochondria are the main place of glycolysis, which is very important for the proliferation of cancer cells. Therefore, the malignant potential of tumor cells can be reduced by regulating cuproptosis to inhibit glucose metabolism [11]. Besides, the therapeutic effect of copper and its complex has also been confirmed in cancer [12,13,14], which includes inducing autophagic cell death by targeting ULK1 in colorectal cancer [15], causing immunogenic cell death of breast cancer stem cells [16] and leading to caspase-independent cell death about diffuse large B cell lymphoma [17]. Thus, it may be meaningful to explore the impact of cuproptosis on tumor microenvironment (TME) and cancer therapy.
Long noncoding RNAs (lncRNAs) with transcription length of more than 200 nucleotides are transcribed by RNA polymerase II [18]. Although there is no potential to encode proteins, lncRNAs play vital roles in tumorigenesis and metastasis through gene transcription and post transcriptional modification [19,20,21]. LncRNAs are considered as promising biomarkers for early-stage detection, diagnosis, prognosis and prediction of drug therapy response in cancers [22, 23], such as lung cancer [24], gastric cancer [25], liver cancer [26], breast cancer [27], colorectal cancer [28] and so on. In addition, studies have shown that the extraction of lncRNAs is related to glucose metabolism tumors [29]. However, whether cuproptosis-related lncRNAs (CRLs) play important roles in COAD has not been explored.
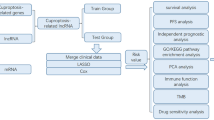
In our study, we analyzed the expression of 18 cuproptosis-related genes (CRGs) to screen the related lncRNAs. The differentially expressed CRLs between COAD tumors and normal tissues were analyzed. All samples were randomly divided into training and test groups at the ratio of 7:3. By univariate Cox regression analysis, 33 survival-associated CRLs were identified in training group. Following, we enrolled 7 CRLs to establish a prognostic model by multivariate Cox regression analysis. The COAD patients were divided into high-risk and low-risk groups according to risk score. The Kaplan–Meier (K–M) survival curves of training and test groups both showed the low-risk group had better outcomes. The 1-, 2-, 3-year ROC curves also verified its accuracy of prognostic prediction. Besides, we constructed a nomogram model based on independent risk factors, including risk score, age and T stage which had more excellent prognosis prediction ability. We also performed functional enrichment analysis of high-risk and low-risk groups by Gene Set Enrichment Analysis (GSEA) software. The correlation of risk score with clinical parameters was also analyzed. Furthermore, the differences of immune cells scores, immune functions scores, immune checkpoints and drug treatment response between two risk groups were explored. Finally, we explored the diagnostic value of CRLs in serum. Our results provided a promising direction for the study of cuproptosis-related lncRNAs in COAD and contributed to the development of personalized immunotherapy for COAD patients.
Materials and methods
Data acquisition and analysis
The transcriptomic data of 473 COAD tumors and 41 normal samples were downloaded from the TCGA database [30]. Then, we separated the expression of 14,056 lncRNAs and 19,573 mRNAs in COAD samples by Strawberry Perl. The clinical information of 421 COAD patients was also obtained from TCGA database after excluding samples with short-term survival (less than 30 days) or missing follow-up days (Table 1). By data merging, 417 COAD patients were finally included in the present analysis. We collected 18 cuproptosis-related mRNAs from previous literature [31,32,33,34,35,36], including FDX1, DLD, PDHA1, PDHB, MTF1, GLS, CDKN2A, DLAT, LIAS, LIPT1, LIPT2, ATP7A, ATP7B, SLC31A1, SLC31A2, DLST, NFE2L2, NLRP3 and extracted the expression of those CRGs from COAD samples accordingly.
Serum of 150 COAD patients and 135 healthy controls was collected. The diagnosis of COAD patients was confirmed by histopathology or biopsy and recruited from the Department of General Surgery, Qilu Hospital of Shandong University, from April 2018 to October 2020. The healthy controls were enrolled from the Department of Physical Examination Center, Qilu Hospital of Shandong University. Serum samples were separated by centrifugation at 6000 g for 10 min followed by another centrifugation at 12,000 g for 10 min and then stored at − 80 °C for further analysis.
