
Abstract
Background
Complex developmental encephalopathy syndromes might be the consequence of unknown genetic alterations that are likely to contribute to the full neurological phenotype as a consequence of pathogenic gene combinations.
Methods
To identify the additional genetic contribution to the neurological phenotype, we studied as a test case a boy, with a KCNQ2 exon-7 partial duplication, by single-nucleotide polymorphism (SNP) microarray to detect copy-number variations (CNVs).
Results
The proband presented a cerebral palsy like syndrome with a severe motor and developmental encephalopathy. The SNP array analysis detected in the proband several de novo CNVs, nine partial gene losses (LRRC55, PCDH9, NALCN, RYR3, ELAVL2, CDH13, ATP1A2, SLC17A5, ANO3), and two partial gene duplications (PCDH19, EFNA5). The biological functions of these genes are associated with ion channels such as calcium, chloride, sodium, and potassium with several membrane proteins implicated in neural cell-cell interactions, synaptic transmission, and axon guidance. Pathogenically, these functions can be associated to cerebral palsy, seizures, dystonia, epileptic crisis, and motor neuron dysfunction, all present in the patient.
Conclusions
Severe motor and developmental encephalopathy syndromes of unknown origin can be the result of a phenotypic convergence by combination of several genetic alterations in genes whose physiological function contributes to the neurological pathogenic mechanism.
Similar content being viewed by others

Background
Children with severe non-hereditary neurodevelopmental delay, and neonatal or early onset of epileptic seizures, recently named as developmental and epileptic encephalopathy [1, 2], frequently have an unknown etiology, which are likely to be very heterogeneous. In this context, genetic alterations can play a relevant pathogenic role. Genomic studies based on whole-exome sequencing (WES) and single-nucleotide polymorphism (SNP) microarray to detect CNVs can identify genetic alterations contributing to the pathogenic mechanism of these complex clinical phenotypes [3]. These underlying genetic alterations may be of different types, and the better known are those associated to dominant point mutations. However, the genetic heterogeneity of complex neurological diseases such as cerebral palsy [4], epilepsy [5], and autism [6, 7] has already been associated to a combination of several different pathogenic gene variants, and to several CNVs in affected individuals, and the combination of CNV changes contributes to the individual variome [8, 9]. CNVs are structural gene alterations deleting or duplicating gene segments. Genomic SNP microarray studies have detected multiple CNV changes, deletions, and duplications, which are consistent with the heterogeneity observed in epileptic patients [10], but no common pattern was identified. The presence of a CNV in a gene can either alter its level of expression, the stability of the RNA, the structure of the protein, or its subcellular localization. Any of these effects will alter their function, which may be even more relevant in proteins associated to the membranes where they can play a role in specific neural functions, ion and neurotransmitter transport, neural cell interactions, or signal transmission.
Neonatal onset epilepsies related to KCNQ2 mutations share a loss of the potassium channel function. However, a common loss of function can cause very heterogeneous neurological phenotypes that accompany the epileptic phenotype [11, 12]. Therefore, it is likely that there are additional unidentified genetic changes that can contribute to the heterogeneity and complexity of the individual neurological phenotype [5, 10, 12]. An alternative approach to detect candidate pathogenic genes is to search for specific CNV alterations in a proband with respect to the family members. Moreover, the analysis of CNVs in these patients has detected a variable number of additional genetic changes, but did not identify a specific or unique pattern of specific gene alterations common to different patients [13]. In neurons, where ion channels and specific cellular interactions are critical for its development and correct functions, CNVs can alter protein level, and their subcellular localization or density. These CNVs can play a pathogenic role if associated to the neurological functions that are altered in the clinical phenotype. The identification of underlying genetic alterations in neuromotor developmental disorders is proving very useful in a significant number of neurological phenotypes [14,15,16].
Human normal diversity is a consequence of different combinations of non-pathogenic gene variants. Therefore, it is likely that heterogeneity in neurological phenotypes can also be a consequence of alternative combinations of genetic changes in genes associated to cellular functions that are involved in the phenotype and constitute the individual variome [8, 9]. Therefore, we tested this hypothesis by searching for CNV variome differences among the four family members of a complex case of a child, as test case, that presented a developmental and epileptic encephalopathy of neonatal onset that has a novel KCNQ2 mutation resulting from a partial KCNQ2 exon 7 duplication that impairs its inhibitory signal [17] and to determine if the proband has a CNV pattern associated to neuronal functions. However, the complexity of the neurological syndrome suggested that, although this mutation is necessary, there has to be additional cooperating genetic alterations. We have approached the identification of the underlying genetic problem by studying CNV alterations and their association to genes whose function can contribute to the neuropathological phenotype reported in the proband and absent in the family [17], as an alternative approach to searching for common variants among unrelated individuals with similar phenotypes. Complex neurodevelopmental delay and epileptic phenotypes might be the result of de novo combination of CNV alterations in genes associated to neurological functions that contribute to the patient syndrome and determine its pathogenic mechanism.
Materials and methods
Standard protocol approvals, registrations, and patient consents
The genomic studies were performed for diagnosis of a pediatric neurological syndrome of unknown etiology. For the diagnostic genomic studies, total DNA was obtained from peripheral blood samples. Written informed consent for the genomic study was obtained from both parents. Research protocol and consent forms were approved by the Institutional Review Boards of Hospital Universitario de Salamanca-Centro de Investigación del Cancer as reported in a previous publication [17].
