
Abstract
Background
Celiac disease is a complex chronic immune-mediated disorder of the small intestine. Today, the pathobiology of the disease is unclear, perplexing differential diagnosis, patient stratification, and decision-making in the clinic.
Methods
Herein, we adopted a next-generation sequencing approach in a celiac disease trio of Greek descent to identify all genomic variants with the potential of celiac disease predisposition.
Results
Analysis revealed six genomic variants of prime interest: SLC9A4 c.1919G>A, KIAA1109 c.2933T>C and c.4268_4269delCCinsTA, HoxB6 c.668C>A, HoxD12 c.418G>A, and NCK2 c.745_746delAAinsG, from which NCK2 c.745_746delAAinsG is novel. Data validation in pediatric celiac disease patients of Greek (n = 109) and Serbian (n = 73) descent and their healthy counterparts (n = 111 and n = 32, respectively) indicated that HoxD12 c.418G>A is more prevalent in celiac disease patients in the Serbian population (P < 0.01), while NCK2 c.745_746delAAinsG is less prevalent in celiac disease patients rather than healthy individuals of Greek descent (P = 0.03). SLC9A4 c.1919G>A and KIAA1109 c.2933T>C and c.4268_4269delCCinsTA were more abundant in patients; nevertheless, they failed to show statistical significance.
Conclusions
The next-generation sequencing-based family genomics approach described herein may serve as a paradigm towards the identification of novel functional variants with the aim of understanding complex disease pathobiology.
Similar content being viewed by others


Background
Celiac disease is a complex chronic immune-mediated disorder of the small intestine. Today, the pathobiology of the disease is unclear, perplexing differential diagnosis, patient stratification, and decision-making in the clinic. Genetics has been reported to play a key role. The HLA-DQ2 gene is identified in up to 95 % of celiac disease patients, while most of the remaining patients have the HLA-DQ8 gene. Notwithstanding, the Chinese and Japanese populations (devoid of HLA-DQ2) are not expected to develop the disease, yet this is not true for the individuals with the HLA-DQ8 gene. Celiac disease is also associated with an extended ancestral haplotype that is defined by class I and II HLAs (A, B, DR, DQ). Notably, HLA-DQ2 and/or HLA-DQ8 expression is necessary but not sufficient for disease development. Thus, other genes are anticipated to be involved. Indeed, genome-wide association studies (GWAS) have revealed 26 non-HLA genetic loci-associated celiac disease and other autoimmune or chronic immune disorders (diabetes mellitus type I, rheumatoid arthritis) [1, 2]. In 2008 to 2011, several new celiac disease risk loci have been identified [3–5], bringing the number of known loci (including the HLA one) to 40 and indicating genes and gene regulatory elements of paramount importance. In 2015, five new genetic loci were identified, being independent of HLA-DQA1 and HLA-DQB1 and associated with celiac disease predisposition [6].
Although a genetic component has been described, disease occurrence has been also associated with environmental factors and gut microbiome. In all cases, gluten has been identified as the environmental trigger of the disease, leading to the stimulation of gluten-specific T cells. Differential diagnosis is still a major issue. Although a gold standard diagnostic approach has been defined (endoscopy with biopsy of the small intestine coupled to positive disease serology), several pathological conditions have been reported sharing similar mucosal transformations with celiac disease as well as other autoimmune disorders (thyroid disease, Addison disease, autoimmune liver disease, Sjögren syndrome) that occur ten times more frequently in celiac disease patients often masking celiac disease symptoms. Disease management options are restricted to a gluten-free lifestyle, which ultimately fails to protect patients from disease symptoms due to its chronic nature. Can we delineate individual variability towards differential diagnosis? Can we highlight the disease mechanisms in question to assist disease management?