Identification of differentially expressed cuproptosis-related LncRNAs
After the Pearson correlation algorithm with the filter of |coefficient|> 0.3 and P < 0.001, we selected lncRNAs that were related with cuproptosis genes and considered as CRLs. Next, we identified differentially expressed lncRNAs (|Log2 fold change (FC)|> 1, false discovery rate (FDR) < 0.05) in COAD tumor tissues comparing with normal tissue using differential analysis by R package “limma” [37].
Establishment and evaluation of CRLs prognostic signature
After the COAD patients were randomly divided into training and test groups, we first performed univariate Cox analysis (P < 0.01) to screen CRLs associated with prognosis. Next, we established a prognostic signature by multivariate Cox regression analysis in the training group. Hence, the risk score of each COAD patient could be calculated according to the coefficient and CRLs expression in our prognostic signature. And the COAD patients were divided into the high-risk and low-risk groups by the median value of the risk score.
We used the Kaplan–Meier (K–M) and receiver operating characteristic (ROC) curves to evaluate the value of the prognostic signature in both training and test groups by R packages “survival,” “survminer” and “timeROC” [38]. Besides, the relationship between the risk score and prognosis of COAD patients was also displayed by heatmap jointly, risk score curve, and survival status diagram. Moreover, whether the risk score was related with clinical parameters was also examined.
Independent prognostic analysis and development of nomogram model
The univariate and multivariate Cox regression analyses were performed to identify the independent risk factors of COAD, including risk score and clinical characteristics. Subsequently, the nomogram model was constructed based on independent risk factors using the R package “rms.” Then, we used calibration curves to estimate the prediction power of the model.
Gene enrichment analysis by GSEA
To identify pathway enrichment in two risk groups, we used GSEA software (4.2.2) to perform the enrichment analysis of the Kyoto Encyclopedia of Genes and Genomes (KEGG) (c2.cp.kegg.v7.5.1.symbols.gmt) [39]. The random sample permutation number was set as 1,000, and the significance threshold was P < 0.05.
Immune infiltration analysis by single sample gene set enrichment analysis (ssGSEA)
The enrichment score of infiltration estimation and immune function of different immune cells between two risk groups was compared using ssGSEA analysis [40] by R packages “GSVA,” “GSEABase” and “Limma.” So, we could easily explore the association between risk score, immune infiltration and immune function. And the significance threshold was FDR < 0.05.
The value of risk score in predicting response of patients to immunotherapy and chemotherapy
We also analyzed the differential expression of 47 immune checkpoint genes, including IDO1, LAG3, CTLA4, TNFRSF9, ICOS, CD80, PDCD1LG2, TIGIT, CD70, TNFSF9, ICOSLG, KIR3DL1, CD86, PDCD1 (PD1), LAIR1, TNFRSF8, TNFSF15, TNFRSF14, IDO2, CD276, CD40, TNFRSF4, TNFSF14, HHLA2, CD244, CD274 (PD-L1), HAVCR2, CD27, BTLA, LGALS9, TMIGD2, CD28, CD48, TNFRSF25, CD40LG, ADORA2A, VTCN1, CD160, CD44, TNFSF18, TNFRSF18, BTNL2, C10orf54, CD200R1, TNFSF4, CD200, NRP1 between two risk groups. Besides, we collected 15 commonly used drugs for the clinical treatment of gastrointestinal tumors, including Epothilone B, Sorafenib, Cisplatin, Doxorubicin, Etoposide, Imatinib, Lapatinib, OSI.906, PHA.665752, ABT.888, Camptothecin, Docetaxel, Mitomycin C, Paclitaxel, and Sunitinib. The half-maximal inhibitory concentration (IC50) of drugs was used to evaluate the therapy response of patients in two risk groups by R package “pRRophetic.” The significance threshold of all the above analyses results was P < 0.05 except for the multiple hypothesis test which used FDR to adjust.