SNP microarray
The SNP microarray analysis of genomic alteration was performed using the matrix chip CytoScan HD array (Affymetrix; Thermo Fisher Scientific, Inc.) following manufacturer’s instructions. The matrix contains 2.696.550 probes that include 743,304 SNPs and 1,953,246 non-polymorphic probes. The mean spacing between probes for RefSeq genes is 800 bp, and 96% of the genes are represented. Briefly, 500 ng of DNA was digested with Nsp1 for 2 h at 37 °C. Digested DNA was purified and ligated to primers/adaptors at 16 °C for 15 h. The products of this ligation were used to generate amplicons by PCR using the primers provided by the manufacturer (Affymetrix). The PCR program was one cycle at 94 °C for 1 min, thirty-five cycles (94 °C for 30 s, 60 °C for 45 s, and 65 °C for 1 min) and a final extension cycle at 68ªC for 7 min. Purified PCR products were digested with DNase I for 35 min at 37ªC, and the fragmented DNA was labeled with biotinylated nucleotides with deoxynucleotide terminal transferase for 4 h at 37ªC. A 250 μg of fragmented DNA was hybridized to the Chip Affymetrix chip Cytoscan HD preequilibrated at 50 °C for 18 h. The matrixes were washed and stained in the GeneChip Fluidics Station 450 (Affymetrix Inc.), and DAT images were acquired with the GeneChip Scanner 3000 7G (Affymetrix Inc.). The data files (archives.cel) were generated with the Affymetrix GeneChip Command Console Software (AGCC) software (Affimetrix Inc., Santa Clara, CA). Data analysis was performed using the analysis program package from Affymetrix Chromosome Analysis Suite (CHAS 4.0). The CGH results were compared with data in ChAs 3.3 NetAffix Build 33.2(Hg19) as reference. In addition, the aCGH results were compared among the four members of the family to confirm their presence in the proband, but not in the parents and sister. Furthermore, the exome assay segments were determined through the Control-Freec program [18], using as reference either the father, mother, and daughter exomes. An example that that shows the overlapping alterations (nCN-LOH, gains, and losses) between the aCGH microarray and WES exome assays for the patient with respect to the other three family members is represented in Supplementary Figure S1. Graphs were depicted through the custom table option in the UCSC Genome Browser for the GRCh37 human genome version.
Whole exome sequence and comparison with SNP microarray
The WES study has been previously published in the report with the KCNQ2 exon 7 partial duplication [17]. CN variant analysis in WES data was performed using VarScan2 [19].
Briefly, raw FASTQ files were mapped against the hg19 version of the human genome using the BWA-MEM aligner. The resulting BAM files were pre-processed following the GATK [20]: workflow, marking PCR duplicates, and correcting errors in the base quality scores. The exome assay CNV segments were determined through both the Control-Freec [18] and the VarScan2 [19] programs, using as a reference either the father, mother, or daughter exomes. The Integrative Genomics Viewer (IGV) [21] was used to inspect the sequencing data and construct the graphs associated to WES-CNV analysis.
Analysis of gene alterations and function associated to clinical phenotypes
All genes alterations or variants detected by either SNP microarray or WES were analyzed using the VarElect program [22] (LifeMap Sciences Inc., Tel Aviv, Israel), to search for a correlations between the biological function of altered genes and the clinical phenotypes of the case, in order to detect a mechanistic implication of the gene contribution to clinical symptoms.
Results
Neuromotor developmental delay phenotype of patient
The patient is a 6-year-old boy presenting a cerebral palsy-like syndrome associated to severe development delay of unknown origin. The patient has a severe axial hypotonia without head control, spastic-dystonic tetraparesis, and peripheral neuro-axonal motor neuropathy, hypertonia of all limbs with dystonic movements of arms, no hand use and is not able to sit or crawl, and is unable to talk. He also presents an epileptic encephalopathy of neonatal onset with seizures well controlled since 4 years of age [17]. The full clinical study has already been reported [17]. The WES study detected a partial KCNQ2 exon 7 duplication (Clinvar ID 617505) that impairs its function [17], but it did not identify any other neuropathogenic mutation or gene variant [17] that could be functionally associated to the complex neurological phenotype of this patient.
Cooperating CNVs in neuro-pathogenic genes
We hypothesized that the proband might have additional candidate genetic alterations, which must occur in genes associated to neurological functions, and if they are known to have mutations, these mutants should also be associated to neurological phenotypes. Therefore, the phenotypic convergence is due to a combination of several alterations in genes whose protein biological functions can contribute to different aspects of this complex neurological syndrome. In this context, we reasoned that additional genetic factors have to contribute to this complex clinical phenotype, either in the form of additional genetic mutations or changes in gene copy number that will alter the expression and level of their proteins. Because the WES study did not identify any additional gene variant or mutant associated to the pathology, in addition to the known KCNQ2 exon 7 partial duplication [17], and in order to detect additional cooperating gene alterations that contribute to the pathogenesis of the patient complex neurological syndrome, the genome of the proband and family members was further studied by SNP microarrays to detect CNVs.
The SNP microarray study of the CNV variome in the patient can detect changes genes related to the altered neurological functions, and therefore, genes located in them are candidates to be involved in the clinical phenotype. The number of CNVs was similar in the four members of the family (Table 1), most of them were already known, and a small proportion of them were de novo CNVs; however, they were not shared among the four family members. The microarray study detected in the proband, compared to the other three family members, several genomic de novo CNVs larger than 3 kb (Table 2), and its markers are detailed in the Supplementary Table S1. Larger de novo loss of heterozygosity (LOH) genomic regions in the patient are detailed in Supplementary Table S2. The genes included within these de novo CNVs unique to the proband child are indicated in Supplementary Tables S1 and S2. In five genes, there is a deletion affecting several exons (SLC17A5, RYR3, ATP1A2, ELAVL2, ANO3), one gene (PCDH19) has an exon duplication, three genes have intronic deletions (NALCN, CDH13, LRCC55), and two genes have an intronic duplication (EFNA5, PCDH9). Intronic alterations can alter the processing of the RNA or its stability.