So far, findings account for 49 % of the genetic basis of the disease. As in other immune-mediated diseases, genetic predisposition to celiac disease remains unresolved as we still need to explain the remaining major fraction of heritability, including rare as well as additional common risk variants. Causal variants and genes still need to be identified and/or more finely localized. In this context, the Immunochip Consortium was developed to explore comprehensive datasets containing common, low-frequency, and rare variants in related diseases (autoimmune thyroid disease, ankylosing spondylitis, Crohn disease, celiac disease, IgA deficiency, multiple sclerosis, primary biliary cirrhosis, psoriasis, rheumatoid arthritis, systemic lupus erythematosus, type 1 diabetes mellitus, and ulcerative colitis) [7].
As expected, the advent of technology and, in particular, next-generation sequencing has provided unprecedented opportunities to delineate disease pathobiology as well as inter-individual differences [8, 9]. Herein, we propose a multi-step next-generation sequencing-based family genomics approach, piloted in a celiac disease trio of Greek descent to identify novel genomic variants of functional significance with the aim of understanding disease pathobiology.
Methods
Case selection, DNA isolation, and whole-genome sequencing
A seven-member Greek family has been recruited (informed consents have been obtained), and a trio analysis (III-1, III-2, IV-3) has been performed using the celiac disease model (Additional file 1: Figure S1). A family-based design was employed rather than a population-based design, as the former is generally considered to be robust against population admixture and stratification and may yield both within- and between-family information [10]. Genomic DNA isolation was performed from saliva using the Oragene collection kit (DNA Genotek, Ontario, Canada) (Serbian cohort) and peripheral blood using an automated system (MagNA Pure Compact, Roche, Basel, Switzerland) (Greek cohort). Whole-genome sequencing was performed using Complete Genomics’ (CA, USA) DNA nanoarray platform [11]. DNA sequencing coverage was 110×. Only high-quality call variants were included in the analysis (>93 %). Genomes were aligned with the hg19 reference genome.
Bioinformatics and in silico analyses
Next-generation sequencing data (Complete Genomics Inc., CA) were analyzed using Ingenuity Variant Analysis version 3.1.2 (Ingenuity® Systems, www.ingenuity.com). This is a well-established software that identifies associations between phenotypes, defined by the user by classification of the tested individuals, and variants in the sequenced genome. Upon classification of the family members by phenotype (celiac vs. normal), a number of variants were listed; the output was filtered into a smaller variant list upon classification of the family members by those being celiac patients vs. those who were healthy and known not to be celiac disease subjects. The genetic model used for this comparison was of an autosomal dominant model, since it traces the genetic inheritance from mother (III-2) to daughter (IV-3) in a highly penetrant form. A total of 263 genes followed an autosomal dominant pattern, and 227 variants were identified in the genetic model. Out of these, 6 genes and 7 variants were identified in the biological/phenotype pattern of celiac disease, due to either the past association of the genes considered or via the biological significance as determined by the IVA software.
Due to the large amount of variants which are normally present in the genome, even when excluding intronic sequences, several filtering steps are used in data analysis to include only the genes which are likely to be causative in the final output connected with just the disease of interest, in this case—celiac disease. All variants were filtered according to the analysis required, using custom scripts and Complete Genomics Analysis Tools (CGA™ Tools). The filtering cascade utilized in the present data analysis is as follows:
-
1.
Confidence: Only variants with call quality at least 20.0 in cases or at least 20.0 in controls (i.e., 99 % call accuracy) were included.
-
2.
Common variants: All variants observed to have an allele frequency ≥3.0 % of the genomes in the 1000 genomes project OR ≥3.0 % of the public Complete Genomics genomes OR ≥3.0 % of the NHLBI ESP exomes were excluded.
-
3.
Predicted deleterious: Only variants that are experimentally observed to be associated with a phenotype (Pathogenic, Possibly Pathogenic, Unknown Significance OR established gain of function in the literature OR gene fusions OR inferred activating mutations by Ingenuity OR predicted gain of function by BSIFT OR in a microRNA binding site OR Frameshift, in-frame indel, or stop codon change OR Missense and not predicted to be innocuous by SIFT OR disrupt splice site up to 2.0 bases into intron OR deleterious to a microRNA OR structural variant) were kept.
-
4.