RNA extraction and RT-qPCR
The total RNA was extracted from serum samples using TRIzol LS Reagent (Invitrogen, Eugene, OR, USA). The concentration of RNA was measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). RNA was reverse transcription into cDNA using SureScript RTase Mix and RT Reaction Buffer, and qPCR was performed using Blaze Taq qPCR Mix (GeneCopoeia, Guangzhou, China). The relative expression of target lncRNAs was normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and calculated 2 − ΔΔCt. The primer sequences are shown in Additional file 1: Table S1.
Results
Identification of CRLs and differentially expressed CRLs in COAD
A total of 2003 lncRNAs were identified as CRLs according to the filters mentioned in the method section (Additional file 2: Table S2). The correlationship between CRGs and lncRNAs in COAD is shown in Additional file 3: Fig. S1. Then, the differential expression of CRLs between 473 COAD tumor and 41 normal tissues was compared and 1042 differentially expressed CRLs were obtained as shown in volcano plot (Fig. 1A, Additional file 4: Table S3). The top 100 CRLs were displayed by heatmap which could successfully separate tumor and non-tumor tissues (Fig. 1B).

CRLs in COAD patients. A The volcano plot of CRLs, in which the red dots indicated upregulated lncRNAs in COAD tumors, the green dots indicated down-regulated lncRNAs, and black dots indicated lncRNAs that were no significant difference. B The heatmap of top 100 differentially expressed CRLs. C The Sankey diagram displayed the regulation of CRGs and CRLs
Establishment of the prognostic signature based on CRLs
We obtained 33 prognosis-related CRLs as shown in Additional file 5: Table S4. The Sankey diagram of CRGs and CRLs was plotted to display their correlationship in Fig. 1C. Then, we selected 7 CRLs to construct a prognostic signature by multivariate Cox regression analysis. The risk score was derived as follows: risk score = (1.817 × AC010997.4) + (0.727 × AP003555.1) + (1.03 × FALEC) + (0.908 × AC083967.1) + (0.822 × AC005841.1) + (1.366 × AP001619.1)—(0.767 × ZKSCAN2-DT). The patients were divided into high-risk and low-risk groups based on the median risk score. K–M curve showed that patients in the low-risk group had better outcomes than those in the high-risk group in both training and test groups (P < 0.05, Fig. 2A, B). Figure 2C–H illustrates the heatmap of the prognostic signature, distribution of risk score and survival status diagrams, respectively. The 1-, 2-, and 3-year AUC of the ROC curves in the training group was 0.73, 0.737, and 0.761, and that in the test group was 0.707, 0.775, and 0.766, respectively (Fig. 3A, B). The above results implied that the risk score performed well in predicting the prognosis of COAD patients. Besides, we also noticed that there was significant difference in both stage I–II (P = 0.002) and stage III–IV (P = 0.001) (Fig. 3C, D). So, our prognostic model was effective in predicting outcomes not only for advanced patients, but also for early patients in COAD.

Establishment of prognostic signature based on CRLs in COAD. K–M curve for OS in training group (A) and test group (B), heatmap of 7 CRLs enrolled in the prognostic signature (C, D), the risk score curve (E, F) and survival status diagrams (G, H) of COAD patients in training group and test group

Evaluation of prognostic signature. The 1-, 2-, and 3-year ROC curves of prognostic signature in the training (A) and test (B) groups. K–M curve for OS of two risk groups in stage I–II patients (C) and stage III–IV patients (D)
The correlation between the risk score and clinical characteristics was analyzed. Our results demonstrated that the risk score was associated with survival status (P < 0.001), clinical stages (P = 0.042), T stage (P = 0.002) and N stage (P = 0.011) (Fig. 4C–F).