Next, we performed a search for a pathogenic association of all the genes comprised within these CNVs detected in the patient, by either SNP microarray or WES, and correlated their functions with different aspects of the clinical phenotype [17]. For this aim, the VarElect program was used [22]. All changes in the proband were also normalized with respect to the genome of the other family members, both parents and sister. The search was performed to identify functional and mechanistic correlations between gene functions and components of the clinical phenotype such as seizures, dystonia, epilepsy, neurotransmission, and motor neuron function. The neuropathogenicity of candidate genes within these genomic regions was determined by their previous association of their known mutations to a neurological phenotype. All the genes identified in the patient within CNVs that have a correlation with the clinical phenotype are expressed in neurons, have specific functions associated to the nervous system, or its known mutations have been associated to a neurological phenotype (Table 2). Functionally, most of these genes codify for several ion channel proteins or membrane proteins implicated in neuronal cell interactions, which can affect synaptic transmission and cell polarization. This indicates that individually these genes by themselves are not sufficient to cause the phenotype, but can contribute to the disease when they are combined with other genetic alterations in a unique individual. The affected boy presents a combined haploinsuficiency, mainly of CNV losses, that are likely to contribute to the pathogenic mechanism and the severity of the syndrome based on the expression level and subcellular localization of these proteins, the neurological functions associated to these proteins, and to the neurological pathogenic phenotypes associated to their genetic mutations in other patients.
One gene implicated in neurotransmission, SLC17A5, has a CNV loss that affects exons 7 to 9 (Table 3, Fig. 1). The SLC17A5 protein is required for the transport of aspartate and glutamate into synaptic vesicles, which are driven by the membrane potential [30]. However, it is not known whether a SLC17A5 haploinsuficiency, expressing, and aberrant protein might behave in a manner similar to its mutants regarding symptoms such as dystonia, hypotonia, or seizure crisis and share some symptoms such as hypotonia, ataxia, epilepsy, nystagmus, and findings of cerebral and cerebellar atrophy detected in patients with Salla disease [33].

Exon deletion in SLC17A5 gene in the proband. The genomic region comprising the SLC17A5 gene in the four family members is shown. The region deleted in the proband is marked by a box and a red line. The arrow indicates the direction of transcription
RYR3 (ryanodine receptor type 3) codes for a presynaptic endoplasmic reticulum ryanodine receptor-mediated Ca2+ and forms a voltage-independent, nonselective, non-inactivating cation channel permeable to Na+, K+, and Ca2+, which regulates the neuronal background sodium leak conductance [70]. Functionally, RyRs proteins regulate the generation of plateau potentials in motor neurons and also affect vesicle mobilization and synaptic plasticity [70]. In motor neurons, the RYR3 protein regulates intracellular calcium, in which AMPA-type GluR (glutamate receptor) channels regulate the intracellular calcium homeostasis that is altered in neurodegenerative diseases and can play an important role in the pathogenesis of motor neuron disorders (MND) [71]. The patient has a CNV loss that includes exon 2 (Table 3, Supplementary Fig. S2). Furthermore, haploinsuficiency of RYR3 might cooperate in an indirect way with several membrane proteins coded by genes implicated in sodium or calcium voltage channels, including SCN1A that is also implicated in epilepsy [54].
The NALCN protein is a sodium leak channel [72] expressed in neurons of the substantia nigra, and its reduction impairs the spontaneous firing required for the inhibition of downstream brain areas [73]. The NALCN protein interacts with UNC80 and pathogenic variants in both genes have been associated to dystonia [44, 74]. NALCN pathogenic variants have been associated to Neuroaxonal Dystrophy (INAD) patients, as well as to patients with severe hypotonia, speech impairment, cognitive delay, epilepsy, and mental disability [48]. The patient has a CNV loss, an intronic deletion in NALCN that alters its mRNA. Therefore, a reduction in the NALCN protein level might mimic a defective NALCN-UNC80 complex in the pathogenesis of dystonia. NALCN deficiency has been associated to channelopathies and cervical dystonia [47].
The SNP microarray analysis detected a large deletion that contains the ANO3 (Anoctamine 3) gene coding for a protein belonging to the TMEM16 family that functions as a Ca (2+)-activated chloride channel. ANO3 pathogenic variants have a dominant effect on dystonia [58], and some have been associated to a complex neurological syndrome combining dystonia and myoclonus phenotypes [60]. In the proband, ANO3 has an LOH that incudes exons 3 to 5 (Table 3, Supplementary Fig. S3), and its combination with pathogenic variants and CVNs in other genes is likely to contribute to the syndrome.
Genes implicated in seizures and epileptic-like phenotypes present alterations in EEG patterns. The CNV analysis identified four genes with de novo alterations, three with loses (ATP1A2, SLC17A5, and NALCN) and one with a gain (PCDH19), which have a direct relation with epileptic-like phenotypes (Table 1). ATP1A2 is highly expressed in the brain and codes for an integral membrane protein responsible for establishing and maintaining the electrochemical gradients of Na and K ions across the plasma membrane. ATP1A2 has been associated, in several studies, to rare forms of epilepsy and seizures [25]. The proband has a loss comprising exons 2 to 7 (Table 3, Supplementary Fig. S4). Other genes with CNV losses that have an indirect relation to epilepsy are RYR3, CDH13, PCDH9, and LRRC55 (Table 2).
Three of the affected genes in the proband, PCDH19, PCDH9, and CDH13, code for members of the cadherin protein family, which are implicated in neural intercellular interactions. PCDH19 mediates cell adhesion in neural tissues and regulates signaling at synaptic junctions. Pathogenic PCDH19 variants and CNV changes affect this gene in epilepsy [75]. More than one hundred different PCDH19 pathogenic variants, located in the extracellular domain of the protein, have been associated with epilepsy, mostly in females, and recently were also detected in males [65]. Truncation mutations in PCDH19 have also been associated to seizures [76]. PCDH19 is located on chromosome X and the patient has a CNV duplication that includes exon 3 (Table 3, Supp. Fig. S5) that might alter its gene expression and protein stability or level and modify local interactions among neural cells. CDH13 is a negative regulator of axon growth during neural differentiation [43], and a change in its protein level can also alter neuronal interactions and network organization. Both CDH13 and PCDH9 have an intronic deletion and a duplication, respectively, and both have been associated to epileptic encephalopathy [75]. Some of these cadherins are expressed in other organs such as the colon, kidney, heart, liver, and lung. However, the function of all these organs was normal in the proband.