Genetic analysis: Only variants with the following genotype characteristics were kept: associated with gain of function OR heterozygous_alt OR haploinsufficient OR heterozygous OR homozygous OR heterozygous_amb OR compound_heterozygous OR hemizygous AND occur in at least 2 of the case samples at the gene level in the Case samples AND not which are associated with gain of function OR heterozygous_alt OR haploinsufficient OR heterozygous OR homozygous OR heterozygous_amb OR compound_heterozygous OR hemizygous AND occur in at least 1 of the control samples at the variant level in the control samples.
-
5.
Biological context: Only variants that are within 1 hop (direct targets) of upstream regulators or downstream regulatory targets of such genes and that are known or predicted to affect celiac disease.
Variants of interest were annotated with Annovar in Galaxy [12] and compared with NCBI dbSNP build 137 (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi), 69 reference genomes from Complete Genomes (http://www.completegenomics.com/publicdata/69Genomes/), and GWAS (http://www.genome.gov/gwastudies) to determine their novelty or otherwise. To obtain a list of variants of potential functional significance, we employed protein variation effect analyzer (PROVEAN) v1.1.3 (PROVEAN human genome variants tool) that provides both scale-invariant feature transform (SIFT) [13] and PROVEAN [14] predictions for a given list of human genome variants as well as accessory information (dbSNP rs IDs, gene description, PFAM domain, GO terms, etc.). PROVEAN is able to make predictions for any type of protein sequence alteration, including single or multiple amino acid substitutions, deletions, and insertions [15]. Additionally, Variant Effect Predictor [16] and RegulomeDB [8] were employed to allow further data interrogation.
Downstream molecular analysis
Selected variants were subsequently validated in pediatric celiac disease patients of Greek (n = 109) [17] and Serbian (n = 73) [18] descent and their healthy counterparts (n = 111 and n = 32, respectively). The diagnosis of celiac disease was based on the criteria of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) [19]. For children diagnosed prior to 1990, the “Interlaken criteria” were applied. The Ethics Committee of University Children’s Hospital, University of Belgrade, and the Review Board of “Aghia Sophia” Children’s Hospital have approved the study.
Amplification was carried out according to the KAPA2G Fast HotStart protocol (KAPABIOSYSTEMS, MA, USA); detailed information per SNP amplification conditions is available upon request. For SLC9A4 c.1919G>A, an allele-specific polymerase chain reaction (PCR) assay was developed (two alternative reverse primers hybridizing exclusively either to the wild-type or the mutant allele). For NCK2 c.745_746delAAinsG, PCR products were subjected to XcmI (New England Biolabs, MA, USA) restriction endonuclease analysis at 37 °C for 1.15 h and subsequent enzyme deactivation (65 °C, 20 min). Restriction fragments were visualized by 3 % agarose gel electrophoresis following ethidium bromide or Midori Green staining. For HoxD12 c.418G>A, HoxB6 c.668C>A, and KIAA1109 c.2933T>C and c.4268_4269delCCinsTA, a PCR-based conventional Sanger resequencing approach was employed. Capillary electrophoresis was performed on the ABI Prism 3130xl DNA Analyzer (Applied Biosystems). Sanger sequencing was also employed to ensure PCR-RFLP and allele-specific PCR method verification.
Statistical analysis
Herein, a thorough statistical review and analysis has been attempted, having always in mind that we used the celiac disease model (trio analysis) as a reason to conduct a more refined approach in searching/genotyping the resulted variants in a selective population of celiac disease of Greek and Serbian descent. We tested for deviations from HWE using the chi-square goodness of fit test and principal component analysis. Considering that the χ 2 approximation can be poor when there are low genotype counts, a Fisher exact test (R genetics package) was also used, as it does not rely on the χ 2 approximation [20]. Tests were performed as two-tailed, and differences were considered statistically significant when P < 0.05. Focusing on case-control phenotypes, we tested the null hypothesis of no association between rows and columns of the 2 × 3 matrix that contains the counts of the three genotypes (the two homozygotes and the heterozygote) among cases and controls. Again, a Fisher exact test was preferred (to evaluate genotype and allele frequencies), as it is computationally more demanding, but it is easily implemented in R. We also performed the Armitage test (Monte Carlo method; it obtains results that are closer to an exact test, since the classical Cochran-Armitage trend test is based on approximation) [20].