The relationship between the risk score and clinical parameters
Independent prognostic analysis and construction of a nomogram model
By univariate and multivariate Cox regression analyses, we evaluated the independent predictors of COAD. The results showed that age (P = 0.045), clinical stage (P < 0.001), T stage (P < 0.001), N stage (P < 0.001), M stage (P < 0.001) and risk score (P < 0.001) were prognostic indicators (Fig. 5A). Furthermore, we found that only age (P = 0.009), T stage (P = 0.002) and risk score (P = 0.004) were independent prognostic indicators (Fig. 5B). Then, a nomogram model was constructed based on independent prognostic indicators which had excellent prognosis prediction ability (Fig. 5C). The accuracy of the nomogram model was estimated by the calibration plots which showed good consistency with the actual observation (Fig. 5D).

The nomogram model based on independent prognostic factors. Independent prognostic analysis by univariate Cox (A) and multivariate Cox regression (B). The nomogram model that integrated the risk score, age and T stage predicted the probability of the 1-, 2-, and 3-year OS (C) and the calibration plots (D)
KEGG pathways enrichment analysis by GSEA
The results showed that enriched KEGG pathways had significant difference between high-risk group (Additional file 6: Table S5) and low-risk group (Additional file 7: Table S6). Interestingly, pathways of citrate cycle TCA cycle, glycolysis gluconeogenesis, oxidative phosphorylation, glutathione metabolism and so on were enriched in the low-risk group which might be the reason of different survival outcomes of two groups (Fig. 6).

The KEGG signal pathway enriched analysis of high- and low-risk groups by GSEA
The difference of immune infiltration between two risk groups
The immune scores of immune cells (aDCs, B cells, iDCs, Mast cells, pDCs, and so on.) and immune functions (APC co-stimulation, CCR, checkpoint, cytolytic activity, parainflammation, and so on) were higher in the low-risk group (Fig. 7A, B). The results showed that the risk score was negatively correlated with the infiltration of most anti-tumor immune cells.

The investigation of tumor immune factors and drug sensitivity between two risk groups. The difference of immune cells (A), immune functions scores (B) and expression of immune checkpoints genes (C) between high- and low-risk groups. The drug sensitivity of two risk groups to OSI.906 (D), ABT.888 (E), Sorafenib (F). FDR: 0.05 > * > 0.01 > ** > 0.001 > ***
The relationship of risk score with immune checkpoints expression and drug treatment response
The expression of 4 immune checkpoints (HHLA2, CD160, TNFRSF14 and TNFRSF25) was different between two risk groups (Fig. 7C). Among them, the expression of HHLA2 was higher in the low-risk group. However, the expression of CD160, TNFRSF14 and TNFRSF25 was on the contrary. In the term of IC50 of 15 common drugs for gastrointestinal cancer, patients in the high-risk group were more sensitive to OSI.906, ABT.888 (Fig. 7D, E), while patients in the low-risk group were more sensitive to Sorafenib (Fig. 7F). The 95% CI of drug sensitivity analysis is shown in Additional file 8: Table S7.
Differential expression of serum CRLs between COAD patients and healthy controls
To further explore the possible diagnostic values of CRLs, the expression of above 7 CRLs in serum between COAD patients and healthy controls was analyzed. Our results showed that three lncRNAs including FALEC, AC083967.1 and AC010997.4 were significantly increased in serum of COAD patients (Fig. 8A–C) which were consistent with RNA sequencing data of tissues. The AUC of FALEC, AC083967.1 and AC010997.4 was 0.772, 0.726 and 0.714, respectively, which indicated that they might be used as diagnostic markers (Fig. 8D–F). However, the other 4 CRLs were really low expressed in serum of both COAD patients and healthy controls which were hardly to accurately detected by RT-qPCR.

The expression of FALEC (A), AC083967.1 (B) and AC010997.4 (C) in serum of COAD patients and healthy controls by RT-qPCR. The ROC curves of FALEC (D), AC083967.1 (E) and AC010997.4 (F) to diagnosis COAD patients
Discussion
An appropriate amount of copper in cells is essential for life. It will impact the function of important metal binding enzymes if the content is too little, while too much of copper may lead to cell death [41]. Cuproptosis is a form of cell death caused by excessive copper that induces the aggregation of lipoylated dihydrolipoamide S-acetyltransferase (DLAT) and results in protein toxic stress [31, 42]. As critical regulators of gene expression, lncRNAs have the ability to affects the occurrence and development of cancers through multiple mechanisms including regulating gene–environment interaction of diseases [43, 44]. Studies have shown that epigallocatechin-3-gallate (EGCG) can regulate SLC31A1 (CTR1, copper transporter 1) expression through upregulated lncRNA nuclear, which effects cisplatin sensitivity for the treatment of non-small cell lung cancer cells [45]. Therefore, it is not surprising to speculate that related lncRNAs can regulate the process of cuproptosis to affect prognosis and treatment of COAD patients.