Additional genes with CNV changes that can contribute to epileptic crisis and seizures were detected. LRRC55 (Leucine Rich Repeat Containing or BK Channel Auxiliary Gamma Subunit) has a CNV loss deleting the 3′ untranslated region of the mRNA in the patient. LRRC55 gene is expressed in the cortex, cerebellum, and spinal cord, and its protein is a regulator of large-conductance, voltage, and calcium-activated potassium channel (BK alpha) that modulates its gating properties [37]. Also there is a deletion of exon 3 in the ELAVL2 gene (Supplementary Figure S6) coding for a neural-specific RNA-binding protein that binds to several 3′ UTRs and is expressed in early neuronal progenitors to mature neurons [35] and also regulates co-expression networks of neurodevelopmental and synaptic genes [36]. The EFNA5 coding for ephrin5A also has an intronic duplication that can affect its transcription or RNA stability. Ephrin5 is implicated in neurodevelopment and axon bundling [62].
Discussion
In patients with neurodevelopmental problems, there is a large genetic heterogeneity implicating several regions that present CNV changes. CNVs indicate a change in gene dosage, which pathogenically means that there is a lower, or higher, level of the protein in cells, depending on whether the type of change is a loss or a gain. Cellular functions are performed by proteins, and therefore, their levels and subcellular distribution are an important factor. The nervous system membrane-associated proteins, such as those that participate in ion and neurotransmitter channels, or in cell-cell interactions, are very likely to be affected depending of their level of expression and alteration in the number of functional complexes in the cell. Functionally, a change in protein levels is a mechanism different from mutant proteins. The change in protein level can affect its distribution and location in neuronal cell surface, leading to the alteration of complexes in which they participate. This is particularly relevant for membrane proteins that can affect cellular interactions, ion transport, or vesicle release, such as in synapsis. All these roles are essential for a normal neural development and its associated functions.
The clinical heterogeneity reflects a very complex situation in which an unknown combination of alterations in gene dosage and pathogenic variants is likely to contribute to the pathogenesis of complex neurological phenotypes. However, for a clinical phenotype, the cooperation of genes related to that particular phenotype are necessary, but the specific gene combination may vary from case to case and result in a neurological pathway perturbation that is pathogenic [77].
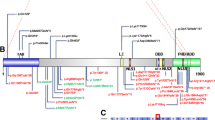
The contribution of individual genes with CNV changes to the patient phenotype is not known, but most of them occur in genes coding for proteins that participate in ion channels or neural cell interactions, which can regulate ion transport and neurotransmission. Thus, it is likely that an imbalance among several of these proteins and their functions contribute to complex neurological phenotypes. In the case of this patient, functions of the OMIM genes identified in the CNV analysis cluster by their mechanisms of action with the functional characteristics of the clinical phenotype (Fig. 2). The detection that several of the altered genes, directly or indirectly, are implicated in ionic (calcium, sodium) or glutamate transport and consequently are likely contributors to alterations in EEG, seizures, neurotransmission, epileptic-like crisis, or dystonia. Moreover, protocadherins and cadherin CNVs by altering their protein level and distribution on the cell surface can affect neural cell-cell interactions. Therefore, it is not possible to attribute a single phenotype to any individual gene, but the combination of several haplo-insufficient genes can generate a pathological situation related to their function. In this particular case, it is striking that four of the genes with a haploinsuficiency (LRRC5, NALCN, RYR3, and ANO3) affect calcium channels. CNVs in one of the implicated genes, PCDH19 [75, 78], were already known to contribute to epileptic and neurodevelopmental delay syndromes [75].

Pathogenic convergence between de novo altered genes and the clinical phenotype. The diagram illustrates the connection between OMIM genes with a pathogenic variant or CNV change that has a functional connection with the clinical phenotype of the patient. The lines indicate a direct association between a gene alteration (CNV or pathogenic variant) and the phenotype that has also been individually reported in other patients. The phenotypic associations were detected using the VarElect program to identify functional relations between genes and phenotypes. Dup: duplication
The simultaneous alterations of several gene coding for different ion channels and cell-cell interaction proteins create a compound genetic haplo-insufficiency, which due to its complexity is likely to occur in patients presenting complex neurodevelopmental delays with an unknown etiology. What is relevant regarding the affected genes is their functional pathogenic combination rather than the implication of specific individual genes. It is important to consider that these neurodevelopmental phenotypes are likely to be the consequence of a complex pattern of alteration in gene dosage and expression levels of their proteins, rather that resulting from a unique monogenic defect, as it occurs with human variation in multiple phenotypes, normal or pathological. However, these complex clinical phenotypes can also be modulated by additional gene variants. It is likely that the clinical phenotype and its evolution will be conditioned by additional genetic alterations. Therefore, it is likely that alternative gene combinations can also cause related and similar clinical phenotypes.
Nowadays, the cause of cerebral palsy is changing from its origin in childbirth problems to an unknown origin, which is likely to be genetic and heterogeneous. It is possible that when unraveled, they might share common pathogenic pathways, although they might involve different genes that affect the same functions. The identification of gene/protein networks associated to the clinical neurological phenotype can set the bases for designing novel therapeutic approaches to manage these patients and minimize, or compensate, the functional consequences of neuropathogenic gene combinations.
Conclusion
We conclude that the genetic heterogeneity of early severe neurodevelopmental delays, cerebral palsy-like with dystonia and epileptic encephalopathy, will have to be characterized in the context of the initiating pathogenic variant that is modulated by several additional genetic changes coding for proteins associated to neuronal functions.
Availability of data and materials
SNP microarray data sets have been submitted to GEO with the identifier GSE122584. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122584
Whole Exome Sequence (WES) raw data files are available in:
https://www.ncbi.nlm.nih.gov/sra/PRJNA629061
Abbreviations
- PCDH19:
-
Protocadherin 19
- CDH13:
-
Cadherin 13
- PCDH9:
-
Protocadherin 9
- ELAVL2:
-
ELAV-like neuron-specific RNA binding protein 2
- NALCN:
-
Na+ leak channel
- ANO3:
-
Ca2+-activated Cl- channel
- ATP1A2:
-
ATPase Na+/K+ transporting subunit alpha 2
- SLC17A5:
-
Sodium-dependent Asp/Glu transporter
- RYR3:
-
Brain ryanodine receptor-calcium release channel
- LRRC55:
-
Auxiliary protein of the large-conductance, voltage, and Ca2+-activated K+ channel
- EFNA5:
-
Ephrin A5
References
McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–16. https://doi.org/10.1016/S1474-4422(15)00250-1.