Results and discussion
Whole-genome sequencing analysis of trio reveals newly identified genetic risk variants for pediatric celiac disease
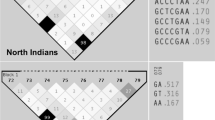
Our multi-step next-generation sequencing-based family genomics approach revealed six genomic variants of prime interest: SLC9A4 c.1919G>A, HoxD12 c.418G>A, KIAA1109 c.2933T>C and c.4268_4269delCCinsTA, HoxB6 c.668C>A, and NCK2 c.745_746delAAinsG. Susceptibility to celiac disease (CELIAC6) and to autoimmunity (AIS5) has been previously mapped to the 4q27 region, within a linkage disequilibrium block encompassing KIAA1109, TENR, IL2, and IL21 genes [21]. So far, the most significant linkage outside the HLA region refers to rs13119723 (P = 2.0 × 10−7 in the KIAA1109 gene on chromosome 4q27). Zhernakova and coworkers [2] hypothesized that the KIAA1109/TENR/IL2/IL21 susceptibility region reported by van Heel and coworkers [21] might represent a general risk locus for multiple autoimmune diseases. Even though several CELIAC6 genomic variants have been reported [1, 2, 4, 21], this is the first time such a disease association is revealed for KIAA1109 c.2933T>C and c.4268_4269delCCinsTA. SLC9A4 c.1919G>A, HoxD12 c.418G>A, HoxB6 c.668C>A, and NCK2 c.745_746delAAinsG are also reported for the first time as celiac disease risk variants (Fig. 1).

Distribution of newly identified risk variants in Greek and Serbian populations. a HoxD12 c.418G>A is more abundant in pediatric celiac disease patients of Greek and Serbian descent, reaching statistical significance (**P < 0.01) in the Serbian population. b NCK2 c.745_746delAAinsG is more abundant in healthy individuals of Greek and Serbian descent, reaching statistical significance (*P = 0.03) in the Greek population
NCK2 c.745_746delAAinsG has not been annotated in either dbSNP or the 1000 Genomes Project/exome variant server data, and hence, it may be considered to be novel. NCK2 codes for Human Nck2 (hNck2), a 380-residue adapter protein consisting of three SH3 domains and one SH2 domain. Nck2 plays a pivotal role in connecting and integrating signaling networks constituted by transmembrane receptors, such as ephrinB and effectors critical for cytoskeletal dynamics and remodeling [22–25]. A transient Nck2/PINCH-1 association process has been also reported that may trigger rapid focal adhesion turnover during integrin signaling, mediating cell shape change and migration [26, 27]. We showed that the novel variant identified herein results in a single amino acid change (p.K249E), being this exact amino acid that is reported of fundamental importance during the abovementioned Nck2 SH3 domain protein-protein interactions (Fig. 2a) [22, 26, 27]. p.K249E could severely alter this network of polar interactions and affect the interaction between the two proteins (Fig. 2b). In 2014, Nadalutti et al. [28] observed that celiac patient IgA antibodies disturb the extracellular protein cross-linking function of transglutaminase 2, thus altering cell-extracellular matrix interactions and thereby affecting endothelial cell adhesion, polarization, and motility.

a View of the SH3-3/LIM4 binding interface as previously shown by NMR spectroscopy [26]. The Nck2 DH3 domain interface is shown in gray and the PINCH-1 LIM4 domain in green. K249 interacts with E233 and is part of a network connecting R192 of LIM4 and N250 of SH3. b Structural model of the mutant p.K249E and the potential effect in the interactions between SH3 and LIM4
In silico analyses
To ascertain whether the variants of interest have functional significance, in silico analysis was performed using the SIFT and PROVEAN algorithms [13, 14], as well as Variant Effect Predictor [16] and Regulome DB [8]. As summarized in Table 1, analyses yielded that all variants share an effect to protein structure, which in the case of NCK2 c.745_746delAAinsG is considered to be damaging. Regulome DB questioned a possible role of the variants of interest in terms of transcription factor binding sites, chromatin states, eQTLs, differentially methylated regions, validated functional SNPs and DNase sensitivity. All variants had minimum or no impact (Additional file 2: Table S1).