In the present study, we select 7 CRLs (AC010997.4, AP003555.1, FALEC, AC083967.1, AC005841.1, AP001619.1 and ZKSCAN2-DT) to establish a prognostic signature. Then, the K–M and ROC curves confirmed that the model had good predictive value for the prognosis of COAD patients. In fact, many lncRNAs in the signature have been proved to play the role in the prognosis of cancers. For example, FALEC can promote colorectal cancer progression via regulating miR-2116-3p-targeted PIWIL1 [46]. AC083967.1, AP001619.1 and AP003555.1 can be used as the prognostic marker for colorectal cancer [47,48,49,50]. The above studies are consistent with our results. Meanwhile, we also found that our prognostic signature was equally effective for stage I-II and stage III-IV of patients, which avoided the poor prediction of the prognosis for early COAD patients and increased the practicability of the signature. Besides, the risk score, age and T stage were identified as independent prognostic factors. So, we also built a nomogram model to comprehensively predict 1-year, 2-year, and 3-year OS of COAD patients based on independent factors. This is of great significance to evaluate the condition of COAD patients more accurately by combining risk score and clinical characteristics.
Tumor microenvironment (TME) that tumor cells depend on for growth and survival plays an important role in tumor progression. With the further understanding of the diversity and complexity of TME, its role in tumor progression, immune escape and immunotherapy response has been paid more and more attention [51]. By GSEA and ssGSEA analyses, we found that patients in the high-risk group had a significant low degree of immune infiltration. We believed that these patients had obvious immunosuppression and belonged to immune-desert phenotype which was characterized by immune tolerance, immune ignorance and associated with poor clinical outcomes [52]. Interestingly, patients in the low-risk group were enriched in glucose-related metabolic pathways, which were closely related to cuproptosis and had high level of immune cell infiltration and immune function. Therefore, we speculated that these patients were immune-inflamed phenotype which was characterized by adaptive immune cell infiltration and immune activation [53,54,55]. Also, cuproptosis might inhibit the progression of COAD partly, which requires further research to explore the potential mechanism.
Immunotherapy represented by immune checkpoint blockade (ICB) has shown amazing clinical efficacy and durable responses. But it is only for a small number of cancer patients, and most patients have little benefit, far from a met clinical need [56]. Analyzing the expression of immune checkpoint genes in COAD patients will help to improve the effect of immunotherapy. We found that the expression of CD160, TNFRSF14, TNFRSF25 was higher in high-risk group, yet the expression of HHLA2 was higher in low-risk group. Therefore, ICBs for the above genes could implement more accurate treatment for different patients. Besides, we also collected 15 commonly used drugs for gastrointestinal tumors and analyzed the sensitivity differences between two risk groups. We found that patients in the high-risk group were more sensitive to OSI.906 and ABT.888, while patients in the low-risk group were more sensitive to Sorafenib. These results could provide further guidance for the personalized drug treatment of patients with COAD based on our signature.
To further explore whether the above 7 CRLs also have diagnostic value, we detected the expression of CRLs in serum by RT-qPCR. Compared with healthy controls, FALEC, AC083967.1 and AC010997.4 were highly expressed in serum of COAD patients and ROC curves also verified their good diagnostic value. Interestingly, the diagnostic value of plasma FALEC in cervical cancer has been reported and considered as a promising diagnostic marker [57].