El Kosseifi C, Cornet MC, Cilio MR. Neonatal developmental and epileptic encephalopathies. Semin Pediatr Neurol. 2019;32:100770. https://doi.org/10.1016/j.spen.2019.08.006.
Borlot F, Regan BM, Bassett AS, Stavropoulos DJ, Andrade DM. Prevalence of pathogenic copy number variation in adults with pediatric-onset epilepsy and intellectual disability. JAMA Neurol. 2017;74:1301–11. https://doi.org/10.1001/jamaneurol.2017.1775.
McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, van Bon BW, van Eyk CL, Broadbent J, Reynolds C, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatry. 2015;20:176–82. https://doi.org/10.1038/mp.2014.189.
Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, Franke A, Malafosse A, Genton P, Thomas P, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962. https://doi.org/10.1371/journal.pgen.1000962.
Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, McCague P, Dimiceli S, Pitts T, et al. A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet. 1999;65:493–507. https://doi.org/10.1086/302497.
Liu X, Shimada T, Otowa T, Wu YY, Kawamura Y, Tochigi M, Iwata Y, Umekage T, Toyota T, Maekawa M, et al. Genome-wide association study of autism spectrum disorder in the East Asian populations. Autism Res. 2015;9:340–9. https://doi.org/10.1002/aur.1536.
Burn J, Watson M. The human variome project. Hum Mutat. 2016;37:505–7. https://doi.org/10.1002/humu.22986.
Iourov IY, Vorsanova SG, Yurov YB. The variome concept: focus on CNVariome. Mol Cytogenet. 2019;12:52. https://doi.org/10.1186/s13039-019-0467-8.
Fahey MC, Maclennan AH, Kretzschmar D, Gecz J, Kruer MC. The genetic basis of cerebral palsy. Dev Med Child Neurol. 2017;59:462–9. https://doi.org/10.1111/dmcn.13363.
Weckhuysen S, Mandelstam S, Suls A, Audenaert D, Deconinck T, Claes LR, Deprez L, Smets K, Hristova D, Yordanova I, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012;71:15–25. https://doi.org/10.1002/ana.22644.
Lee IC, Yang JJ, Li SY. KCNQ2-Associated epilepsy: a review of variable phenotypes and neurodevelopmental outcomes. Neuropsychiatry. 2018;8:318–23. https://doi.org/10.4172/Neuropsychiatry.1000353.
Segel R, Ben-Pazi H, Zeligson S, Fatal-Valevski A, Aran A, Gross-Tsur V, Schneebaum-Sender N, Shmueli D, Lev D, Perlberg S, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. 2015:84, 1660–1668. https://doi.org/10.1212/WNL.0000000000001494.
Rosenfeld JA, Patel A. Chromosomal microarrays: understanding genetics of neurodevelopmental disorders and congenital anomalies. J Pediatr Genet. 2017;6:42–50. https://doi.org/10.1055/s-0036-1584306.
Zarrei M, Burton CL, Engchuan W, Young EJ, Higginbotham EJ, MacDonald JR, Trost B, Chan AJS, Walker S, Lamoureux S, et al. A large data resource of genomic copy number variation across neurodevelopmental disorders. NPJ Genom Med. 2019;4:26. https://doi.org/10.1038/s41525-019-0098-3.
Zarrei M, Fehlings DL, Mawjee K, Switzer L, Thiruvahindrapuram B, Walker S, Merico D, Casallo G, Uddin M, MacDonald JR, et al. De novo and rare inherited copy-number variations in the hemiplegic form of cerebral palsy. Genet Med. 2018;20:172–80. https://doi.org/10.1038/gim.2017.83.
Lazo PA, Garcia JL, Gomez-Puertas P, Marcos-Alcalde I, Arjona C, Villarroel A, Gonzalez-Sarmiento R, Fons C. Novel dominant KCNQ2 Exon 7 partial in-frame duplication in a complex epileptic and neurodevelopmental delay syndrome. Int J Mol Sci. 2020;21:E4447. https://doi.org/10.3390/ijms21124447.
Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, Barillot E. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28:423–5. https://doi.org/10.1093/bioinformatics/btr670.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. https://doi.org/10.1101/gr.129684.111.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 10 11–33. https://doi.org/10.1002/0471250953.bi1110s43.
Robinson JT, Thorvaldsdottir H, Wenger AM, Zehir A, Mesirov JP. Variant review with the integrative genomics viewer. Cancer Res. 2017;77:e31–4. https://doi.org/10.1158/0008-5472.CAN-17-0337.
Stelzer G, Plaschkes I, Oz-Levi D, Alkelai A, Olender T, Zimmerman S, Twik M, Belinky F, Fishilevich S, Nudel R, et al. VarElect: the phenotype-based variation prioritizer of the GeneCards Suite. BMC Genomics. 2016;17, 444(Suppl 2). https://doi.org/10.1186/s12864-016-2722-2.
Kinoshita PF, Leite JA, Orellana AM, Vasconcelos AR, Quintas LE, Kawamoto EM, Scavone C. The influence of Na(+), K(+)-ATPase on glutamate signaling in neurodegenerative diseases and senescence. Front Physiol. 2016;7:195. https://doi.org/10.3389/fphys.2016.00195.
Talsma AD, Chaves JF, LaMonaca A, Wieczorek ED, Palladino MJ. Genome-wide screen for modifiers of Na (+) /K (+) ATPase alleles identifies critical genetic loci. Mol Brain. 2014;7:89. https://doi.org/10.1186/s13041-014-0089-3.