Replication analyses
Replication analyses were carried out in two cohorts to account for population differences. We found that NCK2 c.745_746delAAinsG is a novel variant that is more abundant in healthy individuals, reaching statistical significance (P = 0.03) in the Greek population. Moreover, HoxD12 c.418G>A, a frameshift variant, was more abundant in pediatric celiac disease patients, reaching statistical significance (P < 0.01) in the Serbian population. When celiac disease risk assessment is considered, it is important to note that apart from genotype data, records related to family history along with gender and country of residence should not be disregarded [29]. In both Greek and Serbian patients, SLC9A4 c.1919G>A and KIAA1109 c.2933T>C and c.4268_4269delCCinsTA were more abundant in patients; nevertheless, they failed to show statistical significance, possibly due to small sample sizes. All the studied variants satisfied the Hardy-Weinberg equilibrium. All variants were verified as non-frequent ones in agreement with PROVEAN’s scoring scheme, separating disease-associated variants from common ones [14]. Genotype frequencies (%) are summarized in Fig. 1 and Table 2.
Conclusions
In relation to disease diagnosis and prognosis, data interpretation requires an understanding of the variation in risk-associated variants. In celiac disease, in particular, this knowledge is still largely lacking. As whole-genome and/or whole-exome sequencing approaches begin to be employed in clinical care, the understanding of detected sequence variations on diagnosis (and prognosis) is still not a trivial undertaking. We envisage that the clinical implementation of next-generation sequencing will play a crucial role in delineating inter-individual variability and identification of novel variants towards improved therapeutic modalities. Herein, we propose a multi-step next-generation sequencing-based family genomics approach, similar to our previous conducted cancer genomics study [30], but piloted towards a complex genetic disease, such as celiac disease, to analyze a family trio of Greek descent to identify novel genomic variants of functional significance with the aim of understanding complex disease pathobiology. Recently, we have outlined the paradigm of pharmacometabolomics-aided pharmacogenomics in autoimmune diseases to address the interplay of genomic and environmental influences with information technologies to facilitate data analysis as well as sense- and decision-making on the basis of synergy between artificial and human intelligence. We propose that better-informed, rapid, and cost-effective “omics” studies need the implementation of holistic and multidisciplinary approaches [31].
References
Burton PR, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–78.
Zhernakova A, et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. 2011;7(2):e1002004.
Hunt KA, et al. Novel celiac disease genetic determinants related to the immune response. Nat Genet. 2008;40(4):395–402.
Romanos J, et al. Six new coeliac disease loci replicated in an Italian population confirm association with coeliac disease. J Med Genet. 2009;46(1):60–3.
Trynka G, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43(12):1193–201.
Gutierrez-Achury J, et al. Fine-mapping in the MHC region accounts for 18 % additional genetic risk for celiac disease. Nat Genet. 2015;47(6):577–8.
Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther. 2011;13(1):1.
Boyle AP, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22(9):1790–7.
Mizzi C, et al. Personalized pharmacogenomics profiling using whole-genome sequencing. Pharmacogenomics. 2014;15(9):1223–34.
Laird NM, Lange C. Family-based designs in the age of large-scale gene-association studies. Nat Rev Genet. 2006;7(5):385–94.
Drmanac R, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327(5961):78–81.
Hiltemann S, et al. CGtag: complete genomics toolkit and annotation in a cloud-based Galaxy. GigaScience. 2014;3:1.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(8):1073–81.
Choi Y, et al. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688.
Boycott KM, et al. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14(10):681–91.
McLaren W, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–70.