Conclusions
Overall, although our prognostic signature performed well in predicting the diagnosis, prognosis and treatment responses of COAD, our current study inevitably had some limitations. On the one hand, it is hard for our team to follow-up large amount of COAD patients to acquire survival information and verify the signature. On the other hand, the research on cuproptosis is still in the early stage at present, and the fewer CRGs limit the scope of research. Our results provided new insights into understanding the relationship of COAD tumorigenesis mechanism and cuproptosis, and further molecular and clinical trials are still needed to confirm our findings. We believe that with the further research on cuproptosis in the future, it will have more comprehensive and in-depth understanding to improve relevant research.
Availability of data and materials
Statistical analyses were performed using the R 4.1.2 software, Strawberry Perl (64-bit) 5.30.0.1, GSEA 4.2.2. The Wilcox test was used to compare difference between COAD tumors and normal tissues, serum expression of CRLs between COAD patients and healthy controls and differences of immune cells, immune functions between two risk groups. Chi-squared test was used to perform K–M survival analysis of the prognostic model. Differences were considered statistically significant when P value or FDR was < 0.05. All R scripts and data generated or analyzed during this study are available from the corresponding author on reasonable request.
Abbreviations
- CRGs:
-
Cuproptosis-related genes
- CRLs:
-
Cuproptosis-related lncRNAs
- lncRNAs:
-
Long noncoding RNAs
- CRC:
-
Colorectal cancer
- CC:
-
Colon cancer
- RC:
-
Rectal cancer
- COAD:
-
Colon adenocarcinoma
- TCGA:
-
The Cancer Genome Atlas
References
Siegel RL, Miller KD, Goding Sauer A, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(3):145–64.
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69(5):363–85.
Sakata S, Larson DW. Targeted therapy for colorectal cancer. Surg Oncol Clin N Am. 2022;31(2):255–64.
Boukouris AE, Theochari M, Stefanou D, et al. Latest evidence on immune checkpoint inhibitors in metastatic colorectal cancer: a 2022 update. Crit Rev Oncol Hematol. 2022;173:103663.
Zhang X, Yang Z, An Y, et al. Clinical benefits of PD-1/PD-L1 inhibitors in patients with metastatic colorectal cancer: a systematic review and meta-analysis. World J Surg Oncol. 2022;20(1):93.
Liu Y, Zhang H, Cui H, et al. Combined and targeted drugs delivery system for colorectal cancer treatment: conatumumab decorated, reactive oxygen species sensitive irinotecan prodrug and quercetin co-loaded nanostructured lipid carriers. Drug Deliv. 2022;29(1):342–50.
Liu XZ, Xiong Z, Xiao BY, et al. Multicenter real-world study on safety and efficacy of neoadjuvant therapy in combination with immunotherapy for colorectal cancer. Zhonghua wei chang wai ke za zhi = Chin J Gastrointest Surg. 2022;25(3):219–27.
Ding PR. Immunotherapy for microsatellite-instability-high advanced colorectal cancer. Zhonghua wei chang wai ke za zhi = Chin J Gastrointest Surg. 2022;25(3):199–204.
Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32:417–8.
Ge EJ, Bush AI, Casini A, et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. 2022;22(2):102–13.
da Silva DA, De Luca A, Squitti R, et al. Copper in tumors and the use of copper-based compounds in cancer treatment. J Inorg Biochem. 2022;226: 111634.
Passeri G, Northcote-Smith J, Suntharalingam K. Delivery of an immunogenic cell death-inducing copper complex to cancer stem cells using polymeric nanoparticles. RSC Adv. 2022;12(9):5290–9.
Jiang Y, Huo Z, Qi X, Zuo T, Wu Z. Copper-induced tumor cell death mechanisms and antitumor theragnostic applications of copper complexes. Nanomedicine. 2022;17(5):303–24.
Hu Y, Qian Y, Wei J, et al. The disulfiram/copper complex induces autophagic cell death in colorectal cancer by targeting ULK1. Front Pharmacol. 2021;12: 752825.
Kaur P, Johnson A, Northcote-Smith J, Lu C, Suntharalingam K. Immunogenic cell death of breast cancer stem cells induced by an endoplasmic reticulum-targeting copper(ii) complex. ChemBioChem. 2020;21(24):3618–24.