Wilbur C, Buerki SE, Guella I, Toyota EB, Evans DM, McKenzie MB, Datta A, Michoulas A, Adam S, Van Allen MI, et al. An infant with epilepsy and recurrent hemiplegia due to compound heterozygous variants in ATP1A2. Pediatr Neurol. 2017;75:87–90. https://doi.org/10.1016/j.pediatrneurol.2017.06.003.
Prontera P, Sarchielli P, Caproni S, Bedetti C, Cupini LM, Calabresi P, Costa C. Epilepsy in hemiplegic migraine: genetic mutations and clinical implications. Cephalalgia. 2017;38:361–73. https://doi.org/10.1177/0333102416686347.
Al-Bulushi B, Al-Hashem A, Tabarki B. A wide clinical phenotype spectrum in patients with ATP1A2 mutations. J Child Neurol. 2014;29:265–8. https://doi.org/10.1177/0883073813504623.
Costa C, Prontera P, Sarchielli P, Tonelli A, Bassi MT, Cupini LM, Caproni S, Siliquini S, Donti E, Calabresi P. A novel ATP1A2 gene mutation in familial hemiplegic migraine and epilepsy. Cephalalgia. 2014;34:68–72. https://doi.org/10.1177/0333102413498941.
Pisano T, Spiller S, Mei D, Guerrini R, Cianchetti C, Friedrich T, Pruna D. Functional characterization of a novel C-terminal ATP1A2 mutation causing hemiplegic migraine and epilepsy. Cephalalgia. 2013;33:1302–10. https://doi.org/10.1177/0333102413495116.
Miyaji T, Omote H, Moriyama Y. Functional characterization of vesicular excitatory amino acid transport by human sialin. J Neurochem. 2011;119:1–5. https://doi.org/10.1111/j.1471-4159.2011.07388.x.
Lodder-Gadaczek J, Gieselmann V, Eckhardt M. Vesicular uptake of N-acetylaspartylglutamate is catalysed by sialin (SLC17A5). Biochem J. 2013;454:31–8. https://doi.org/10.1042/BJ20130300.
Zhao J, Ramadan E, Cappiello M, Wroblewska B, Bzdega T, Neale JH. NAAG inhibits KCl-induced [(3)H]-GABA release via mGluR3, cAMP, PKA and L-type calcium conductance. Eur J Neurosci. 2001;13:340–6.
Tarailo-Graovac M, Drogemoller BI, Wasserman WW, Ross CJ, van den Ouweland AM, Darin N, Kollberg G, van Karnebeek CD, Blomqvist M. Identification of a large intronic transposal insertion in SLC17A5 causing sialic acid storage disease. Orphanet J Rare Dis. 2017;12:28. https://doi.org/10.1186/s13023-017-0584-6.
Carvalho DP, Dupuy C. Thyroid hormone biosynthesis and release. Mol Cell Endocrinol. 2017;458:6–15. https://doi.org/10.1016/j.mce.2017.01.038.
Yano M, Hayakawa-Yano Y, Okano H. RNA regulation went wrong in neurodevelopmental disorders: the example of Msi/Elavl RNA binding proteins. Int J Dev Neurosci. 2016;55:124–30. https://doi.org/10.1016/j.ijdevneu.2016.01.002.
Berto S, Usui N, Konopka G, Fogel BL. ELAVL2-regulated transcriptional and splicing networks in human neurons link neurodevelopment and autism. Hum Mol Genet. 2016;25:2451–64. https://doi.org/10.1093/hmg/ddw110.
Li Q, Fan F, Kwak HR, Yan J. Molecular basis for differential modulation of BK channel voltage-dependent gating by auxiliary gamma subunits. J Gen Physiol. 2015;145:543–54. https://doi.org/10.1085/jgp.201511356.
Adler EM. Of BK regulation, painful connections, and versatile neuropeptide signaling. J Gen Physiol. 2015;145:457–8. https://doi.org/10.1085/jgp.201511430.
Zhang J, Yan J. Regulation of BK channels by auxiliary gamma subunits. Front Physiol. 2014;5:401. https://doi.org/10.3389/fphys.2014.00401.
Yan J, Aldrich RW. BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc Natl Acad Sci U S A. 2012;109:7917–22. https://doi.org/10.1073/pnas.1205435109.
Redies C, Hertel N, Hubner CA. Cadherins and neuropsychiatric disorders. Brain Res. 2012;1470:130–44. https://doi.org/10.1016/j.brainres.2012.06.020.
Kim SY, Yasuda S, Tanaka H, Yamagata K, Kim H. Non-clustered protocadherin. Cell Adh Migr. 2011;5:97–105. https://doi.org/10.4161/cam.5.2.14374.
Lesca G, Rudolf G, Labalme A, Hirsch E, Arzimanoglou A, Genton P, Motte J, de Saint Martin A, Valenti MP, Boulay C, et al. Epileptic encephalopathies of the Landau-Kleffner and continuous spike and waves during slow-wave sleep types: genomic dissection makes the link with autism. Epilepsia. 2012;53:1526–38. https://doi.org/10.1111/j.1528-1167.2012.03559.x.
Perez Y, Kadir R, Volodarsky M, Noyman I, Flusser H, Shorer Z, Gradstein L, Birnbaum RY, Birk OS. UNC80 mutation causes a syndrome of hypotonia, severe intellectual disability, dyskinesia and dysmorphism, similar to that caused by mutations in its interacting cation channel NALCN. J Med Genet. 2016;53:397–402. https://doi.org/10.1136/jmedgenet-2015-103352.
Cochet-Bissuel M, Lory P, Monteil A. The sodium leak channel, NALCN, in health and disease. Front Cell Neurosci. 2014;8:132. https://doi.org/10.3389/fncel.2014.00132.
Mok KY, Schneider SA, Trabzuni D, Stamelou M, Edwards M, Kasperaviciute D, Pickering-Brown S, Silverdale M, Hardy J, Bhatia KP. Genomewide association study in cervical dystonia demonstrates possible association with sodium leak channel. Mov Disord. 2014;29:245–51. https://doi.org/10.1002/mds.25732.