Krini M, et al. HLA class II high-resolution genotyping in Greek children with celiac disease and impact on disease susceptibility. Pediatr Res. 2012;72(6):625–30.
Stanković B, et al. HLA genotyping in pediatric celiac disease patients. Bosn J Basic Med Sci. 2014;14(3):171.
Walker-Smith J, et al. Revised criteria for diagnosis of coeliac disease. Report of working group of ESPGAN. Arch Dis Child. 1990;65:909–11.
Balding DJ. A tutorial on statistical methods for population association studies. Nat Rev Genet. 2006;7(10):781–91.
van Heel DA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007;39(7):827–9.
Liu J, et al. Structural insight into the binding diversity between the human Nck2 SH3 domains and proline-rich proteins. Biochemistry. 2006;45(23):7171–84.
Mayer BJ. SH3 domains: complexity in moderation. J Cell Sci. 2001;114(7):1253–63.
Koytiger G, et al. Phosphotyrosine signaling proteins that drive oncogenesis tend to be highly interconnected. Mol Cell Proteomics. 2013;12(5):1204–13.
Ran X, Song J. Structural insight into the binding diversity between the Tyr-phosphorylated human ephrinBs and Nck2 SH2 domain. J Biol Chem. 2005;280(19):19205–12.
Vaynberg J, et al. Structure of an ultraweak protein-protein complex and its crucial role in regulation of cell morphology and motility. Mol Cell. 2005;17(4):513–23.
Velyvis A, et al. Structural and functional insights into PINCH LIM4 domain-mediated integrin signaling. Nat Struct Mol Biol. 2003;10(7):558–64.
Nadalutti CA, et al. Celiac disease patient IgA antibodies induce endothelial adhesion and cell polarization defects via extracellular transglutaminase 2. Cell Mol Life Sci. 2014;71(7):1315–26.
Liu E, et al. Risk of pediatric celiac disease according to HLA haplotype and country. N Engl J Med. 2014;371(1):42–9.
Karageorgos I, et al. Identification of cancer predisposition variants in apparently healthy individuals using a next-generation sequencing-based family genomics approach. Hum Genomics. 2015;9(1):1.
Katsila T, et al. Pharmacometabolomics-aided pharmacogenomics in autoimmune disease. EBioMedicine. 2016;5:40–5.
Acknowledgements
Not applicable
Funding
This study was funded by the European Commission (RD-CONNECT; FP7-304555) research grant to GPP.
Availability of data and materials
The datasets during and/or analyzed during the current study are available from the corresponding author on request.
Authors’ contributions
TK and GPP conceived and designed the study. AB, AP, BS, GN, AJ, EM, AS, AIL, VS, AT, NG, SG, ZZ, MK, MK, KS, and TK carried out the PCR-based conventional Sanger resequencing approach. BAP and RD supervised the next-generation sequencing analyses. TK carried out the in silico analyses. BRA, NC, ER, JB, KP, GC, BZ, NR, RD, SP, TK, and GPP drafted the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare the following competing interests: BAP and RD are employees of the Complete Genomics Inc. (Mountain View, CA, USA).
Consent for publication
Not applicable
Ethics approval and consent to participate
A family trio of Greek descent was recruited for this study (informed consents have been obtained). The Ethics Committee of University Children’s Hospital, University of Belgrade, and the Review Board of “Aghia Sophia” Children’s Hospital have approved the study.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
A seven-member Greek family has been recruited (informed consents have been obtained), and a trio analysis (III-1, III-2, IV-3) has been performed using the celiac disease model. (PNG 136 kb)
Additional file 2:
Regulome DB questioned a possible role of the variants of interest in terms of transcription factor binding sites, chromatin states, eQTLs, differentially methylated regions, validated functional SNPs and DNase sensitivity. All variants had minimum or no impact. (XLSX 3.62 mb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Balasopoulou, A., Stanković, B., Panagiotara, A. et al. Novel genetic risk variants for pediatric celiac disease. Hum Genomics 10, 34 (2016). https://doi.org/10.1186/s40246-016-0091-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-016-0091-1