Lee K, Hart MR, Briehl MM, Mazar AP, Tome ME. The copper chelator ATN-224 induces caspase-independent cell death in diffuse large B cell lymphoma. Int J Oncol. 2014;45(1):439–47.
Wang DQ, Fu P, Yao C, et al. Long non-coding RNAs, novel culprits, or bodyguards in neurodegenerative diseases. Mol Ther Nucleic Acids. 2018;10:269–76.
Liu SJ, Dang HX, Lim DA, Feng FY, Maher CA. Long noncoding RNAs in cancer metastasis. Nat Rev Cancer. 2021;21(7):446–60.
Gao J, Wang F, Wu P, Chen Y, Jia Y. Aberrant LncRNA expression in leukemia. J Cancer. 2020;11(14):4284–96.
Du XH, Wei H, Qu GX, Tian ZC, Yao WT, Cai QQ. Gene expression regulations by long noncoding RNAs and their roles in cancer. Pathol Res Pract. 2020;216(6): 152983.
Beylerli O, Gareev I, Sufianov A, Ilyasova T, Guang Y. Long noncoding RNAs as promising biomarkers in cancer. Noncoding RNA Res. 2022;7(2):66–70.
Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77(15):3965–81.
Zhao T, Khadka VS, Deng Y. Identification of lncRNA biomarkers for lung cancer through integrative cross-platform data analyses. Aging. 2020;12(14):14506–27.
Lei L, Li N, Yuan P, Liu D. A new risk model based on a 11-m(6)A-related lncRNA signature for predicting prognosis and monitoring immunotherapy for gastric cancer. BMC Cancer. 2022;22(1):365.
Chen Y, Xi L, Wei L, Sun D, Zeng T. Immune-related lncRNA signature delineates an immune-excluded subtype of liver cancer with unfavorable clinical outcomes. J Clin Lab Anal. 2022;36(3): e24244.
Chen F, Yang J, Fang M, Wu Y, Su D, Sheng Y. Necroptosis-related lncRNA to establish novel prognostic signature and predict the immunotherapy response in breast cancer. J Clin Lab Anal. 2022;36(4): e24302.
Zhou Z, Wang W, Deng J, et al. A long noncoding RNA, LncRNA-LOC100127888, is associated with poor prognosis in colorectal cancer patients. Bull Cancer. 2022;109(3):258–67.
Balihodzic A, Barth DA, Prinz F, Pichler M. Involvement of long non-coding RNAs in glucose metabolism in cancer. Cancers. 2021;13(5):977.
Weinstein JN, Collisson EA, Mills GB, et al. The Cancer Genome Atlas pan-cancer analysis project. Nat Genet. 2013;45(10):1113–20.
Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–61.
Cronan JE. Progress in the enzymology of the mitochondrial diseases of lipoic acid requiring enzymes. Front Genet. 2020;11:510.
Lutsenko S. Dynamic and cell-specific transport networks for intracellular copper ions. J Cell Sci. 2021;134(21):jcs240523.
Shen N, Korm S, Karantanos T, et al. DLST-dependence dictates metabolic heterogeneity in TCA-cycle usage among triple-negative breast cancer. Commun Biol. 2021;4(1):1289.
Li Q, Liao J, Zhang K, et al. Toxicological mechanism of large amount of copper supplementation: Effects on endoplasmic reticulum stress and mitochondria-mediated apoptosis by Nrf2/HO-1 pathway-induced oxidative stress in the porcine myocardium. J Inorg Biochem. 2022;230: 111750.
Deigendesch N, Zychlinsky A, Meissner F. Copper regulates the canonical NLRP3 inflammasome. J Immunol. 2018;200(5):1607–17.
Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Blanche P, Dartigues JF, Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med. 2013;32(30):5381–97.
Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–12.
Kahlson MA, Dixon SJ. Copper-induced cell death. Science. 2022;375(6586):1231–2.
Wang Y, Zhang L, Zhou F. Cuproptosis: a new form of programmed cell death. Cell Mol Immunol. 2022;4.