Bend EG, Si Y, Stevenson DA, Bayrak-Toydemir P, Newcomb TM, Jorgensen EM, Swoboda KJ. NALCN channelopathies: distinguishing gain-of-function and loss-of-function mutations. Neurology. 2016;87:1131–9. https://doi.org/10.1212/WNL.0000000000003095.
Wang Y, Koh K, Ichinose Y, Yasumura M, Ohtsuka T, Takiyama Y. A de novo mutation in the NALCN gene in an adult patient with cerebellar ataxia associated with intellectual disability and arthrogryposis. Clin Genet. 2016;90:556–7. https://doi.org/10.1111/cge.12851.
Gal M, Magen D, Zahran Y, Ravid S, Eran A, Khayat M, Gafni C, Levanon EY, Mandel H. A novel homozygous splice site mutation in NALCN identified in siblings with cachexia, strabismus, severe intellectual disability, epilepsy and abnormal respiratory rhythm. Eur J Med Genet. 2016;59:204–9. https://doi.org/10.1016/j.ejmg.2016.02.007.
Zhou Q, Yang J, Cao B, Chen Y, Wei Q, Ou R, Song W, Zhao B, Wu Y, Shang H. Association analysis of NALCN polymorphisms rs1338041 and rs61973742 in a Chinese population with isolated cervical dystonia. Parkinsons Dis. 2016;2016:9281790. https://doi.org/10.1155/2016/9281790.
Dayanithi G, Mechaly I, Viero C, Aptel H, Alphandery S, Puech S, Bancel F, Valmier J. Intracellular Ca2+ regulation in rat motoneurons during development. Cell Calcium. 2006;39:237–46. https://doi.org/10.1016/j.ceca.2005.10.011.
Takeshima H, Ikemoto T, Nishi M, Nishiyama N, Shimuta M, Sugitani Y, Kuno J, Saito I, Saito H, Endo M, et al. Generation and characterization of mutant mice lacking ryanodine receptor type 3. J Biol Chem. 1996;271:19649–52.
Lu B, Su Y, Das S, Liu J, Xia J, Ren D. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. 2007;129:371–83. https://doi.org/10.1016/j.cell.2007.02.041.
Steel D, Symonds JD, Zuberi SM, Brunklaus A. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia. 2017. https://doi.org/10.1111/epi.13889.
Helbig I, Tayoun AA. Understanding genotypes and phenotypes in epileptic encephalopathies. Mol Syndromol. 2016;7:172–81. https://doi.org/10.1159/000448530.
Domingo A, Erro R, Lohmann K. Novel dystonia genes: clues on disease mechanisms and the complexities of high-throughput sequencing. Mov Disord. 2016;31:471–7. https://doi.org/10.1002/mds.26600.
Pedemonte N, Galietta LJ. Structure and function of TMEM16 proteins (anoctamins). Physiol Rev. 2014;94:419–59. https://doi.org/10.1152/physrev.00039.2011.
Charlesworth G, Bhatia KP, Wood NW. The genetics of dystonia: new twists in an old tale. Brain. 2017-2037;2013:136. https://doi.org/10.1093/brain/awt138.
Charlesworth G, Plagnol V, Holmstrom KM, Bras J, Sheerin UM, Preza E, Rubio-Agusti I, Ryten M, Schneider SA, Stamelou M, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet. 2012;91:1041–50. https://doi.org/10.1016/j.ajhg.2012.10.024.
Lohmann K, Klein C. Update on the genetics of dystonia. Curr Neurol Neurosci Rep. 2017;17:26. https://doi.org/10.1007/s11910-017-0735-0.
Zech M, Boesch S, Jochim A, Weber S, Meindl T, Schormair B, Wieland T, Lunetta C, Sansone V, Messner M, et al. Clinical exome sequencing in early-onset generalized dystonia and large-scale resequencing follow-up. Mov Disord. 2017;32:549–59. https://doi.org/10.1002/mds.26808.
Davy A, Gale NW, Murray EW, Klinghoffer RA, Soriano P, Feuerstein C, Robbins SM. Compartmentalized signaling by GPI-anchored ephrin-A5 requires the Fyn tyrosine kinase to regulate cellular adhesion. Genes Dev. 1999;13:3125–35.
Cooper SR, Jontes JD, Sotomayor M. Structural determinants of adhesion by Protocadherin-19 and implications for its role in epilepsy. Elife. 2016;5:e18529. https://doi.org/10.7554/eLife.18529.
de Lange IM, Rump P, Neuteboom RF, Augustijn PB, Hodges K, Kistemaker AI, Brouwer OF, Mancini GMS, Newman HA, Vos YJ, et al. Male patients affected by mosaic PCDH19 mutations: five new cases. Neurogenetics. 2017;18:147–53. https://doi.org/10.1007/s10048-017-0517-5.
Tsuchida N, Nakashima M, Kato M, Heyman E, Inui T, Haginoya K, Watanabe S, Chiyonobu T, Morimoto M, Ohta M, et al. Detection of copy number variations in epilepsy using exome data. Clin Genet. 2017;93:577–87. https://doi.org/10.1111/cge.13144.
Terracciano A, Trivisano M, Cusmai R, De Palma L, Fusco L, Compagnucci C, Bertini E, Vigevano F, Specchio N. PCDH19-related epilepsy in two mosaic male patients. Epilepsia. 2016;57:e51–5. https://doi.org/10.1111/epi.13295.
Kurian M, Korff CM, Ranza E, Bernasconi A, Lubbig A, Nangia S, Ramelli GP, Wohlrab G, Nordli DR Jr, Bast T. Focal cortical malformations in children with early infantile epilepsy and PCDH19 mutations: case report. Dev Med Child Neurol. 2017;60:100–5. https://doi.org/10.1111/dmcn.13595.
Perez D, Hsieh DT, Rohena L. Somatic Mosaicism of PCDH19 in a male with early infantile epileptic encephalopathy and review of the literature. Am J Med Genet A. 2017:173, 1625–1630. https://doi.org/10.1002/ajmg.a.38233.