Karlsson O, Baccarelli AA. Environmental health and long non-coding RNAs. Curr Environ Health Rep. 2016;3(3):178–87.
Ghafouri-Fard S, Hussen BM, Gharebaghi A, Eghtedarian R, Taheri M. LncRNA signature in colorectal cancer. Pathol Res Pract. 2021;222: 153432.
Chen A, Jiang P, Zeb F, et al. EGCG regulates CTR1 expression through its pro-oxidative property in non-small-cell lung cancer cells. J Cell Physiol. 2020;235(11):7970–81.
Jiang H, Liu H, Jiang B. Long non-coding RNA FALEC promotes colorectal cancer progression via regulating miR-2116-3p-targeted PIWIL1. Cancer Biol Ther. 2020;21(11):1025–32.
Xu S, Zhou Y, Luo J, et al. Integrated analysis of a ferroptosis-related LncRNA signature for evaluating the prognosis of patients with colorectal cancer. Genes. 2022;13(6):1094.
Liang Y, Sun HX, Ma B, Meng QK. Identification of a genomic instability-related long noncoding RNA prognostic model in colorectal cancer based on bioinformatic analysis. Dis Mark. 2022;2022:4556585.
Huang QR, Pan XB. Prognostic lncRNAs, miRNAs, and mRNAs form a competing endogenous RNA network in colon cancer. Front Oncol. 2019;9:712.
Li R, Gao X, Sun H, Sun L, Hu X. Expression characteristics of long non-coding RNA in colon adenocarcinoma and its potential value for judging the survival and prognosis of patients: bioinformatics analysis based on The Cancer Genome Atlas database. J Gastrointest Oncol. 2022;13(3):1178–87.
Zhang B, Wu Q, Li B, Wang D, Wang L, Zhou YL. m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol Cancer. 2020;19(1):53.
Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27(8):1492–504.
Gajewski TF. The next hurdle in cancer immunotherapy: overcoming the non-T-cell-inflamed tumor microenvironment. Semin Oncol. 2015;42(4):663–71.
Joyce JA, Fearon DT. T cell exclusion, immune privilege and the tumor microenvironment. Science. 2015;348(6230):74–80.
Salmon H, Franciszkiewicz K, Damotte D, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Naizhaer G, Kuerban A, Kuerban R, Zhou P. Up-regulation of lncRNA FALEC indicates prognosis and diagnosis values in cervical cancer. Pathol Res Pract. 2019;215(8):152495.
Acknowledgements
Not applicable for that section.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82172347) and the Major Scientific and Technological Innovation Project of Shandong Province (2021CXGC010603).
Author information
Authors and Affiliations
Contributions
SCL and YJL analyzed most of the data, interpreted the results and wrote the manuscript. SCZ and XRY downloaded the TCGA data and analyzed part of the data. GXZ conceived the concept and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Qilu Hospital of Shandong University, and written informed consent was obtained from each subject (Approval Number: KYLL-2021(KS)-114). Written informed consent was obtained from every participant for the use of venous blood sample. All research methods were implemented following relevant guidelines and regulations.
Consent for publication
Not applicable for that section.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The primer sequences of CRLs
Additional file 2: Table S2.
The cuproptosis-related lncRNAs
Additional file 3: Fig. S1.
The network between CRGs and CRLs
Additional file 4: Table 3.
The differential expressed CRLs between COAD tumor and normal tissues
Additional file 5: Table 4.
The 33 prognosis-related CRLs about COAD
Additional file 6: Table 5.
The results of GSEA analysis in high-risk group
Additional file 7: Table 6.
The results of GSEA analysis in low-risk group
Additional file 8: Table 7.
The 95 % CI of drug sensitivity analyzed by Wilcox test
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, S., Zhang, S., Liu, Y. et al. Comprehensive analysis of cuproptosis-related long noncoding RNA for predicting prognostic and diagnostic value and immune landscape in colorectal adenocarcinoma. Hum Genomics 17, 22 (2023). https://doi.org/10.1186/s40246-023-00469-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-023-00469-5