Perucca P, Scheffer IE, Harvey AS, James PA, Lunke S, Thorne N, Gaff C, Regan BM, Damiano JA, Hildebrand MS, et al. Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 2017;131:1–8. https://doi.org/10.1016/j.eplepsyres.2017.02.001.
Meissner G. The structural basis of ryanodine receptor ion channel function. J Gen Physiol. 2017;149:1065–89. https://doi.org/10.1085/jgp.201711878.
Jahn K, Grosskreutz J, Haastert K, Ziegler E, Schlesinger F, Grothe C, Dengler R, Bufler J. Temporospatial coupling of networked synaptic activation of AMPA-type glutamate receptor channels and calcium transients in cultured motoneurons. Neuroscience. 2006;142:1019–29. https://doi.org/10.1016/j.neuroscience.2006.07.034.
Kschonsak M, Chua HC, Noland CL, Weidling C, Clairfeuille T, Bahlke OØ, Ameen AO, Li ZR, Arthur CP, Ciferri C, et al. Structure of the human sodium leak channel NALCN. Nature. 2020. https://doi.org/10.1038/s41586-020-2570-8.
Lutas A, Lahmann C, Soumillon M, Yellen G. The leak channel NALCN controls tonic firing and glycolytic sensitivity of substantia nigra pars reticulata neurons. Elife. 2016;5:e15271. https://doi.org/10.7554/eLife.15271.
Obeid T, Hamzeh AR, Saif F, Nair P, Mohamed M, Al-Ali MT, Bastaki F. Identification of a novel homozygous UNC80 variant in a child with infantile hypotonia with psychomotor retardation and characteristic facies-2 (IHPRF2). Metab Brain Dis. 2018;33:869–73. https://doi.org/10.1007/s11011-018-0200-z.
Lindy AS, Stosser MB, Butler E, Downtain-Pickersgill C, Shanmugham A, Retterer K, Brandt T, Richard G, McKnight DA. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59:1062–71. https://doi.org/10.1111/epi.14074.
Butler KM, da Silva C, Alexander JJ, Hegde M, Escayg A. Diagnostic yield from 339 epilepsy patients screened on a clinical gene panel. Pediatr Neurol. 2017;77:61–6. https://doi.org/10.1016/j.pediatrneurol.2017.09.003.
Li Y, McGrail DJ, Latysheva N, Yi S, Babu MM, Sahni N. Pathway perturbations in signaling networks: linking genotype to phenotype. Semin Cell Dev Biol. 2020;99:3–11. https://doi.org/10.1016/j.semcdb.2018.05.001.
Stosser MB, Lindy AS, Butler E, Retterer K, Piccirillo-Stosser CM, Richard G, McKnight DA. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet Med. 2018;20:403–10. https://doi.org/10.1038/gim.2017.114.
Funding
Grants from Agencia Estatal de Investigación, Ministerio de Ciencia e Innovación (SAF2016-75744-R, PID2019-105610RB-I00) and Consejería de Educación-Junta de Castilla y León (CSI264P20, CLC-2017-01, and UIC-258) to P.A.L. Agencia Estatal de Investigación-Ministerio de Economía y Competitividad-FEDER-Fondo Social Europeo (RTC-2017-6494-1 and RTI2018-094434-B-I00) to P. G-P. Departament de Salut de la Generalitat de Catalunya-URDCat Project (SLT002/16/00174) to C.F.
The study sponsors have no role in study design, in analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Author information
Authors and Affiliations
Contributions
JLGH and LAC performed and analyzed the SNP microarray. IM-A and P-GP performed whole exome sequence study. CS coordinated the clinical characterization of the patient. PAL coordinated and integrated genetic and neurological studies and wrote the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All participants were provided with an explanation of the genetic study research, and written informed consent for the diagnostic genetic study was obtained from the parents of the affected child. The genomic diagnostic study was approved by the ethics committee of the Hospital Universitario de Salamanca and Hospital Sant Joan de Deu.
Consent for publication
Written informed consent for publication was obtained from the participant’s legal guardian/next of kin.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure S1.
CNV from WES studies in the SLC17A5 gene. CNV alterations (Gains [red] and losses [blue]) retrieved by WES assays through the VarScan2 algorithm for the patient with respect to the other three family members. Graphs were depicted using the integrative genome viewer (IGV) tool for the GRCh37 human genome version.
Additional file 2: Supplementary Figure S2.
Exon deletion in RYR3 gene in the proband. The genomic region comprising the RYR3 gene in the four family members is shown. The region deleted in the proband is marked by a box and a red line. The arrow indicates the direction of transcription.
Additional file 3: Supplementary Figure S3.
Exon deletion in ANO3 gene in the proband. The genomic region comprising the ANO3 gene in the four family members is shown. The region deleted in the proband is marked by a box and a red line. The arrow indicates the direction of transcription.
Additional file 4: Supplementary Figure S4.
Exon deletion in ATP1A2 gene in the proband. The genomic region comprising the ATP1A2 gene in the four family members is shown. The region deleted in the proband is marked by a box and a red line. The arrow indicates the direction of transcription.
Additional file 5: Supplementary Figure S5.
Duplication encompassing exon 3 in the PCDH19 gene. The genomic region comprising the PCDH19 gene in the four family members is shown. The region deleted in the proband is marked bya box and a red line. The arrow indicates the direction of transcription.
Additional file 6: Supplementary Figure S6.
Deletion encompassing exon 3 in the ELAVL2 gene. The genomic region comprising the ELAVL2 gene in the four family members is shown. The region deleted in the proband is marked by a box and a red line. The arrow indicates the direction of transcription.
Additional file 7: Supplementary Table S1.
de novo CNVs (> 1Kb) identified in patient.
Additional file 8: Supplementary Table S2.
Regions with loss of heterozygosity (LOH)> 3 Mb in the patient.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
García-Hernández, J.L., Corchete, L.A., Marcos-Alcalde, Í. et al. Pathogenic convergence of CNVs in genes functionally associated to a severe neuromotor developmental delay syndrome. Hum Genomics 15, 11 (2021). https://doi.org/10.1186/s40246-021-00309-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-021-00309-4